

Abstract
The analysis of large-scale genomic information (such as sequence data or expression patterns) frequently involves grouping genes on the basis of common experimental features. Often, as with gene expression clustering, there are too many groups to easily identify the functionally relevant ones. One valuable source of information about gene function is the published literature. We present a method, neighbor divergence, for assessing whether the genes within a group share a common biological function based on their associated scientific literature. The method uses statistical natural language processing techniques to interpret biological text. It requires only a corpus of documents relevant to the genes being studied (e.g., all genes in an organism) and an index connecting the documents to appropriate genes. Given a group of genes, neighbor divergence assigns a numerical score indicating how “functionally coherent” the gene group is from the perspective of the published literature. We evaluate our method by testing its ability to distinguish 19 known functional gene groups from 1900 randomly assembled groups. Neighbor divergence achieves 79% sensitivity at 100% specificity, comparing favorably to other tested methods. We also apply neighbor divergence to previously published gene expression clusters to assess its ability to recognize gene groups that had been manually identified as representative of a common function.
The availability of genomic sequence and genome-scale data sets for expression, regulation, and proteomics is shifting the focus of data analysis from individual genes to families of genes. Frequently, the analysis of genome-scale experiments results in the definition of gene groups. For example, gene expression (Eisen et al. 1998), protein sequence (Altschul et al. 1990, 1997), deletion phenotypes (Winzeler et al. 1999; Hughes et al. 2000), and yeast-2-hybrid screens (Uetz et al. 2000) can all be used to produce sets of related genes. Given a set of genes, it is important to recognize if there is a common functional feature, or if the set is in some way entirely novel. The large number of genes and their multiple functions prohibit easy manual assessment of common function. A computational method that detects common function in a set of genes would be useful, therefore, for assessing the significance of an experimentally derived gene set and prioritizing those groups that deserve follow-up. For example, such a method could be used to rapidly screen large numbers of gene expression clusters and identify functionally interesting ones.
The published literature contains virtually every important biological development, and much of the literature is accessible in electronic form—often as full text, and almost always in abstract form (http://www.ncbi.nlm.nih.gov/PubMed/). Article abstracts about genes can be exploited to predict biological function (Raychaudhuri et al. 2002). We assert that the biological literature (here we use PubMed abstracts) contains the necessary information for assessing whether a group of genes represents a common biological function.
In this paper we propose a novel computational method, neighbor divergence, that rapidly assesses whether a set of genes shares a common biological function by automatic analysis of scientific text. It requires only a corpus of articles relevant to all of the genes being studied (e.g., all genes appearing on an expression array) and an index associating the articles to appropriate genes. Such reference lists are often available from genomic databases (Gelbart et al. 1997; Cherry et al. 1998; Bairoch and Apweiler 1999; Blake et al. 2002) or can be compiled automatically by scanning titles and abstracts of articles for gene names (Jenssen et al. 2001).
An alternative approach to assessing the functional coherence of a gene group is to cross-reference it against predefined groups of related genes that have been compiled automatically from the literature or by manual annotation. Jenssen and colleagues used co-occurrence of gene names in abstracts to create networks of related genes automatically form literature (Jenssen et al. 2001). They showed that those groups were useful in gene expression analysis. The Gene Ontology (GO) Consortium and Munich Information Center for Protein Sequences (MIPS) provide vocabularies of function and assign the relevant terms to genes from multiple organisms (Ashburner et al. 2000; Mewes et al. 2000). Genes that are assigned the same term constitute a functional group of genes. However, such resources may not be comprehensive and up to date at any given time, and it is also laborious to maintain the vocabulary and the gene assignments. Our approach requires only a set of references associated with genes. It requires no precompiled lexicons of biological function, previous annotations, or co-occurrence in the literature. It is kept current and up to date if it is provided a current literature base. Furthermore, this method can be applied to any arbitrary set of genes, as long as an index of gene–article associations is provided.
Recognizing coherent gene groups from the literature is a difficult problem because some genes have been extensively studied, whereas others have only been recently discovered. In addition, most genes have multiple functions. The literature about genes reflects these differences. A given gene may have many relevant documents or none, and the documents about it may cover a wide spectrum of functions. Consequently, the available text can skew performance of text analysis algorithms. However, individual articles tend to address functions very specifically; it is this specificity that we exploit in our approach.
We use statistical natural language processing (NLP) methods to access and interpret biological text (Manning and Schütze 1999). Statistical NLP techniques have already been shown to be useful in annotating individual genes (Tamames et al. 1998; Eisenhaber and Bork 1999; Fleischmann et al. 1999; Raychaudhuri et al. 2002), determining gene or protein interactions (Blaschke et al. 1999; Thomas et al. 2000; Jenssen et al. 2001; Stephens et al. 2001), and assigning keywords to genes or groups of genes (Andrade and Valencia 1997; Shatkay et al. 2000; Masys et al. 2001).
The intuition behind neighbor divergence involves recognizing articles that are about the function represented in the group. If a group of genes shares some specific function, such as “autophagy”, an article germane to that function will refer to at least one of the genes in the group. Furthermore, other similar articles that pertain to the same function will tend to refer to the same gene or to other genes in the group.
Neighbor divergence assigns a functional coherence score to a group of genes on the basis of the literature. It uses semantic neighbors; two articles are semantic neighbors if there is similar word usage in each of them (Manning and Schütze 1999). First, semantic neighbors are precomputed for each article in the corpus. Given a gene group, each article's relevance to the group is scored by counting the number of neighbors that have references to genes in the group. If the group represents a coherent biological function, articles that discuss that function will have many referring neighbors and therefore will score high (see Fig. 1). Articles that address biological functions that are irrelevant to the group function will score low. If there are many high-scoring articles, the group likely represents genes with shared function. Neighbor divergence determines whether a function is represented in a gene group from the distribution of article scores. Specifically, the neighbor divergence measure of functional coherence of a gene group is an information-theoretic measure of the difference between the empirical distribution of article scores and a theoretical distribution of scores that would be expected with a noncoherent group of genes.
Figure 1.

Scoring articles relative to gene groups. We graphically depict a small gene group of three autophagy genes (boxes with dotted boundaries). The genes are connected to their respective article references (boxes with solid boundaries). Articles about autophagy are dark boxes with white lettering. Notice that, for all genes, only a few of the referenced articles are about autophagy, the critical function that unites these genes in the group. The arrows are used to indicate the semantic neighbors of article B.2, an autophagy article. The significance of this article to the group's unifying function becomes apparent when we notice that many of its neighbors, also autophagy articles, are references for other genes in the same group.
To evaluate neighbor divergence and to compare it with other approaches, we used 19 groups of yeast genes, each representing a different function. We also devised 1900 decoy random yeast gene groups. We tested methods by scoring all groups. A good method should assign high scores to functional groups and low scores to random groups. We report the percentile of the functional groups relative to the 1900 groups as a measure of success; a score that exceeds all random group scores is in the 100th percentile. Also, we calculate the precision and recall of a method at different score cutoff levels. The precision is the number of functional groups scoring above the cutoff divided by the number of total groups scoring above the cutoff. The recall is the number of functional groups scoring above the cutoff divided by the total number of functional groups. A good method achieves 100% recall at 100% precision.
We also examined how removing legitimate genes and replacing them with irrelevant genes in the gene group affects the score. If the score falls off monotonically, then the score is well behaved and even partial groups have some signal. The neighbor divergence method can then also be used to refine gene groups, by adding and replacing genes to increase the functional coherence score.
Gene expression clustering algorithms generate a large number of clusters, many of which are spurious. We tested our method's ability to recognize 10 yeast gene expression clusters that were manually recognized by investigators as representative of a common function (Eisen et al. 1998). This is a real-world test of the sensitivity of neighbor divergence in detecting meaningful groupings derived from experimental data.
RESULTS AND DISCUSSION
Gold Standard for Benchmarking Method Performance
To assess the performance of neighbor divergence and other methods, we selected 19 functional yeast gene groups as a gold standard that were defined by an independent body (Ashburner et al. 2000). These groups varied in size and content (Table 1A). This diversity is representative of gene groups that experimental procedures may derive. Also, many of the genes were members of more than a single functional gene group (Table 1B), which underscores the multiple functionality that many genes have. We created 1900 random yeast gene groups as a negative set. This may be a poor negative set because experimentally derived gene sets are rarely completely random. However, this set is sufficient to use in comparing the different methods and establishing a performance baseline for neighbor divergence.
Performance of Neighbor Divergence
Neighbor divergence achieves 79% recall (15 of 19 functional groups) at 100% precision; this is equivalent to 79% sensitivity at 100% specificity. In Figure 2 we have plotted the precision and recall at different cutoff levels for neighbor divergence and other methods for comparison. Because the cutoff score is selected to be more stringent, some functional groups are not obtained and therefore recall is lower. However, most random groups fail to make the cutoff and the precision is higher. In Table 2 we have listed the percentile of the score assigned by the method for the different functional groups relative to the 1900 random groups. Neighbor divergence assigned 15 of the 19 functional groups scores that exceeded all of the 1900 random groups; another 3 functional groups had scores exceeding 98% of the random groups (Table 2).
Figure 2.

Precision-recall plot for each of the functional coherence scoring methods. We used each method to score the functional coherence of the 19 functional gene groups and the 1900 random gene groups. We calculated and plotted precision and recall at cutoff scores of different stringency. There is a trade-off between precision and recall. More stringent cutoff values select fewer true functional groups, and recall (or sensitivity) is compromised; however, less stringent cutoff values cause many random groups to be selected inappropriately and precision is compromised. An ideal precision-recall plot achieves 100% precision for every value of recall. The neighbor divergence method is closest to the optimal curve.
Table 2.
Percentile Scores Achieved by the Neighbor Divergence Scoring Method
| Functional classification | Percentilea |
|---|---|
| Signal transduction | 100.0% |
| Cell adhesion | 99.7% |
| Autophagy | 100.0% |
| Budding | 100.0% |
| Cell cycle | 100.0% |
| Biogenesis | 100.0% |
| Shape size control | 100.0% |
| Cell fusion | 100.0% |
| Ion homeostasis | 100.0% |
| Membrane fusion | 98.8% |
| Sporulation | 100.0% |
| Stress response | 100.0% |
| Transport | 100.0% |
| Amino acid metabolism | 100.0% |
| Carbohydrate metabolism | 100.0% |
| Electron transport | 89.3% |
| Lipid metabolism | 100.0% |
| Nitrogen metabolism | 98.6% |
| Nucleic acid metabolism | 100.0% |
Relative to the scores of 1900 random groups. A good method assigns a score that exceeds that of all random groups; such a score is in the 100th percentile.
Neighbor divergence performance is robust to different size gene groups. Smaller groups usually contain fewer genes, fewer articles, and consequently are more difficult to discover. Despite that, neighbor divergence is able to assign relatively high scores to these groups (Table 2).
In Figure 3 we have plotted the distribution of neighbor divergence scores for the 1900 random gene groups and the 19 functional gene groups. Although there is some overlap, most functional groups have scores that are about an order of magnitude higher than the highest score assigned to a random gene group.
Figure 3.

Histogram of neighbor divergence scores. Each open square represents (≤) the count of random gene group scores in the range indicated on the horizontal axis; each closed diamond represents the count of functional gene group scores in the range on the horizontal axis. There is little overlap between the two histograms. None of the random gene groups score above .16; most of the functional gene groups score well above .16.
Calculating Neighbor Divergence Scores With Article Score Distributions
The neighbor divergence measure of functional coherence in a gene group is a measure of the disparity between the empirical distribution of article scores and a theoretical distribution of article scores. We use a Poisson distribution to approximate this theoretical distribution of article scores for a noncoherent gene group. As an example, we have scored all of the articles against one functional gene group and plotted the resulting empirical distribution of scores (see Fig. 4A). If the score distribution is different from the Poisson, then the gene group likely represents a biological function. The log ratio of probability in both distributions is plotted for each article score in Figure 4B. Very high scoring articles are relevant to the group's function and are overrepresented relative to the Poisson distribution.
Figure 4.

Observed and expected distribution of article scores. (A) The bar graph in the figure represents the observed empirical distribution of article scores for the “signal transduction” gene group. The line on the figure is the Poisson distribution; it is the expected distribution of scores for a random gene group of the same size. (B) The ratio in log scale of observed (bars in Fig. 4A) to expected (line in Fig. 4A) distribution of article scores. The X-axis is drawn at a ratio of one, where observed is equal to expected. Because the gene group represents a well-defined biological function, the distributions are very different. High-scoring articles that discuss signal transduction and low-scoring articles that discuss functions besides signal transduction are overrepresented.
Performance of Naïve Word Divergence Method
For purposes of comparison, we developed and tested a naïve word divergence method that is based on an intuitive statistical NLP strategy. Abstracts are divided into those that refer to group genes and those that do not. A probability distribution of words in abstracts referring to group genes is calculated from counts and compared with the distribution of words in the other articles. Word divergence is an information-theoretic measure of the disparity between the two word distributions. If a subset of rare words is used significantly more inside the group than it is outside the group, then these words may be indicative of some biological function within the gene group. Therefore, word divergence should be sensitive to the presence of biological function in the gene group.
Word divergence only achieves 10.5% recall (2 of 19 functional groups) at 8.3% precision on the same data set (Fig. 2); this is equivalent to 10.5% sensitivity at 98.9% specificity. This method performs relatively poorly. Although an individual article may address a single aspect of a gene's function, different articles referring to the same gene may discuss many different biological functions (Fig. 1). Consequently, pooling all of the articles referring to a gene results in an uninformative distribution of words. If all articles written about a gene addressed the same function, this method would have been more successful.
Performance of Other Article-Scoring Approaches
The best article score and best article p-value are similar to neighbor divergence in that all articles are scored for relevance against the gene group by counting the number of referring semantic neighbors. In these methods, however, only the single “best” article score is used as a score for the group. These methods perform better than word divergence because they do not combine signals from many different articles, but rather consider the articles individually. Best article score achieves 58% recall at 8.3% precision (93.7% specificity), and best article p-value performs comparably, achieving 58% recall at 9.1% precision (94.2% specificity) (Fig. 2).
These methods search for articles that have semantic content that is relevant to the group. The advantage of this approach is that articles are treated as individuals. This approach is more appropriate for the problem because genes are often multifaceted, but scientific articles tend to be focused on the subject they are addressing. The best article score method is limited because large groups would be expected to have larger scores on average. To correct for this, we have tried computing a p-value for the best score instead. The p-values seem to overcompensate for larger groups, however.
Both methods are limited by their use of scores of only a single article; this ignores other high-scoring articles that should be abundant if the gene group represents a function. The neighbor divergence method relies on the referring neighbor principle also, but in contrast obtains greater statistical power by considering the scores of all articles and not just the extreme-valued ones.
Performance of Neighbor Divergence Without Filter
Abstracts referring to well-studied genes often have semantic neighbors that refer to the same gene. If such a gene is in the group, an abstract referring to the gene may receive a spuriously high score because many of its article neighbors refer to the same gene. That abstract may not, however, be relevant to the group function. A sufficient number of such high-scoring abstracts can increase the neighbor divergence score. To reduce potential false-positive gene groups produced by this effect, our implementation of neighbor divergence includes a filter in determining the semantic neighbors. When calculating semantic neighbors for an article, only articles that refer to different genes are considered. Without the filter (neighbor divergence–no filter), performance is reduced to 68% recall at 100% precision (Fig. 2).
Understanding the Gene Group's Function
Neighbor divergence determines whether a group of genes has a coherent function. It does not tell us the function. Because all of the articles are scored by neighbor divergence for a given gene group, the easiest way to determine a group's function is to examine the higher-scoring articles manually or automatically. For example, in the ion homeostasis functional group, the highest scoring article is titled “Resolution of subunit interactions and cytoplasmic subcomplexes of the yeast vacuolar proton-translocating ATPase” (Tomashek et al. 1996). The highest scoring article for the autophagy gene group is titled “Structural and functional analyses of APG5, a gene involved in autophagy in yeast” (Kametaka et al. 1996). Both of these articles contain clues to the nature of the gene group. These and other high-scoring articles indicate the shared function. The high-scoring articles could be collected and examined manually to determine group function.
Alternatively, keywords for the group that describe the function of the group could be determined automatically. Investigators have already developed algorithms to find keywords in collections of documents that could be applied to these high-scoring articles to determine functional keywords (Andrade and Valencia 1997).
Corrupting Functional Groups
We examined the robustness of the scores to removal of genes and replacement with random genes. As this procedure is conducted, scores slowly decrease. About half of the genes for the two functional groups examined can be removed while still maintaining a reasonably strong signal (see Fig. 5). Incomplete gene functional sets can be detected, although their scores will be lower. Therefore, partial functional groups derived from experimental screens are still discernable.
Figure 5.

Replacing functional genes with random genes reduces neighbor divergence scores gracefully. We replaced genes in two functional gene groups (“autophagy” and “ion homeostasis”) with random genes, and scores were recalculated for the corrupted groups. Each point represents 10 scores; error bars indicate 95% confidence interval of scores for that many genes replaced. Neighbor divergence scores above .1 are very significant (see Fig. 3). Neighbor divergence scores remain significant despite replacement of about 38% (6 of 16 genes) of the “autophagy” genes and 60% (26 of 43 genes) of the “ion homeostasis” genes.
Furthermore, the more representative a gene group is of a specific function, the greater the neighbor divergence score. This indicates that, as scores are optimized by addition and removal of genes, more ideal functional gene groups can be obtained. There is then the possibility of using neighbor divergence in bioinformatics algorithms to automatically define gene groups in the context of experimental data.
Application of Functional Coherence Scoring to Manually Labeled Gene Expression Clusters
Eisen and colleagues (1998) collected expression measurements on yeast genes under 79 diverse conditions. They used a hierarchical clustering algorithm to identify groups of genes with coherent gene expression patterns. A few of the gene clusters contained many genes with similar function. These published clusters were manually identified and labeled with a summary label. We hypothesized that our method could rapidly identify the functionally coherent groups of genes. We reevaluated the functional coherence of these clusters automatically with neighbor divergence. Our results are presented in Table 3. We found that 7 of the 10 clusters had very high functional coherence scores.
Table 3.
Assigning Neighbor Divergence Scores to Experimentally Obtained Gene Expression Clusters
| Functional label assigned to expression clustera | Number of genes | Neighbor divergence score | Score percentileb |
|---|---|---|---|
| ATP synthesis | 14 | 0.1358 | 99.9% |
| Chromatin structure | 8 | 0.1456 | 100.0% |
| DNA replication | 5 | 0.1867 | 100.0% |
| Glycolysis | 17 | 0.2118 | 100.0% |
| Mitochondrial ribosome | 22 | 0.0269 | 53.3% |
| mRNA splicing | 14 | 0.0248 | 48.3% |
| Proteasome | 27 | 0.3007 | 100.0% |
| Ribosome and translation | 125 | 0.2224 | 100.0% |
| Spindle pole body assembly and function | 11 | 0.0272 | 53.8% |
| Tricarboxylic acid cycle and respiration | 16 | 0.1249 | 99.8% |
Eisen et al. (1998) clustered genes from diverse experimental conditions. They labeled 10 of the clusters in Fig. 2 of their paper as containing genes that represent some consistent biological function. Each row represents a gene cluster.
Relative to 1900 random gene groups.
For three of the clusters, the functional coherence score was poor. The “spindle pole body assembly and function” cluster contained 11 yeast genes; we found that only 3 of these genes were among the 32 listed “spindle pole” genes in the Comprehensive Yeast Genome Database (CYGD) (Mewes et al. 2000). Similarly, the “mitochondrial ribosome” cluster contained 22 genes; only 10 of these genes were among the 49 “mitochondrial ribosome” genes listed by CYGD. Also, the “mRNA splicing” cluster contained 14 genes; we found only 3 of these genes among the 38 listed “mRNA splicing” yeast genes in CYGD. Many of the genes in these clusters do not represent the annotated function. Although these clusters are suggestive, they are not coherent functional groups based on our scoring criteria; they contain less than half of the genes with the reported function. Accordingly, the functional coherence scores are low. In fact, it may be that these clusters represent a novel association of genes that should be pursued and validated for their functional implications.
Future Directions
There is growing interest in enhancing biological data analysis by using the published literature as a knowledge source to guide bioinformatics algorithms. Inclusion of literature has been shown to directly augment biological data analysis, such as sequence homology searches, sequence-based assignment of cellular compartment, and gene expression analysis (MacCallum et al. 2000; Shatkay et al. 2000; Chang et al. 2001; Jenssen et al. 2001; Stapley et al. 2002). Many analytical approaches, such as those based on supervised and unsupervised machine learning, aim to define groups of genes based on patterns in experimental data (Raychaudhuri et al. 2001). Neighbor divergence can be a critical piece in connecting such data analysis algorithms to the scientific literature. New algorithms can be written that search for groups with consistent signal in the experimental data that also have high functional coherence. For example, a clustering algorithm can be rewritten to identify groups of genes with similarities in expression and also similarities in function as assessed from the literature; the solution is to modify the objective function in gene group searches to include similarity of the literature for a group as well as experimental similarity. The neighbor divergence score may have other applications in defining new functional groups, annotating genes, and organizing genes in a functional hierarchy.
The work that we have presented here is limited in that it only uses article abstracts and not the whole text of articles. A more complete implementation of this method would leverage the full text of articles; these are now becoming available on line (Roberts et al. 2001). Our method relies on abstracts focusing on specific subjects. Inclusion of full text articles will probably be most effective if the text is broken into smaller, more specific semantic units, perhaps individual paragraphs.
METHODS
Neighbor Divergence Algorithm
Data Types: Document Corpus and Reference List
The neighbor divergence calculation for a gene group requires a corpus of documents relevant to all genes in the organism and a reference list indicating the articles that are germane to each gene. Here, all documents are PubMed abstracts. Only the title and abstract fields in the PubMed records are used. From these documents, we find unique tokens by tokenizing on white space, punctuation, and common nonalphanumeric characters such as hyphens and parentheses. Only those tokens that were present in >4 abstracts and <10,000 abstracts were considered as vocabulary words. Abstracts are converted into vectors of word counts in which each dimension represents a specific word.
Identifying Semantic Neighbors for Corpus Articles
For each article, the k most similar articles, including the original article, are precomputed. Here we use k = 20. To quantify the similarity between two documents, we used the cosine between the two weighted document word vectors. Word vectors of articles were first converted into inverse document frequency-weighted word vectors (Manning and Schütze 1999):
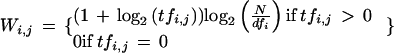 |
where Wi,jis the weighted count of word i in document j, tfi,jis the number of times word i is in document j, dfiis the number of documents that word i is present, and N is the total number of documents. Inverse document frequency weighting is commonly used to reduce the impact of very common words. Article similarity is calculated as the cosine of the angle between these two weighted article vectors.
In the selection of the 20 similar articles for each article, we applied a simple filter as discussed earlier. Except for the seed article, all other articles that referred to a subset of genes referred to in the seed article were removed from consideration.
Scoring Article Relative to Gene Groups
Given a gene group, neighbor divergence then assigns a score, Si, to each article i. The score is a count of semantic neighbors that refer to group genes. Groups that represent genetic functions will induce many articles to have high scores.
Practically, most articles in the data set refer to multiple genes rather than a single one. Neighboring articles with some genes referring to gene groups are counted fractionally.
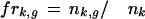 |
where nk,gis the number of genes in the gene group g that the neighboring article k refers to, nkis the number of genes that article k refers to, and frk,g is the fractional reference for article k to group g.
To obtain the article score, we sum the referring fractions of the 20 neighbors and round to the nearest integer.
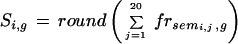 |
where Si,gis the score for an article i for a group g calculated by rounding and summing the fractional reference of its 20 neighbor articles whose indices are semi,j. Si,gis an integer that ranges between 0 and 20.
Calculating a Theoretical Distribution of Scores
If the gene group has no coherent functional structure, the semantic neighbors of any given article should refer to group genes independently with a probability q. Each of these trials should be independent. A Poisson distribution estimates this distribution accurately for small values of q. In this case:
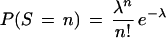 |
where λ = 20*q. For a given gene group, we estimate q, the fraction of articles referring to group genes, by summing all of the fractional references, fr, of all articles and dividing by the number of articles, N.
Quantifying the Difference Between the Empirical Score Distribution and the Theoretical One
An empirical distribution of the article scores is computed for the gene group. If the gene group contains no functional coherence, the distribution of scores should be similar to the Poisson distribution. The functional coherence of the gene group is scored as the Kullback-Leiber (KL) divergence between the empirical distribution and the Poisson distribution.
KL Divergence
To quantify the difference between two distributions, we use KL divergence or relative entropy (Manning and Schütze 1999). Given two distributions, a theoretical one, h, and an observed one, g, we calculate divergence:
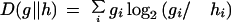 |
If two distributions are the same, the divergence is zero; the more disparate the two distributions, the larger the divergence.
Other Methods to Score Functional Coherence
Word Divergence: A Baseline Method for Comparison
As a baseline, we test an alternate method, word divergence. This method requires calculation of two distributions of words. The first distribution is computed from words in abstracts referring to genes within the group; counts of each word are divided by the total number of words these abstracts. A second distribution is computed similarly for all abstracts referring to genes outside the group. Both distributions are smoothed with Dirichlet priors, assuming 300 prior words distributed according to a baseline distribution; the baseline distribution of each word is computed by dividing its count in all abstracts by the total count of all words in all abstracts. The KL divergence of these two distributions of words is then computed as a measure of functional coherence; the gene group distribution is treated as the observed distribution.
Best Article Score and Best P-Value
These scoring schemes are also based on scoring articles against gene groups as describedearlier. Here, we used the highest article score as a measure of a gene group's functional coherence (best article score). In a different approach, we used the negative log of the p-value for the best article score (best article p-value). To calculate the p-value of an article, we use the Poisson distribution. The p-value of an article is the summed probability of an article having equal or more referring neighbors than it has.
Neighbor Divergence–No Filter
This method is identical to neighbor divergence except the filter applied in selection of semantic neighbors is not used.
Evaluation
Data Types
All experiments described in the following section are conducted in Saccharomyces cerevisiae.
We used a reference list that contained PubMed abstract references to yeast genes from the Saccharomyces Genome Database (Cherry et al. 1998). The reference list included 20,101 articles with 50,860 references to 4205 genes; the article records were obtained from the National Center for Biotechnology Information in Medline format. A total of 12,301 words were selected for the vocabulary. All documents were converted into 12,301 dimensional vectors of word counts.
Assembling the Functional Gene Groups
To test our method, we assembled gold standard functional gene groups from GO (Ashburner et al. 2000). We focused on “gene process” GO terms. We selected 19 diverse process GO terms relevant to yeast biology that had at least three genes. A functional group included genes assigned the listed term by the GO consortium or a more specific child of the listed term. The GO terms and properties of the groups they correspond to are described in Table 1A. Many genes were assigned to multiple gene groups (see Table 1B). We used the 2 Nov 2001 release of the GO process ontology and the 17 October 2001 GO gene associations for yeast.
Assembling the Decoy Random Gene Groups
We assembled 1900 random gene groups as decoy gene groups. For each gold standard functional gene group, 100 random gene groups of the same size were created.
Evaluating Methods to Identify Common Biological Function
In this study we evaluated five different methods to score the functional coherence of a gene group: (1) word divergence, (2) best article score, (3) best article p-value, (4) neighbor divergence, and (5) neighbor divergence–no filter. Each method was used to score the 1900 decoy gene groups and the 19 functional gene groups. The percentile for the score of each of the 19 functional groups relative to the 1900 random gene groups was calculated. Also, for different cutoff scores, precision and recall values were calculated for the gene groups.
Corruption Studies
For two of the gene groups, “ion homeostasis” and “autophagy”, we sequentially removed genes in random order and swapped in other genes. This process was repeated until only one original gene remained. Neighbor divergence score was calculated after each swap. This procedure was repeated 10 times, and the results were averaged together.
Computation
PubMed database queries and data preprocessing were implemented using perl (Schwartz and Christianson 1997), Python (Lutz and Ascher 1999), and the biopython toolkit (www.biopython.org). All mathematical computations were performed with Matlab (Mathworks).
WEB SITE REFERENCES
http://www.ncbi.nlm.nih.gov/PubMed/; of open–source bioinformatics Python modules.
http://www.biopython.org/; repository.
Table 1A.
Gold Standard Functional Gene Groups
| Functional classification | Gene ontology code | Genes | Total articles referenced | Unique articles referenced |
|---|---|---|---|---|
| Signal transduction | GO:0007165 | 94 | 3484 | 1944 |
| Cell adhesion | GO:0007155 | 6 | 82 | 59 |
| Autophagy | GO:0006914 | 16 | 110 | 55 |
| Budding | GO:0007114 | 74 | 1692 | 979 |
| Cell cycle | GO:0007049 | 341 | 8399 | 4438 |
| Biogenesis | GO:0016043 | 459 | 6439 | 3840 |
| Shape size control | GO:0007148 | 54 | 1629 | 1014 |
| Cell fusion | GO:0006947 | 89 | 2495 | 1470 |
| Ion homeostasis | GO:0006873 | 43 | 667 | 363 |
| Membrane fusion | GO:0006944 | 6 | 212 | 209 |
| Sporulation | GO:0007151 | 27 | 646 | 553 |
| Stress response | GO:0006950 | 94 | 2603 | 1866 |
| Transport | GO:0006810 | 313 | 4559 | 2708 |
| Amino acid metabolism | GO:0006519 | 78 | 1594 | 1221 |
| Carbohydrate metabolism | GO:0005975 | 90 | 2719 | 1855 |
| Electron transport | GO:0006118 | 8 | 205 | 187 |
| Lipid metabolism | GO:0006629 | 90 | 1035 | 715 |
| Nitrogen metabolism | GO:0006807 | 15 | 264 | 229 |
| Nucleic acid metabolism | GO:0006139 | 676 | 12345 | 6674 |
Table 1B.
Genes in Multiple Groups
| Number of GO codes/gene | 0 | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|---|
| Number of genes | 2412 | 1242 | 386 | 113 | 40 | 9 | 3 |
| Total genes | 4205 | ||||||
| Total GO code assignments | 2576 |
Most genes are not in a single functional group. Some genes are in as many as six functional groups.
Acknowledgments
R.B.A. is supported by NIH LM06244, GM61374, NSF DBI-9600637, and a grant from the Burroughs-Wellcome Foundation; S.R. is supported by NIH GM-07365. The authors also thank Kara Dolinski of SGD for providing a curated data set of yeast gene associated articles, and Patrick D. Sutphin, Joshua M. Stuart, and Meenakshy Chakravorty for assistance in manuscript preparation.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL russ.altman@stanford.edu; FAX (650) 725-7944.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.116402.
REFERENCES
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrade MA, Valencia A. Automatic annotation for biological sequences by extraction of keywords from MEDLINE abstracts. Development of a prototype system. Proc Int Conf Intell Syst Mol Biol. 1997;5:25–32. [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bairoch A, Apweiler R. The SWISS-PROT protein sequence data bank and its supplement TrEMBL in 1999. Nucleic Acids Res. 1999;27:49–54. doi: 10.1093/nar/27.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blake JA, Richardson JE, Bult CJ, Kadin JA, Eppig JT. The Mouse Genome Database (MGD): The model organism database for the laboratory mouse. Nucleic Acids Res. 2002;30:113–115. doi: 10.1093/nar/30.1.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaschke C, Andrade MA, Ouzounis C, Valencia A. Automatic extraction of biological information from scientific text: Protein-protein interactions. Proc Int Conf Intell Syst Mol Biol. 1999;2:60–67. [PubMed] [Google Scholar]
- Chang JT, Raychaudhuri S, Altman RB. Including biological literature improves homology search. Pac Symp Biocomput. 2001;14:374–383. doi: 10.1142/9789814447362_0037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherry JM, Adler C, Ball C, Chervitz SA, Dwight SS, Hester ET, Jia Y, Juvik G, Roe T, Schroeder M, et al. SGD: Saccharomyces Genome Database. Nucleic Acids Res. 1998;26:73–79. doi: 10.1093/nar/26.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenhaber F, Bork P. Evaluation of human-readable annotation in biomolecular sequence databases with biological rule libraries. Bioinformatics. 1999;15:528–535. doi: 10.1093/bioinformatics/15.7.528. [DOI] [PubMed] [Google Scholar]
- Fleischmann W, Moller S, Gateau A, Apweiler R. A novel method for automatic functional annotation of proteins. Bioinformatics. 1999;15:228–233. doi: 10.1093/bioinformatics/15.3.228. [DOI] [PubMed] [Google Scholar]
- Gelbart WM, Crosby M, Matthews B, Rindone WP, Chillemi J, Russo Twombly S, Emmert D, Ashburner M, Drysdale RA, Whitfield E, et al. FlyBase: A Drosophila database. The FlyBase Consortium. Nucleic Acids Res. 1997;25:63–66. doi: 10.1093/nar/25.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes TR, Marton MJ, Jones AR, Roberts CJ, Stoughton R, Armour CD, Bennett HA, Coffey E, Dai H, He YD, et al. Functional discovery via a compendium of expression profiles. Cell. 2000;102:109–126. doi: 10.1016/s0092-8674(00)00015-5. [DOI] [PubMed] [Google Scholar]
- Jenssen TK, Laegreid A, Komorowski J, Hovig E. A literature network of human genes for high-throughput analysis of gene expression. Nat Genet. 2001;28:21–28. doi: 10.1038/ng0501-21. [DOI] [PubMed] [Google Scholar]
- Kametaka S, Matsuura A, Wada Y, Ohsumi Y. Structural and functional analyses of APG5, a gene involved in autophagy in yeast. Gene. 1996;178:139–143. doi: 10.1016/0378-1119(96)00354-x. [DOI] [PubMed] [Google Scholar]
- Lutz M, Ascher D. Learning Python (help for programmers). Sebastopol, CA: O'Reilly; 1999. [Google Scholar]
- MacCallum RM, Kelley LA, Sternberg MJ. SAWTED: Structure assignment with text description—enhanced detection of remote homologues with automated SWISS-PROT annotation comparisons. Bioinformatics. 2000;16:125–129. doi: 10.1093/bioinformatics/16.2.125. [DOI] [PubMed] [Google Scholar]
- Manning CM, Schütze H. Foundations of statistical natural language processing. Cambridge, MA: The MIT Press; 1999. [Google Scholar]
- Masys DR, Welsh JB, Lynn Fink J, Gribskov M, Klacansky I, Corbeil J. Use of keyword hierarchies to interpret gene expression patterns. Bioinformatics. 2001;17:319–326. doi: 10.1093/bioinformatics/17.4.319. [DOI] [PubMed] [Google Scholar]
- Mewes HW, Frishman D, Gruber C, Geier B, Haase D, Kaps A, Lemcke K, Mannhaupt G, Pfeiffer F, Schuller C, et al. MIPS: A database for genomes and protein sequences. Nucleic Acids Res. 2000;28:37–40. doi: 10.1093/nar/28.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raychaudhuri S, Sutphin PD, Chang JT, Altman RB. Basic microarray analysis: Grouping and feature reduction. Trends Biotechnol. 2001;19:189–193. doi: 10.1016/s0167-7799(01)01599-2. [DOI] [PubMed] [Google Scholar]
- Raychaudhuri S, Chang JT, Sutphin PD, Altman RB. Associating genes with gene ontology codes using a maximum entropy analysis of biomedical literature. Genome Res. 2002;12:203–214. doi: 10.1101/gr.199701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts RJ, Varmus HE, Ashburner M, Brown PO, Eisen MB, Khosla C, Kirschner M, Nusse R, Scott M, Wold B. Information access. Building a ‘GenBank‘ of the published literature. Science. 2001;291:2318–2319. doi: 10.1126/science.291.5512.2318b. [DOI] [PubMed] [Google Scholar]
- Schwartz RL, Christianson T. Learning Perl. Sebastopol, CA: O’Reilly; 1997. [Google Scholar]
- Shatkay H, Edwards S, Wilbur WJ, Boguski M. Genes, themes and microarrays: Using information retrieval for large-scale gene analysis. Proc Int Conf Intell Syst Mol Biol. 2000;8:317–328. [PubMed] [Google Scholar]
- Stapley BJ, Kelley LA, Sternberg MJ. Predicting the sub-cellular location of proteins from text using support vector machines. Pac Symp Biocomput. 2002;7:374–385. doi: 10.1142/9789812799623_0035. [DOI] [PubMed] [Google Scholar]
- Stephens M, Palakal M, Mukhopadhyay S, Raje R, Mostafa J. Detecting gene relations from Medline abstracts. Pac Symp Biocomput. 2001;56:483–495. doi: 10.1142/9789814447362_0047. [DOI] [PubMed] [Google Scholar]
- Tamames J, Ouzounis C, Casari G, Sander C, Valencia A. EUCLID: Automatic classification of proteins in functional classes by their database annotations. Bioinformatics. 1998;14:542–543. doi: 10.1093/bioinformatics/14.6.542. [DOI] [PubMed] [Google Scholar]
- Thomas J, Milward D, Ouzounis C, Pulman S, Carroll M. Automatic extraction of protein interactions from scientific abstracts. Pac Symp Biocomput. 2000;5:541–552. doi: 10.1142/9789814447331_0051. [DOI] [PubMed] [Google Scholar]
- Tomashek JJ, Sonnenburg JL, Artimovich JM, Klionsky DJ. Resolution of subunit interactions and cytoplasmic subcomplexes of the yeast vacuolar proton-translocating ATPase. J Biol Chem. 1996;271:10397–10404. doi: 10.1074/jbc.271.17.10397. [DOI] [PubMed] [Google Scholar]
- Uetz P, Giot L, Cagney G, Mansfield TA, Judson RS, Knight JR, Lockshon D, Narayan V, Srinivasan M, Pochart P, et al. A comprehensive analysis of protein-protein interactions in Saccharomyces cerevisiae. Nature. 2000;403:623–627. doi: 10.1038/35001009. [DOI] [PubMed] [Google Scholar]
- Winzeler EA, Shoemaker DD, Astromoff A, Liang H, Anderson K, Andre B, Bangham R, Benito R, Boeke JD, Bussey H, et al. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science. 1999;285:901–906. doi: 10.1126/science.285.5429.901. [DOI] [PubMed] [Google Scholar]