

Abstract
The interaction between proteins is one of the most important features of protein functions. Behind protein–protein interactions there are protein domains interacting physically with one another to perform the necessary functions. Therefore, understanding protein interactions at the domain level gives a global view of the protein interaction network, and possibly of protein functions. Two research groups used yeast two-hybrid assays to generate 5719 interactions between proteins of the yeast Saccharomyces cerevisiae. This allows us to study the large-scale conserved patterns of interactions between protein domains. Using evolutionarily conserved domains defined in a protein–domain database called PFAM (http://PFAM.wustl.edu), we apply a Maximum Likelihood Estimation method to infer interacting domains that are consistent with the observed protein–protein interactions. We estimate the probabilities of interactions between every pair of domains and measure the accuracies of our predictions at the protein level. Using the inferred domain–domain interactions, we predict interactions between proteins. Our predicted protein–protein interactions have a significant overlap with the protein–protein interactions (MIPS: http://mips.gfs.de) obtained by methods other than the two-hybrid assays. The mean correlation coefficient of the gene expression profiles for our predicted interaction pairs is significantly higher than that for random pairs. Our method has shown robustness in analyzing incomplete data sets and dealing with various experimental errors. We found several novel protein–protein interactions such as RPS0A interacting with APG17 and TAF40 interacting with SPT3, which are consistent with the functions of the proteins.
[Supplementary material is available online at http://www.genome.org and http://www-hto.usc.edu/∼msms/ProteinInteraction.]
With the advancement of genomic technology and genome-wide analysis of organisms, more and more organisms are being studied extensively for gene expression on a global scale. Expression profiling is now being used increasingly to analyze gene functions or to functionally group genes on the basis of their expression profiles (Lockhart and Winzeler 2000). After the completion of the genome sequence of Saccharomyces cerevisiae (Goffeau et al. 1996), a budding yeast, many researchers have undertaken the task of functionally analyzing the yeast genome, comprising ∼6280 proteins (YPD), of which roughly one-third do not have known functions (Mewes et al. 2002). Genes can be clustered on the basis of similar expression profiles. This makes it possible to assign a biological function to genes, depending on the functions of other genes in the cluster (Eisen et al. 1998). However, expression profiling gives an indirect measure of a gene product's biological and cellular function. A more complete study of an organism could possibly be achieved by looking at not only the mRNA levels but also the proteins they encode. It is well known that mRNA levels alone are not sufficient to group genes into different functions, because not all mRNAs end up being translated. Most biological functions within a cell are carried out by proteins and most cellular processes and biochemical events are ultimately achieved by interactions of proteins with one another. Thus, it is important to look at protein expression and their interactions simultaneously.
Affinity chromatography, two-hybrid assay, copurification, coimmunoprecipitation, and cross-linking are some of the tools used to verify proteins that are associated physically with one another. Among these techniques, the two-hybrid assay has been used widely to analyze protein–protein interactions in Saccharomyces cerevisiae (Ito et al. 2000, 2001a; Uetz et al. 2000). Their protein interaction profiles have made it possible to look at the interaction networks comprising a large number of proteins and to also functionally classify proteins of unknown function. Uetz et al. (2000) used two different approaches in their two-hybrid experiments. The first was a protein array approach with 192 yeast proteins as bait, Gal4–DNA-binding domain fusions, and ∼6000 yeast transformants as prey, Gal4-activation domain fusions. The second, an interaction sequence tag (IST) approach, used high-throughput screens of an activation domain library encoding ∼6000 yeast genes that were pooled. All yeast proteins were cloned into DNA-binding domain vectors. Of the 6144 yeast ORF PCR products, 5345 were successfully cloned. Their first approach revealed 281 interactions, with less stringent selection criteria, using HIS3. The second approach revealed 692 interactions with the more stringent URA3 selection method. Ito et al. (2001a) used a similar method and reported 4549 interactions among 3278 proteins. Some interactions in both data sets were repeated (bait and prey exchanged). They imposed a more rigorous selection criterion including four reporter genes, ADE2, HIS3, URA3, and MEL1, to minimize false positives due to promoter-specific activation. All of these genes have Gal4-responsive promoter.
Computational methods have been developed to predict protein–protein interactions. Those approaches include the Rosetta stone/gene fusion method (Enright et al. 1999; Marcotte et al. 1999a), the phylogenetic profile method (Pellegrini et al. 1999) and the method combining multiple sources of data (Marcotte et al. 1999b). Other computational methods to predict protein–protein interaction have been presented on the basis of different principles, including the interaction domain pair profile method (Rain et al. 2001; Wojcik and Schachter 2001) and the support vector machine learning method (Bock and Gough 2001). Gomez et al. (2001) developed probabilistic models for protein–protein interactions. Sprinzak and Margalit (2001) analyzed over-represented sequence-signature pairs among protein–protein interactions.
In our study, we use the protein–protein interaction (PPI) data sets of Uetz and Ito to predict domain–domain interactions (DDI) in yeast proteins. The protein-domain information is obtained from a protein-domain family database called PFAM (Bateman et al. 2000). Because every protein can be characterized by either a distinct domain or a combination of domains, understanding domain interactions is crucial to understanding the nature and extent of biomolecular interactions. Our study predicts probable domain–domain interactions solely on the basis of the information of protein–protein interactions. Because proteins interact with one another through their specific domains, predicting domain–domain interactions on a global scale from the entire protein interaction data set make it possible to predict previously unknown protein–protein interactions from their domains. Thus, domain interactions extend the functional significance of proteins and present a global view of the protein–protein interaction network within a cell responsible for carrying out various biological and cellular functions.
It is known that the yeast two-hybrid assay is not accurate in determining protein–protein interactions, and the interaction data used in our study certainly contain many false positive and false negative errors (Legrain and Selig 2000; Hazbun and Fields 2001; Mrowka et al. 2001). Taking into account these errors, we apply the Maximum Likelihood approach to estimate the probability of domain–domain interactions. We have also taken into account multiplicity of observations in the two data sets as evidenced by exchanged baits and preys, repeated interactions, and synonymously used gene names. To assess the accuracy of our method, we predict protein–protein interactions using the inferred domain–domain interactions, and compare them with the observed interactions. The following results are obtained: (1) Our method has shown robustness in analyzing incomplete data sets and dealing with various experimental errors, and we achieve 42.5% specificity and 77.6% sensitivity using the combined Uetz and Ito data. The relative low specificity may be caused by the fact that the observed protein–protein interactions in the Uetz and Ito combined data represent only a small fraction of all of the real interactions. (2) Comparing our predicted protein–protein interactions with the MIPS protein–protein interactions obtained by methods other than the two-hybrid assays, we show that the prediction rate of our method is about 100 times better than that of a random assignment. (3) We also compare the gene expression profile correlation coefficients of our predictions with those of random protein pairs, and our predictions have a higher mean correlation coefficient. (4) Finally, we check for biological significance of our novel predictions, and find several interesting interactions such as RPS0A interacting with APG17 and TAF40 interacting with SPT3, which are consistent with the functions of the proteins. A complete description of our model and the results are given in the sections below.
RESULTS
The two sources of protein–protein interactions are listed in Table 1. The domains include PFAM domains, superdomains, and merged domains. A protein without any domain information is treated as a superdomain. If two or more PFAM domains always coexist in proteins, they are merged into one domain.
Table 1.
Number of Proteins, Domains, and PPI in the Uetz, the Ito, the Uetz and Ito Combined, and the Overlap Data Sets
| Proteins | Domains | PPI | |
|---|---|---|---|
| Uetz | 1337 | 1643 | 1445 |
| Ito | 3277 | 3685 | 4475 |
| Combined | 3729 | 4131 | 5719 |
| Overlap | 855 | 1179 | 201 |
A domain is a Pfam domain, a super-domain, or a merged domain.
We apply both the Association method and the MLE method to estimate domain–domain interactions. However, it is difficult to estimate the accuracies of our prediction at the domain level, because very few domain–domain interactions are known. We use the inferred domain–domain interactions to predict protein–protein interactions and assess the prediction accuracies at the protein level. The accuracies of the predictions are measured by specificity and sensitivity. The specificity, denoted as SP, is defined as the ratio of the number of matched interactions between the predicted set and the observed set over the total number of predicted interactions. The sensitivity, denoted as SN is defined as the ratio of the number of matched interactions over the total number of observed interactions.
Results of the Association Method
Two proteins are predicted to be interacting if there exist two domains, one from each protein, whose association value is greater than a predefined threshold. We achieve 55.5% specificity and 55.0% sensitivity by setting the threshold at 0.65 using the combined data sets.
Results of the MLE Method
We apply the EM algorithm recursively to derive domain–domain interaction probabilities from the combined data of Uetz and the Ito with fixed false positive (fp) and false negative (fn). It was estimated in Hazbun and Fields (2001) that each protein interacts with about t = 5 to 50 proteins. For N = 6359 yeast proteins and t = 5, it gives a total number of 15,898 real interaction pairs.
Therefore,
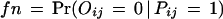 |
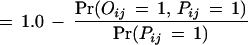 |
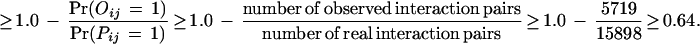 |
Similarly, we can estimate fp. There are a total of N(N + 1)/2 ≈ 2 E7 protein pairs of which about t × N/2 are potentially interacting pairs. Therefore,
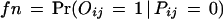 |
 |
 |
Mrowka et al. (2001) estimated that perhaps up to 90% of the total 5719 protein interactions in Uetz's and Ito's combined data are not correct interactions. That gives a false positive rate of about fp = 2.5E – 4. Two proteins are predicted to interact if their interaction probability is greater than a certain threshold. Using the combined data with fp = 2.5E – 4 and fn = 0.80, we achieve SP = 42.5% and SN = 77.6% by setting the threshold at 0.80. The reason for the relatively low specificity is that the protein–protein interactions in the Uetz and Ito combined data set contain only a very small fraction of the potential protein–protein interactions. This can be seen from the small overlap between the Uetz's data set and the Ito data set. Also, many interactions in the MIPS database are not in the combined data set. A reasonable program should predict more interactions than the number of observed interactions, which results in relatively low specificity.
Figure 1 shows the relationship between sensitivity and specificity for both the association method and the MLE method with fp = 2.5E – 4 and fn = 0.80. The MLE approach outperforms the association method. For a given specificity, the sensitivity of the MLE approach is always higher than that of the association method.
Figure 1.

Comparison of specificity and sensitivity of the prediction rates for the association method and the maximum likelihood method.
Figure 2 shows that the specificity and the sensitivity are quite similar for various combinations of fp and fn values. This feature indicates that the MLE method is robust with respect to experimental errors, and is capable of predicting the core interactions in the data.
Figure 2.

Comparison of specificity and sensitivity of the prediction of protein–protein interactions by the maximum likelihood method for four different values of fp and fn.
Validations of the MLE Predictions
The statistical significance of the predictions can be measured by comparing the predicted interactions with the protein–protein interactions in the MIPS database and the gene expression profiles.
Comparing With MIPS
We use the MIPS physical interaction pairs (Mewes et al. 2002) to test our predictions. There are 2575 entries in the MIPS protein physical interaction table. Excluding those interactions overlapping with the Uetz and Ito interaction data, we obtain a test data set of 1417 MIPS interactions. We then measure whether the MLE method can predict them.
Our method gives the probability of interaction for each protein pair. The larger the probability, the more likely the interaction is real. Table 2 shows the matching numbers between the 1417 interactions and our predicted interactions with probability greater than some threshold. If our approach is reasonable, a real interaction should more likely be in the high probability categories than random pairs are. To measure this excess, we calculate the ratio of the fraction of the predicted protein pairs in the test data set with those in all protein pairs. We denote this quantity by Fold:
 |
in which L is the total number of protein pairs, n is the number of protein pairs with interaction probability greater than some threshold, K = 1417, and k0 is the number of matching protein pairs between the 1417 interactions in the test data set and the n predicted interactions.
Table 2.
Number of Matched Protein Pairs Between the Predictions
| Threshold | Prediction | Train | MIPS | MIPS1 | Fold | Z score | P value |
|---|---|---|---|---|---|---|---|
| All | 20221620 | 5719 | 2575 | 1417 | 1 | — | |
| >0.00 | 125435 | 5717 | 1263 | 106 | 12.63 | 33.70 | 3.88e-249 |
| ≥0.20 | 23182 | 5154 | 1074 | 51 | 40.36 | 44.25 | — |
| ≥0.40 | 16287 | 4847 | 993 | 47 | 58.61 | 51.59 | — |
| ≥0.60 | 12748 | 4647 | 933 | 43 | 75.73 | 56.31 | — |
| ≥0.80 | 10441 | 4437 | 882 | 40 | 95.05 | 61.01 | — |
| ≥0.975 | 9413 | 4289 | 845 | 35 | 97.45 | 57.80 | — |
(Prediction) fp = 2.5E-4, fn = 0.80, the training set (Train), the MIPS data (MIPS), and the MIPS excluding the training data (MIPS1), respectively. The corresponding statistics (Fold, Z score, and P value) are also given.
Table 2 shows that the fold number increases as the threshold increases. This is consistent with our prediction. The 1417 protein pairs in the test data set are ∼97 times more likely to have interacting probability >0.975 than random pairs.
To statistically test whether the 1417 protein pairs in the test data set are more likely to have interaction probability greater than the threshold, we use the standard Z-score
 |
where
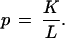 |
Z has an approximate standard normal distribution under the null hypothesis. Both Z-scores and P-values are given in Table 2.
It should be noted that setting the threshold to 0.975 gives 9413 – 4289 = 5124 novel protein–protein interactions. The matches between these interactions and the 1417 MIPS interactions excluding the Uetz and Ito PPIs are a mere 35. However, the small number of overlaps is probably due to the large size of the whole-yeast protein interactions and errors in the two-hybrid experiments.
Comparing With Gene Expression Profiles
Recently, it was noted that genes with similar expression profiles are likely to encode interacting proteins (Ge et al. 2001; Grigoriev 2001). We study the distribution of correlation coefficients for protein pairs with predicted interaction probability greater than a certain threshold. We use the gene expression data of Eisen et al. (1998), which contain 2467 ORFs with 79 data points. Figure 3 gives the distributions of the pairwise correlation coefficients for all gene pairs, our predicted protein pairs with probability ⩾0.975, the Ito and Uetz original data, and the MIPS interaction data.
Figure 3.

Distributions of the pairwise correlation coefficients of gene expression profiles for interaction proteins in all gene pairs, the predicted interactions with threshold, the combined Uetz and Ito data, and the MIPS data.
To test whether the mean expression correlation coefficients for gene pairs in our predicted and experimentally verified interacting protein pairs are significantly higher than that for all the gene pairs, we calculate the T-score and the P-value for the null hypothesis of no difference between the sample (the MIPS and our prediction) mean and the mean of all gene pairs. The results are listed in Table 3. The T-scores are calculated as the standard two sample T-test statistics:
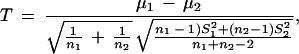 |
where μ is the mean of samples, and
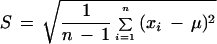 |
is the standard deviation of the samples.
Table 3.
Summary Statistics of Distribution of the Correlation Coefficient Between the Expression Profiles of Two Interacting Proteins (With Gene Expression Profiles From Different Data Sets and Predictions (fp = 2.5E-4, fn = 0.80) With Different Probability Thresholds
| Pairs | Pairs | Mean | Std | T-score | P value |
|---|---|---|---|---|---|
| All ORFs | 3036880 | 0.0410 | 0.2444 | 0.0000 | 5.000e-01 |
| ≥0.20 | 5333 | 0.0657 | 0.2529 | 7.3985 | 7.186e-14 |
| ≥0.40 | 3692 | 0.0774 | 0.2549 | 9.0661 | 6.541e-20 |
| ≥0.60 | 2764 | 0.0858 | 0.2613 | 9.6445 | 2.766e-22 |
| ≥0.80 | 2205 | 0.0832 | 0.2609 | 8.1043 | 2.788e-16 |
| ≥0.975 | 1959 | 0.0841 | 0.2628 | 7.8134 | 2.917e-15 |
| Uetz + Ito | 1307 | 0.0841 | 0.2600 | 6.3775 | 9.292e-11 |
| MIPS | 1100 | 0.1646 | 0.2721 | 16.8504 | 5.860e-64 |
Figure 3 and Table 3 show that the mean correlation coefficient for protein pairs with interacting probability greater than a certain threshold is significantly higher than the mean correlation coefficient for random pairs.
Applying the MLE Method on the MIPS Data
For comparison, we also apply our probabilistic model and the MLE method on the MIPS interaction data. We set fp = 0 and fn = 0.95 because the protein–protein interactions in MIPS are individually verified and thus should have a very small false positive rate. We predict probabilities for domain–domain interactions. To assess the accuracies of the predictions at the protein level, we compute probabilities for protein–protein interactions. Measured by sensitivity and specificity, the MLE method outperforms the Association method. As expected, the overlap between our novel predictions of protein–protein interactions and the yeast two-hybrid data is small. For example, the 4671 novel interactions predicted with a threshold have only 44 matches with the yeast two-hybrid data. Given the small overlap between the Uetz and Ito combined data and the MIPS data, this result is conceivable. All of the results are shown in the supplementary data.
Biological Significance of Novel Predictions
Novel protein–protein interactions are predicted from our probabilistic model. The top 17 predictions with probability >0.95 are listed in Table 4. We observe four interactions in which one of the interactors has unknown function. ORF YOL083W and ORF YNL078W are shown to interact with a transcription factor, TFB1(TFIIH subunit), and a serine/threonine kinase, MRK1. It is possible that the two ORFs have some role to play in the transcription machinery associated with RNA Pol II or DNA repair (TFIIH is also involved in DNA repair) and the kinase pathway of MRK1, respectively.
Table 4.
Some Novel Predictions
| Protein | Function | |
|---|---|---|
| Interactor I | MRK1 | Ser/Thr Kinase |
| Interactor II | YNL078W | unknown |
| Interactor I | CTT1 | Catalase T/cytosolic |
| Interactor II | PEX14 | Interacts with peroxisome membrane receptors (docking interactions)/PMP* |
| Interactor I | LAP4 | Lysosomal/vacuolar aminopeptidase |
| Interactor II | YHR113W | similar to Lap4p |
| Interactor I | SPS18 | Transcription factor, sporulation specific/nuclear |
| Interactor II | YIP1 | Vesicular transport, fusion events/G*, IMP* |
| Interactor I | TFB1 | RNA Pol II transcription, subunit of TFIIH |
| Interactor II | YOL083W | Unknown |
| Interactor I | DPM1 | Protein modification/ER*, IMP* |
| Interactor II | SPS18 | Transcription factor, sporulation specific/nuclear |
| Interactor I | SNZ1 | biosynthetic enzyme, role in cell stress |
| Interactor II | SNZ1 | biosynthetic enzyme, role in cell stress |
| Interactor I | APG17 | authophagy, Vesicular transport |
| Interactor II | RPS0A | Ribosomal protein, RNA-binding protein/Cytoplasmic |
| Interactor I | APG17 | authophagy, Vesicular transport |
| Interactor II | RPS0B | Ribosomal protein, RNA-binding protein/Cytoplasmic |
| Interactor I | SNO3 | putative vitamin biosynthetic enzyme, role in cell stress |
| Interactor II | SNZ3 | similar function as SNZ1 |
| Interactor I | SNO2 | similar function as SNO3 |
| Interactor II | SNZ1 | biosynthetic enzyme, role in cell stress |
| Interactor I | SIW14 | Ser/Thr phosphatase, cell cycle control |
| Interactor II | YCR095C | unknown |
| Interactor I | SIW14 | Ser/Thr phosphatase, cell cycle control |
| Interactor II | SIW14 | as above |
| Interactor I | PRS5 | amino acid and nucleotide metabolism/cytoplasmic |
| Interactor II | RPS3 | similar function as PRS5 |
| Interactor I | PRS5 | as above |
| Interactor II | PRS5 | as above |
| Interactor I | PRS5 | as above |
| Interactor II | PRS1 | similar function as PRS5 |
| Interactor I | TAF40 | RNA Pol II transcription, TFIID component, TBP associated |
| Interactor II | SPT3 | component of nucleosomal HAT complex, TBP associated |
(HAT) Histone acetyl transferase, (ER) Endoplasmic reticulum, (*G) Golgi, (*PMP) Peripheral membrane protein, (*IMP) Integral membrane protein. The functional annotations are obtained from YPD.
Some of our predictions, such as CTT1-PEX14 and TAF40-SPT3 interactions, are significant. PEX14 facilitates docking interactions at the peroxisomal membrane receptors and catalase T is an oxidative enzyme that degrades hydrogen peroxide. Because peroxisomes release enzymes that reduce oxygen stress in the cell, there is a logical interaction between the two proteins. An interesting finding was the SPT3 interaction with TAF40. SPT3 is a component of the nucleosomal HAT (histone acetyl transferase) complex and is TBP associated. TAF40 is also TBP associated and is a transcription factor in Pol II transcription. Thus, at some point between histone acetylation, which facilitates the transcription machinery to bind to DNA and the recruitment of transcription factors to DNA, we find an interaction between the two processes.
Some predictions may indicate previously unknown protein functions such as SPS18, the sporulation-specific transcription factor interacting with YIP1 and DPM1. Both YIP1 and DPM1 are integral membrane proteins and localized at ER and Golgi. Their functions in vesicular transport and protein modification suggest that the sporulation-specific genes activated by SPS18 may be recruited to membranes to form the spore wall and therefore interact with YIP1 and DPM1. SPS18 may be involved in this process.
The rest of our top novel predictions involve interactions between members of the same gene family, such as PRS5, PRS3, PRS1, or between members of two separate gene families, such as the SNO and SNZ gene family. From literature sources, PRS1 and PRS3 are known to interact strongly with each other and PRS5 has interactions with PRS2 or PRS4. Here, we show interactions between PRS5 and PRS3, PRS5 and PRS1, and PRS5 with itself. PRS is a phosphoribosyl pyrophosphate synthetase involved in amino acid and nucleotide metabolism. Each of the SNZ genes has SNO genes upstream, and members of these two gene families are highly conserved and coregulated. Genes of both families are involved in cellular response to nutrient stress and, hence, interactions between the two families is obvious from the biological point of view.
We also observe interactions of two ribosomal proteins, RPS0A and RPS0B, with APG17, a protein involved in vesicular transport and autophagy. However, because pairwise interactions do not give a complete functional role of a protein, we looked at all interactions of APG17 and RPS0A separately in Table 5 and Table 6. We observe RPS0A interactions with APG17, BBP1, YDL100C, and ILV1 with probability greater than 0.5. BBP1 is a spindle-pole body protein and is known to bind Bfr1p (from literature sources), which is involved in vesicular transport of secretory proteins and is localized on a polyribosome–mRNP complex. APG17 is a component of the APG complex of proteins involved in targeting proteins to vacuoles/lysosomes under starvation conditions. We predict binding of BBP1 and BFR1 (from literature) to RPS0A and also binding of APG17 to RPS0A. Thus, binding of two different vesicular transport proteins to ribosomal proteins may or may not be part of one complex, depending on cellular environment. We predict several interactions for APG17 listed in Table 5. We predict nine ORFs of unknown function to have interaction with APG17. It is possible that these ORFs are involved in a APG protein-dependent vesicular transport system. We predict APG17 interacting with SEC9, another protein functioning in vesicular transport, and SPO20, involved in sporulation, both of which are localized at the plasma membrane. We also predict APG17 interacting with four other proteins, LAT1, DOG1, DOG2, and KGD2, all of which are involved in carbohydrate metabolism and energy generation. Because APG17 targets proteins to vacuoles and lysosomes during nutrient stress conditions, it is possible that it targets some other proteins involved in energy generation (under starvation conditions) to mitochondria and some proteins involved in spore wall formation to plasma membrane. Thus, experimental verifications of some of our significant predictions may throw light on cellular processes and explain the roles of proteins that may be plausible links between distinct pathways.
Table 5.
Novel Predictions for APG17 Interactions With High Probability
| Protein | Localization | Function (cellular role or biochemical) |
|---|---|---|
| APG17 | — | authophagy, Vesicular transport |
| RPS0A | Cytoplasmic | Ribosomal protein, RNA-binding protein |
| RPS0B | Cytoplasmic | Ribosomal protein, RNA-binding protein |
| YBR197C | unknown | unknown |
| YPL077C | unknown | unknown |
| YAP7 | — | Transcription factor (Pol II), leucine zipper family |
| CIN5 | Nuclear | Transcription factor (Pol II), leucine zipper family |
| YMR031C | unknown | unknown |
| YKL050C | unknown | unknown |
| YBR270C | unknown | unknown |
| YJL058C | unknown | unknown |
| YMR124W | unknown | unknown |
| PLO1 | unknown | unknown |
| PLO2 | unknown | unknown |
| SPO20 | Plasma membrane | sporulation |
| LAT1 | Mitochondrial | Carbohydrate metabolism, Energy generation |
| SEC9 | Plasma membrane | Vesicular transport (vesicle docking and secretion) |
| DOG1 | — | Carbohydrate metabolism, Hydrolase |
| DOG2 | — | Carbohydrate metabolism, Hydrolase |
| KGD2 | Mitochondrial | Carbohydrate metabolism, Energy generation, Oxidoreductase |
The functional annotations are obtained from YPD.
Table 6.
Novel Predictions for RPS0A Interactions With High Probability
| Protein | Localization | Function (cellular role or biochemical) |
|---|---|---|
| RPS0A | Cytoplasmic | Ribosomal protein, RNA-binding protein |
| APG17 | — | authophagy, Vesicular transport |
| BBP1 | Nuclear | protein in spindle pole body, mitosis |
| YDL100C | Cytoplasmic | similarity with E. coli ArsA ATPase in small molecule transport |
| ILV1 | Mitochondrial | Amino acid metabolism, biosynthesis of amino acid, lyase |
The functional annotations are obtained from YPD.
DISCUSSION
We apply a probabilistic model to derive domain–domain interactions from protein–protein interactions observed in two-hybrid assays. We predict protein–protein interactions from the derived domain–domain interactions, and assess the accuracy of our model at the protein level in three ways as follows: (1) comparing the prediction results with the original experimental data, (2) comparing the prediction results with the MIPS protein–protein interactions derived by methods other than the two-hybrid assays, and (3) comparing the mean gene expression correlation coefficient for the predicted interacting protein pairs with that for random protein pairs.
Our probabilistic model and the Maximum Likelihood Estimation method are robust in handling experimental errors. The structure of our probabilistic model allows us to incorporate various kinds of protein–protein interaction data, even from different organisms, to infer domain–domain interactions. As more and more protein–protein interactions are experimentally determined, the prediction accuracy of our method will improve substantially.
Statistics show that the prediction rate of our method is ∼100 times better than that of a random assignment in predicting the protein–protein interactions in MIPS. Although the statistics are significant, the prediction ratio, 35/(9413 – 4289) = 0.68% does not seem to be practically useful. A possible reason is that the size of the protein interaction network is huge. It is known that every experimental method is biased to certain kinds of proteins and interactions. For example, the Uetz and the Ito original experimental results have a very small number of overlaps with the interactions from other methods. It is possible that some of our novel predictions are real, bias to particular proteins, and cannot be verified by other methods.
Another explanation for the small overlaps between the MIPS data and the yeast two-hybrid data is that the yeast two-hybrid assays are not reliable and contain high false positives (Mrowka et al. 2001). Even though this may be true, the mean correlation coefficient for our predicted protein pairs is significantly higher than that of random pairs. These studies validate our probabilistic model, and prove that the interaction probability we have derived is a good estimation.
The basic assumptions of our model ignore the following biological factors. First, our model assumes the independence of domain–domain interactions. In fact, whether two domains interact or not may depend on other domains in the same protein or other environmental conditions. Although we have identified domains that coexist in proteins and merged them as one domain, there certainly exist many domains whose functions depend on other domains in the same protein. Second, the idea of using domain–domain interactions to predict protein–protein interactions assumes that some subunits with special structure are essential to protein–protein interactions. These subunits may be different from PFAM domains obtained through multiple alignments. Furthermore, compared with functionally annotated PFAM-A domains, PFAM-B domains are shorter and less known, so the roles that they play in protein interactions may not be the same, but in our model, we use them in the same level as the PFAM-A domains.
It has been known that protein–protein interactions have time and space constraints. Two proteins that contain potentially interacting domains may not interact with each other because they may be expressed at different times during the cell cycle, or may be located at different cell compartments. Protein–protein interactions not only depend on structures, but also depend on other environmental conditions. Even two proteins with the same domain structure may have different interaction behavior with other proteins.
It is believed that the experimental protein–protein interaction data is just a small fraction of the whole protein interaction network. The incompleteness of current data makes it difficult to derive domain interaction information. The comparison of two data sets shows very small overlaps between them. This may explain that the size of the protein interaction network is much bigger than these two experimental data, and thus, they have only a small part of overlaps. On the other hand, it is known that the experimental data contain many errors. The exact error rate has to be assessed by using other techniques.
METHODS
Source of Data
We obtain the protein–domain relationship for yeast proteins from PFAM (Bateman et al. 2000), a protein domain family database. PFAM contains multiple sequence alignments for each domain family and uses profile-hidden Markov models to find domains in new proteins. The latest version, PFAM 6.5 (http://pfam.wustl.edu/) contains alignments for 2929 protein domain families in PFAM-A and 57891 domain families in PFAM-B. The protein sequences are derived from SWISSPROT 39 and TrEMBL 14 databases (Bairoch and Apweiler 2000). Domains in PFAM-A are well defined because the corresponding multiple alignments and hidden Markov models have been checked, and most of the domains have been assigned to functions. PFAM-B was generated automatically by programs and includes ProDom domains (Corpett et al. 2000) not covered by PFAM-A. We download both PFAM-A and PFAM-B from the PFAM ftp site. We extract domains along with the Saccharomyces cerevisiae gene names and obtain domain information from both PFAM-A and PFAM-B. In this process, we associate the yeast gene accession numbers with the corresponding SWISSPROT and TrEMBL accession numbers to locate all yeast genes. Proteins for which no domain information is available are classified as superdomains, and those domains that always coexist in proteins are merged as one domain as well.
Association Method
A simple measure of interaction between domain Dm and domain Dn (Sprinzak and Margalit 2001) is the fraction of interacting protein pairs among all of the protein pairs containing the domain pair (Dm, Dn). Let Imn be the number of interacting protein pairs containing the domain pair (Dm, Dn), and Nmn be the total number of protein pairs containing (Dm, Dn). The association measure is given by
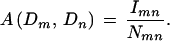 |
The method relies on the accuracy of the observed data, and in this case, the observed interactions are treated as the real interactions. However, this method computes domain–domain interactions locally. By locally, we mean that it ignores other domain–domain interaction information between the protein pairs and, thus, does not make full use of all of the available information.
For example, proteins PI, Pj, and Pk contain domains {Da, Dx}, {Dy, Db}, and {Dy, Dc}, respectively. Domains Dx and Dy do not appear in any other proteins. If we observe Pi interacting with Pj and Pi interacting with Pk, then A(Dx, Dy) = 2/2 = 1. Obviously, this kind of local method ignores other domain–domain interactions such as Da interacting with Db and Dc. In fact, it is possible that Dx and Dy do not interact with each other but Da interacts with both Db and Dc. Therefore, to infer a domain–domain interaction, other related domain–domain interactions have to be taken into account. This means that interactions of other proteins containing domains Da, Db, or Dc are to be included, and thus, more domains and proteins are involved. Iterating this idea, eventually all proteins and all domains are related and need to be taken into account.
The association method also ignores experimental errors. Following, we develop a global approach using a Maximum Likelihood Estimation method to incorporate all of the proteins and domains, as well as experimental errors.
Maximum Likelihood Estimation
Let D1,…, DM denote the M domains, and P1,…, PN denote the N proteins. Let Pij denote the protein pair of Pi and Pj, and Dij denote the domain pair of Di and Dj. Let Pij be the set of domain pairs formed by proteins Pi and Pj. For example, assume that protein P1 contains domains {D1, D2, D3} and protein P2 contains domains {D1, D4}. Then P12 = {D11, D12, D13, D14, D24, D34}.
We treat protein–protein interactions and domain–domain interactions as random variables. Let Pij = 1 if protein Pi and protein Pj interact with each other and Pij = 0 otherwise. Similarly, let Dmn = 1 if domain Dm interacts with domain Dn and Dmn = 0 otherwise. We make the following assumptions throughout the work.
Assumption I
Domain–domain interactions are independent, which means that the event that two domains interact or not does not depend on other domains.
Assumption II
Two proteins interact if and only if at least one pair of domains from the two proteins interact.
Under the above assumptions, we have
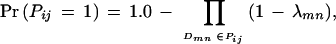 |
1 |
in which λmn = Pr(Dmn =1) denotes the probability that domain Dm interacts with domain Dn.
We consider two types of experimental errors in the two-hybrid assays [another widely used definition of false positives is the ratio of the number of incorrect interactions over the number of predicted interactions (Mrowka et al. 2001)], false positives, in which two proteins do not interact in reality but were observed to be interacting in the experiments, and false negatives, in which two proteins interact in reality but were not observed to be interacting in the experiments. The false positive rate is denoted as fp and the false negative rate is denoted as fn. Let Oij be the variable for the observed interaction result for proteins Pi and Pj: Oij = 1 if the interaction is observed and Oij = 0 otherwise. Then
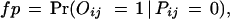 |
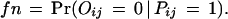 |
Thus, the probability for the observed protein–protein interaction is
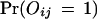 |
2 |
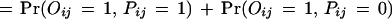 |
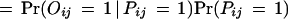 |
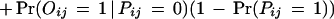 |
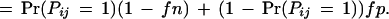 |
The likelihood function, i.e., the probability of the observed whole proteome interaction data is
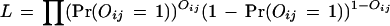 |
3 |
where
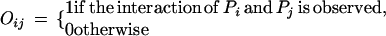 |
The likelihood L is a function of θ = (λmn, fp, fn). In the following, we fix fp and fn.
We estimate θ using a maximum likelihood estimation (MLE) approach. Because of the high dimensionality of θ, it is difficult to maximize L directly. We develop an Expectation-Maximization (EM) algorithm (Dempster et al. 1977) to solve the problem.
The idea of EM algorithms for a general problem is described as follows. To obtain the MLE of the parameters, we supplement the observed data with data that are not observable (called the missing data). The observed data together with the missing data form the complete data. In an EM algorithm, we distinguish the observed data Y from the complete data Z. We can obtain the MLE of the unknown parameters θ on the basis of the complete data Z. We should also be able to calculate the expectation of Z given the observed data. There are two steps in an EM algorithm, the expectation (E) step and the maximization (M) step. In the E step, we calculate the expectation of the complete data Z given the observed data Y, Ẑ = E(Z‖Y,θ(t–1)). In the M-step, we obtain the MLE of θ, θ(t), based on Ẑ. Thus, we obtain a recursive formula to estimate parameters θ.
Next, we adapt the EM algorithm to our problem. The observed data is the experimentally observed interactions O = {Oij = oij, i ⩽ j}. The complete data includes all the domain–domain interactions for each protein–protein pair. Let Am be the set of proteins containing domain Dm. Let Nmn be the total number of protein pairs between Am and An. To estimate λmn, the probability that domain Dminteracts with domain Dn, we need information on the interaction status for protein pairs between Am and An. Define the complete data as (O, D), in which O is given above and D = {D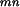 (ij) Pi ε Am, Pj ε An, ∀m, n}. D
(ij) Pi ε Am, Pj ε An, ∀m, n}. D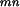 (ij) = 1 if domain Dm and domain Dn interact in the protein pair Pi and Pj and D
(ij) = 1 if domain Dm and domain Dn interact in the protein pair Pi and Pj and D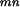 (ij) = 0 otherwise. We derive the EM algorithm as follows.
(ij) = 0 otherwise. We derive the EM algorithm as follows.
The E-step is:
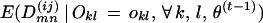 |
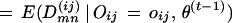 |
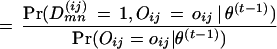 |
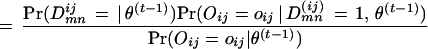 |
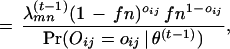 |
where the denominator can be calculated using Equation 2. The MLE of λmn is the fraction of {D(ij)mn, Pi ∈ Am, Pj ∈ An} such that D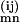 = 1. We thus obtain a recursive formula for the M-step:
= 1. We thus obtain a recursive formula for the M-step:
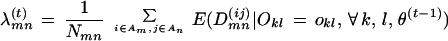 |
4 |
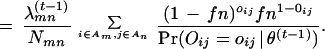 |
The EM algorithm is described as follows: (1) Initialization; choose initial values for {λmn, ∀m, n}, and compute Pr(Pij = 1) by Equation 1 and Pr(Oij = 1) by Equation 2; (2) Update parameters {λmn, ∀m, n} by Equation 4 and compute the likelihood function by Equation 3; (3) Go to step 2, repeat until the value of the likelihood function is unchanged (within certain error).
WEB SITE REFERENCES
http://mips.gfs.de; MIPS database.
http://pfam.wustl.edu; Pfam database.
Acknowledgments
This research was partially supported by National Institutes of Health Grant DK53392, National Science Foundation EIA-0112934, and the University of Southern California.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.153002.
REFERENCES
- Bairoch A, Apweiler R. The SWISSPROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 2000;28:45–48. doi: 10.1093/nar/28.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman A, Birney E, Cerruti L, Durbin R, Etwiller L, Eddy S, Griffiths-Jones S, Howe K, Marshall M, Sonnhammer E. The PFAM Protein Families Database. Nucleic Acids Res. 2002;30:276–280. doi: 10.1093/nar/30.1.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock JR, Gough DA. Predicting protein–protein interactions from primary structure. Bioinformatics. 2001;17:455–460. doi: 10.1093/bioinformatics/17.5.455. [DOI] [PubMed] [Google Scholar]
- Corpett F, Gouzy J, Kahn D. ProDom and ProDom-CG: Tools for protein domain analysis and whole genome comparisons. Nucleic Acids Res. 2000;28:267–269. doi: 10.1093/nar/28.1.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempster AP, Laid NM, Rubin DB. Maximum likelihood from incomplete data via the EM algorithm (with discussion) J Roy Statist Soc Ser B. 1977;39:1–38. [Google Scholar]
- Eisen MB, Spellman PT, Brown PO, Bostein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enright AJ, Iliopoulos I, Kyrpides NC, Ouzounis CA. Protein interaction maps for complete genomes based on gene fusion events. Nature. 1999;402:86–90. doi: 10.1038/47056. [DOI] [PubMed] [Google Scholar]
- Ge H, Liu Z, Church GM, Vidal M. Correlation between transcriptome and interactome mapping data from Saccharomyces cerevisiae. Nat Genet. 2001;29:482–486. doi: 10.1038/ng776. [DOI] [PubMed] [Google Scholar]
- Goffeau A, Barrell BG, Bussey H, Davis RW, Dujon B, Feldmann H, Galibert F, Hoheisel JD, Jacq C, Johnston M, et al. Life with 6000 genes. Science. 1996;274:546–567. doi: 10.1126/science.274.5287.546. [DOI] [PubMed] [Google Scholar]
- Gomez GM, Lo SH, Rzhetsky A. Probabilistic prediction of unknown metabolic and signal-transduction networks. Genetics. 2001;159:1291–1298. doi: 10.1093/genetics/159.3.1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigoriev A. A relationship between gene expression and protein interactions on the proteome scale: Analysis of the bacteriophage T7 and the yeast Saccharomyces cerevisiae. Nucleic Acid Res. 2001;29:3513–3519. doi: 10.1093/nar/29.17.3513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazbun TR, Fields S. Networking proteins in yeast. Proc Natl Acad Sci. 2001;98:4277–4278. doi: 10.1073/pnas.091096398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito T, Tashiro K, Muta S, Ozawa R, Chiba T, Nishizawa M, Yamamoto K, Kuhara S, Sakaki Y. Toward a protein–protein interaction map of the budding yeast: A comprehensive system to examine two-hybrid interactions in all possible combinations between the yeast proteins. Proc Natl Acad Sci. 2000;97:1143–1147. doi: 10.1073/pnas.97.3.1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito T, Chiba T, Ozawa R, Yoshida M, Hattori M, Sakaki Y. A Comprehensive two-hybrid analysis to explore the yeast protein interactome. Proc Natl Acad Sci. 2001a;98:4569–4574. doi: 10.1073/pnas.061034498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito T, Chiba T, Yoshida M. Exploring the protein interactome using comprehensive two hybrid projects. Trends Biotechnol. 2001b;19:S23–S27. doi: 10.1016/S0167-7799(01)01790-5. [DOI] [PubMed] [Google Scholar]
- Legrain P, Selig L. Genome-wide protein interaction maps using two-hybrid systems. FEBS Lett. 2000;480:32–36. doi: 10.1016/s0014-5793(00)01774-9. [DOI] [PubMed] [Google Scholar]
- Lockhart DJ, Winzeler EA. Genomics, gene expression and DNA arrays. Nature. 2000;405:827–836. doi: 10.1038/35015701. [DOI] [PubMed] [Google Scholar]
- Marcotte EM, Pellegrini M, Ng HL, Rice DW, Yeates TO, Eisenberg D. Detecting protein function and protein–protein interactions from genome sequences. Science. 1999a;285:751–753. doi: 10.1126/science.285.5428.751. [DOI] [PubMed] [Google Scholar]
- Marcotte EM, Pellegrini M, Thompson MJ, Yeates TO, Eisenberg D. A combined algorithm for genome-wide prediction of protein function. Nature. 1999b;402:83–86. doi: 10.1038/47048. [DOI] [PubMed] [Google Scholar]
- Mewes HW, Frishman D, Guldener U, Mannhaupt G, Mayer K, Mokrejs M, Morgenstern B, Munsterkotter M, Rudd S, Weil B. MIPS: A database for genomes and protein sequences. Nucleic Acids Res. 2002;30:31–34. doi: 10.1093/nar/30.1.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mrowka R, Patzak A, Herzel H. Is there a bias in proteome research? Genome Res. 2001;11:1971–1973. doi: 10.1101/gr.206701. [DOI] [PubMed] [Google Scholar]
- Pellegrini M, Marcotte EM, Thompson MJ, Eisenberg D, Yeates TO. Assigning protein functions by comparative genome analysis: Protein phylogenetic profiles. Proc Natl Acad Sci. 1999;96:4285–4288. doi: 10.1073/pnas.96.8.4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rain JC, Selig L, Reuse HD, Battaglia V, Reverdy C, Simon S, Lenzen G, Petel F, Wojcik J, Schachter V, et al. The protein–protein interaction map of Helicobacter pylori. Nature. 2001;409:211–215. doi: 10.1038/35051615. [DOI] [PubMed] [Google Scholar]
- Sprinzak E, Margalit H. Correlated sequence-signatures as markers of protein–protein interaction. J Mol Biol. 2001;311:681–692. doi: 10.1006/jmbi.2001.4920. [DOI] [PubMed] [Google Scholar]
- Uetz P, Giot L, Cagney G, Mansfield TA, Judson RS, Knight JR, Lockshon D, Narayan V, Srinivasan M, Pochart P, et al. A Comprehensive analysis of protein–protein interactions in Saccharomyces cerevisiae. Nature. 2000;403:623–627. doi: 10.1038/35001009. [DOI] [PubMed] [Google Scholar]
- Wojcik J, Schachter V. Protein–protein interaction map inference using interacting domain profile pairs. Bioinformatics. 2001;17:S296–S305. doi: 10.1093/bioinformatics/17.suppl_1.s296. [DOI] [PubMed] [Google Scholar]