

Abstract
The Generic Model Organism System Database Project (GMOD) seeks to develop reusable software components for model organism system databases. In this paper we describe the Generic Genome Browser (GBrowse), a Web-based application for displaying genomic annotations and other features. For the end user, features of the browser include the ability to scroll and zoom through arbitrary regions of a genome, to enter a region of the genome by searching for a landmark or performing a full text search of all features, and the ability to enable and disable tracks and change their relative order and appearance. The user can upload private annotations to view them in the context of the public ones, and publish those annotations to the community. For the data provider, features of the browser software include reliance on readily available open source components, simple installation, flexible configuration, and easy integration with other components of a model organism system Web site. GBrowse is freely available under an open source license. The software, its documentation, and support are available at http://www.gmod.org.
Model organism system databases (MODs) are a vital tool for scientific research. They share a common set of tasks: to collect and curate data from the scientific literature such as mutations, alleles, genetic and physical maps, and phenotypes; to integrate this information with the results of large-scale experiments such as microarray studies, SNP screens, and protein-interaction studies; to provide reagent resources such as stocks, genetic constructs, and clones; and, lastly, to provide a common nomenclature for gene symbols, anatomic terms, and other elements of the scientific vocabulary. By integrating, and in some cases reanalyzing, these data, MODs are able to greatly enhance their value. This information is made available to the research community via a Web site that also serves as a nexus for discussions, announcements of interest to the community, and data submissions.
It is important to note that with respect to genomic data, the key role of the model organism databases is to connect genomic features to the classical biology of the organism, a role that is distinct from that of whole genome annotation projects such as EnsEMBL (Hubbard et al. 2002). Also MODs have an interpretative and curatorial role that distinguishes them from GenBank (Benson et al. 2002) and other strictly archival databases.
Four well-established model organism databases are FlyBase, SGD, MGD, and WormBase, which are associated, respectively, with Drosophila melanogaster, Saccharomyces cerevisiae, Mus musculus, and Caenorhabditis elegans. These are four of the five original model organism systems targeted by the NIH component of the Human Genome Project in 1990 (http://www.nhgri.nih.gov/HGP/HGP_goals/5yrplan.html; the fifth was Escherichia coli). As the cost of genome sequencing has come down, an increasing number of organisms are either now in the process of sequencing, such as Anopheles gambiae (Hoffman et al. 2002), Oryza sativa (Sasaki and Burr 2000), Plasmodium falciparum (Gardner 1999), Rattus rattus (Pennisi 2000); or are likely to be sequenced in the near future, such as Dictyostelium discoidium (Kuspa et al. 2001), Leishmania donovani (Blackwell 1997), Zea mays (Bennetzen et al. 2001), honeybee (http://www.nps.ars.usda.gov/menu.htm?newsid=1696), and cow (http://www.hgsc.bcm.tmc.edu/projects/bovine/). There is a clear and present need for new MODs to manage these data sets.
Setting up an MOD is expensive and time-consuming. Recognizing that a major component of the cost of creating a new MOD is the development of database schemata, middleware, and visualization software, the four MODs agreed in the fall of 2000 to pool their resources and to make reusable components available to the community free of charge under an open source license. The goal of this NIH-funded project, christened GMOD for Generic Model Organism Database, is to generate a model organism database construction set that would allow a new model organism to be assembled by mixing and matching various components. Among the components presently under development or in planning are modules for literature curation, tools for classifying genes using the Gene Ontology, management of genetic and physical maps, and an extensible architecture for an MOD Web site. Also part of the GMOD project are a set of standard operating procedures for managing the data gathering, quality control, and community outreach activities of an MOD.
The first large component of the project to be released was the Apollo genome annotation editor, a curator's tool for inspecting and editing genomic annotations (S. Lewis, in prep.). This paper describes the second component of the project, the Generic Genome Browser (GBrowse). GBrowse implements a Web-based display that can be used to display an arbitrary set of features on a nucleotide or protein sequence, and can accommodate genome-scale sequences megabases in length. The browser provides most of the features available in other browsers (Harris 1997; Mural et al. 1999; Kent and Zahler 2000; Hubbard et al. 2002) but was designed from the outset to be portable and extensible. This allows it to integrate well with other components of a MOD Web site in general, and with planned components of the GMOD project in particular. Although GBrowse is targeted at maintainers of model organism databases, it is suitable for any research group that must manage a set of sequence annotations, ranging from those needing to display raw features such as similarity hits through those maintaining high-level genome features such as fully curated gene models.
RESULTS
The GBrowse module is available for download at http://www.gmod.org. It depends only on readily available open source software and runs well on a variety of platforms including such Unix-based systems as Linux, Solaris, FreeBSD, and Macintosh OS X, as well as systems running Windows 2000 and XP. See Methods for a list of server software requirements. On the client side, GBrowse-generated pages are rendered correctly by any Web browser that supports HTML level 4.0 or higher and cascading stylesheets level 2 or higher. This includes Netscape 4.0 and higher, Internet Explorer 5.0 and higher, and many other popular browsers such as Opera and Mozilla.
We first discuss GBrowse from the point of view of the end user accessing it and then from the perspective of the data provider who configures and populates it. For the purposes of illustration, we use a version of GBrowse configured to access the C. elegans genome features available at http://www.wormbase.org/db/seq/gbrowse. Demonstrations of the software running on the human, D. melanogaster, and S. cerevisiae genomes are available at the GMOD Web site (http://www.gmod.org).
Browsing a Region of the Genome
The end user enters GBrowse via the Web page shown in Figure 1. The user selects the feature types he or she is interested in viewing from a set of checkboxes, types a search term or landmark into the text field at the top of the screen, and either hits the enter key or presses the “Go” button. This fetches the region of the genome that spans the landmark, and displays it in an image panel called the “detailed view.”
Figure 1.

The user enters GBrowse by typing a landmark name into the text field at top. Landmarks can be gene names, clone names, accession numbers, or any other identifier configured by the administrator. Once a region is selected, it is displayed in a detailed view that summarizes annotations and other genomic features. An overview panel and a navigation bar together allow the user to move from one place to another.
The detailed view consists of one or more horizontal tracks, each of which contains a particular type of sequence feature. The Figure 1 example shows tracks corresponding to curated gene structures, clone boundaries, cDNA alignments, the location of commercially available PCR primer pairs, and an alignment between C. elegans and an orthologous region in the related nematode Caenorhabditis briggsae. Each track contains glyphs of various sizes, colors, and shapes that provide feature-specific information. For example, the alignment glyph that appears in the third track of Figure 1 is color-coded to indicate the strength of the similarity. A key at the bottom of the detailed view (partly scrolled out of view in the figure) and pop-up “tool tips” that appear when the user hovers the mouse over a glyph both help the user interpret the detailed view. The use of distinctively shaped glyphs distinguishes the GBrowse user interface from the UCSC and EnsEMBL browsers, which rely more heavily on color to distinguish one type of feature from another.
From this display, the researcher can see that the 10-exon structure for the C. elegans gene ace-1 is supported by a full-length cDNA clone (X75331, shown in yellow), that the first four exons and a long 3′-UTR are supported by a set of four EST pairs (shown in green). The exon structure is also supported by alignment with the C. briggsae draft contig cb25.fpc4033 (light and dark blue boxes), but there are intriguing regions of cross-species similarity located just upstream and downstream of the transcript.
Once an interesting region of the genome is in view, the user can navigate through it in a variety of ways. He can jump rapidly from region to region by clicking in the overview panel, a schematic located at the top of the image that shows the entire chromosome (or, in the case of unfinished genomes, a contig) and a number of landmark features such as well-known genetic markers, cytogenetic bands, or sequence scaffolds. He can make finer adjustments by scrolling and zooming with the navigation bar, which appears in the upper-right-hand side of the image in Figure 1. GBrowse provides multiple configurable levels of zoom, and two scroll speeds. For fine adjustment, the user can click on the scale that appears at the top of the detailed view to center the displayed region at that point, or select the “+” and “−” buttons to make fine adjustments in the zoom level. Figure 2 shows the same view after the user has zoomed out to show 200 kb of features surrounding the region of interest.
Figure 2.

The detailed view after zooming out to 200 kb, showing semantic zooming.
Notice the use of semantic zooming in the display. As the user zooms out, GBrowse first inhibits the display of feature labels and descriptions, and then deactivates collision control to allow glyphs to overlap densely (shown by the EST track). Some glyphs will also change appearance during semantic zooming. For example, the gene glyph shows the internal intron/exon structure at high magnification, but at low magnification is rendered as a solid arrow pointing in the direction of transcription. The “dna” glyph shows the literal DNA sequence at very high levels of magnification, and a GC content histogram at lower levels. To get more information about a feature, the user clicks on it. This links the user to another Web page that provides more detailed information about the feature. The destination Web page is not usually generated by GBrowse, but is typically a component of the Web site, such as a static HTML page, a script, or an external destination like NCBI Entrez.
Searching for Regions of Interest
The GBrowse search functionality is very flexible. It will accept a chromosome name using the nomenclature that is appropriate for the organism being displayed, for example “II” for C. elegans, “2L” for D. melanogaster, and “2” for M. musculus. In addition, it can accept a contig name, a clone accession number, a GenBank accession number, a gene symbol, a genetic marker name, an SNP ID, or indeed any other unique feature name that is known to the database. In the case of collision between the IDs of landmarks of different types, the user can qualify the ID using the notation “type:ID”, as in “PCR_Product:sjj_W09B12.1.” The data provider selects which IDs are recognized automatically, the order in which to try different feature types when searching for IDs, and which feature types must always be entered as fully qualified names. To aid in the identification of landmark features, the search system supports wild cards, stem searches, and synonyms.
By default, GBrowse will fit the entire landmark into the detailed view. Advanced users can tweak this by providing start and stop coordinates for the landmark in the format “landmark:start..stop.” For example, when using the C. elegans database, typing “unc-9:1..200” will fetch and display the first 200 bp of the C. elegans gene unc-9. Negative coordinates are acceptable, so that “unc-9:-100..1” will fetch and display the region beginning 100 bp to the left of unc-9.
Should a search term be found in multiple locations, the browser shows the user an intermediate screen that shows the regions graphically and prompts him or her to select one to view. In the case of a region that is too large (where “too large” is an administrator-defined setting), the browser will indicate the region in the “overview panel” (described in more detail below) and ask the user to zoom in.
If the user searches for a landmark that doesn't correspond to a feature name, GBrowse will perform a keyword search on the underlying database, presenting the user with a list of matching features and their genomic coordinates. This enables the administrator to create databases that are searchable for gene function term, name of the submitting author, and so forth. As an example, Figure 3 shows the response to a request for “7 transmembrane receptor” when GBrowse is running on top of the C. elegans database. It is possible to supplement this facility with structured queries using the plug-in mechanism described later.
Figure 3.

A search for the term “7 transmembrane receptor.”
Customizing the Display
The end-user can customize the view in several ways. The simplest way is to select the tracks to display using a list of checkboxes located below the detailed view (scrolled out of view in the screenshots). Checkbox titles also double as documentation; selecting one brings up a text document that gives the citation and other descriptive information for the track. More advanced users can select a “Set Track Options …” button that appears at the bottom of the track checkbox panel. This displays a form that allows the user to adjust, on a track-by-track basis, whether to show the track at all, and if so whether to display it in a compressed format in which overlapping features collide, in expanded format in which overlapping features are offset upward or downward to avoid colliding, or in a format in which each feature is labeled with its name and description. The user can also customize the track order and the width of the image.
When the user is satisfied with the view, he or she can take several actions on it. By selecting the “bookmark” link, the current view and its settings will be added to the user's browser bookmarks, thereby allowing him or her to revisit a set of interesting regions from time to time. This link can also be used to attach the current page to an e-mail message. Other actions are available via a popup menu of plug-ins. One standard plug-in allows the user to format the current view as a FASTA-format file (Pearson 2000), either in plain text, or in an HTML form that highlights a selected set of features using combinations of colors and font changes. Another plug-in generates text dumps of the currently visible features using a number of standard formats including the tab-delimited GFF format (Reese et al. 2000), GenBank (Ouelette 2001), EMBL, GAME/XML (http://www.bioxml.org/Projects/game/game0.1.html), and BSML (http://www.labbook.com/products/xmlbsml.asp). A third plug-in generates restriction maps for the current region in order to assist with cloning constructs.
GBrowse remembers the user's settings between sessions. When the user next visits the GBrowse page, his or her preferences in terms of tracks, track options, track order, display width, and genomic region of interest are automatically restored to their previous values. A reset button located to the right of the navigation bar will restore the standard settings.
Adding Third-Party Features
GBrowse supports third-party features via a set of controls at the bottom of the main browser screen (scrolled out of view in Fig. 1). To add annotations to the genome, a user prepares one or more text files describing the nature and position of his or her annotations and uploads it to GBrowse using a standard file upload field. GBrowse accepts either the full nine-column GFF format or a simplified three-column version. Both formats allow the user to create complex multipart features; to attach names, notes, and links to the features; and to control the way that the features are formatted by specifying their glyph, color, height, and other graphical attributes. Uploaded features appear on the detailed view in specially designated tracks and persist between sessions.
Uploaded features can be expressed using absolute coordinates, or as relative coordinates based on any of the landmarks recognized by GBrowse. This gives the user the choice of annotating in whole chromosome coordinates, relative to the start of a well-known landmark such as a GenBank entry, or relative to another feature, such as the start of a predicted gene. Each time a third-party feature is accessed, GBrowse remaps relative coordinates into absolute ones, a strategy that allows most features to survive updates to the genomic assembly.
Once a feature file is uploaded, it persists on the GBrowse server for a period of time established by the database administrator, typically 60 d since the last time the uploaded file was accessed. The end-user can download his or her features, modify them, and upload them again, or use a simple browser-based editor to change the features directly. Although the uploaded third party feature files are located on a publicly accessible server, they are only accessible via a secret key that is stored in a cookie on the end-user's machine.
To share features with others, end-users can publish them via GBrowse, thereby allowing other researchers to display them alongside other feature tracks. To accomplish this, the end-user places a tab-delimited feature file on an Internet-accessible Web or FTP site, such as a departmental server. He or she then publicizes the existence of this information by e-mailing the file's URL to colleagues or by publishing the URL in a paper. Interested colleagues can now layer these features onto the GBrowse view by entering the URL into a text field in the section labeled “Add Remote Annotations.” Like uploaded files, end-users can add an unlimited number of URL-based tracks to GBrowse, and adjust their order and formatting in the same way as other tracks. GBrowse intelligently caches third-party feature information using a scheme that is described in more detail below. This means that when researchers update their published feature files their changes will immediately become visible to their colleagues.
A user who wishes to make third-party annotations available to a limited number of authorized users may place the annotation information behind a password-protected FTP or Web site. The username and password information can then be entered directly in the URL using the syntax http://username:password@host.name/.
A simplified version of the upload feature can also be activated by invoking GBrowse with a specially formatted URL that directly contains positional information on a feature or features. By taking advantage of this facility, a data provider who wishes to integrate GBrowse into a BLAST search script can modify the links generated by BLAST so as to point to GBrowse, thereby layering users' BLAST hits on top of genomic features. Similar functionality is available for creating in-line images that incorporate third-party features dynamically layered on top of GBrowse images.
Use Case: Visualizing Patterns of DNA-Binding Sites for Drosophila Transcription Factors
As an example of how the Genome Browser can be used by the scientific community, one of us (M. Caudy) used GBrowse to visualize patterns of DNA-binding sites for specific transcription factors (TFs) in relation to potential target genes within the genome (M. Caudy, L. Stein, and J. Tisdall, in prep.).
Transcriptional enhancers and promoters typically contain clusters of multiple binding sites localized in a small region. Perl scripts that recognize clusters patterned on a model promoter were used to identify all similar clusters in the entire D. melanogaster genome. These clusters and their coordinates were then saved as a GFF file and uploaded into GBrowse, thereby allowing the visualization and analysis of cluster positions in relation to potential target genes. Browsing for clusters near potential target genes of interest—in this case neuronal differentiation genes regulated by HLH transcription factors—provided a means of determining which of the many hundreds of such clusters found were likely to be functional enhancers and promoters in vivo. In cases where the function of genes with nearby clusters is unknown to a user, the available functional information for that gene could be rapidly reviewed by following the link for that gene to FlyBase, which makes available such information as expression pattern, phenotype when mutated, and other functional information.
This method has greatly facilitated the identification and prioritization of clusters that should be selected for molecular and genetic analysis in the search for the transcriptional regulatory code embedded within the genome that controls specific target gene expression by HLH transcription factors. This type of genomic pattern analysis would be extremely time-consuming and difficult without the annotation upload, graphical display, and database linkout features provided by GBrowse, and it was accomplished without a heavy investment in bioinformatics mining or visualization software by the researcher.
GBrowse Internals
We now describe the Generic Genome Browser's internal workings. GBrowse consists of several components (Fig. 4). At the top level is a CGI (Common Gateway Interface) script named gbrowse, which is responsible for managing the user interface. This script generates the HTML forms that the end-users interact with, accepts and processes requests, manages the cookies that preserve users' preferences from session to session, and displays the rendered images of annotated regions.
Figure 4.

The Generic Genome Browser is built from multiple software modules. In this illustration, modules that were not produced as part of this project are shown in a lighter color.
BioPerl and Bio::Graphics Software Libraries
Beneath gbrowse are two software libraries. The Bioperl library (Stajich et al., this issue) is responsible for interceding between the CGI script and the underlying database; this abstraction insulates the CGI script from the details of the database schema, and in principle allows the browser to run on top of a variety of database management systems and schemata.
The Bio::Graphics module is responsible for rendering the genome images. It, in turn, makes use of the GD module (http://stein.cshl.org/software/GD/), a Perl library that is capable of generating a variety of image formats, including JPEG, WBMP, and PNG. Although it was developed for use with this project, Bio::Graphics is completely independent of the Generic Genome Browser and can be used to graphically render any type of nucleotide or protein sequence feature. Because of recent patent issues regarding JPEG compression, GBrowse comes configured to generate the open source PNG (Portable Network Graphics) format; this can be altered if desired.
Bio::DB::GFF Database
Underlying these layers is a relational database that is responsible for storing and retrieving features. The current stable release of GBrowse is limited to using the MySQL relational database management system. Within MySQL, however, GBrowse supports two distinct schemata. One schema, called Bio::DB::GFF, is a simple schema that requires minimal preparation on the part of the MOD administrator. It is necessary only for the administrator to prepare a tab-delimited flat file that describes the genome features using the widely used GFF format, and a set of one or more FASTA-format files that contain the DNA sequence itself. The administrator then uses a provided Perl script to load the database from these files, or to update a previously loaded database with new information. Access to the database is typically accomplished via a Perl programmer's API, which translates requests for feature information into the appropriate SQL queries, and then transforms the results into Bioperl objects for inspection and manipulation.
The Bio::DB::GFF schema was designed for simplicity and speed (Fig. 5). The main Bio::DB::GFF table contains one row per feature, and has columns representing the start and end of the feature, its type, and the reference sequence that establishes its coordinate system. The schema allows a single level of hierarchical substructure in features using a “group” field. This allows “gene” features to contain introns, exons, and transcriptional start sites; and “sequence alignment” features to be built up from a series of aligned regions separated by gaps. Bio::DB::GFF allows the maintainer to attach notes, citations, and other information to features using a simply named properties system, but for semantically rich information, it is recommended to either incorporate the Bio::DB::GFF schema into a larger schema, or to use the GadFly schema described below.
Figure 5.

The Bio::DB::GFF database uses a minimal schema to represent features on sequences. The main tables are fdata, which contains the position and type of each feature, fgroup, which tracks the grouping of subfeatures into features, such as high-similarity pairs in a gapped alignment, fdna, which stores the raw DNA sequence, and fattribute_to_feature, which allows attribute information to be attached to features. Attributes are used for storing such textual information as notes, synonyms, and evidence codes. The fattribute and ftype tables, respectively, hold attribute names and the method and source fields. For retrieval efficiency, the fdna table fragments each DNA into small pieces and stores the beginning of each piece in the foffset field.
The Bio::DB::GFF schema is optimized for queries that retrieve features by ID, by their type, or by the region of the genome they overlap. The last query is a challenge, because conventional relational database indexes do not handle positional range queries well. In SQL, the query to find all features that overlap the region on Chromosome 1 between bases 12,010,000 and 12,020,000 looks like this:
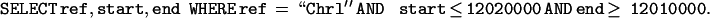 |
If ref, start, and stop are indexed, an SQL optimizer will be able to eliminate half the rows in the feature table, on average, by eliminating all those rows that do not satisfy the criteria for start or end. However, the remainder of the table must then be inspected by a linear search, something that is particularly inefficient when searching for a small feature in the middle of a large chromosome or contig.
The solution that we use is a special case of the R-tree algorithm (Guttman 1984) and was inspired by discussions with Richard Durbin (The Wellcome Trust Sanger Institute). In this optimization, the genome is divided into a hierarchical set of bins. At the top level are bins of size 10 Mb. This is followed by a tier of 1-Mb bins, another of 100 kb, and so on down to 1000 bp. The bins are assigned identifiers using floating point numbers in such a way that adjacent bins in the same tier have adjacent IDs, but bin IDs from different tiers do not overlap. As features are entered into the database, they are assigned to the smallest bin that will entirely contain them. A few very large features, such as entire chromosomes, end up in the large bins, but most features are allocated to the smaller ones.
The Bio::DB::GFF module automatically takes advantage of this binning scheme to optimize the queries. The example positional query given earlier now becomes the following:
SELECT ref,start,end WHERE ref = “Chrl” AND (bin = 100000000.000000 OR bin=10000000.000001 OR bin=1000000.000012 OR bin=100000.000120 OR bin between 10000.001201 and 10000.001202 OR bin between 1000.012010 and 1000.012020)
AND start ≤12010000 AND end ≥12020000
Although this query is more complex than the previous one, it executes much faster. On a 700-MHz Pentium III laptop, searching a 6-million-feature database took 17.4 sec with the unoptimized query, and 0.4 sec using the bin optimization, a performance increase of >400-fold. A version of this indexing scheme has also been adopted for use with the Human Genome Browser at UCSC (Kent et al. 2002).
Although not specifically designed for this purpose, Bio::DB::GFF databases can be used to support genomic analyses. Because it is optimized for positional queries, one can use this database to extract the DNA from arbitrary regions, such as the first intron of every gene, or to find sequence features that intersect others.
GadFly Database
The main limitation of the Bio::DB::GFF schema is that it relies on a flat coordinate system to represent genomic features, and can handle only one level of nesting of sequence features. The flat coordinate system means that for a partially assembled genome, the administrator will have to translate clone- or contig-relative features into chromosome or supercontig coordinates before they can be properly browsed with GBrowse. The restriction on nesting allows a primary transcript to contain multiple features, such as exons, but makes it difficult to represent the fact that multiple primary transcripts belong to one gene (although this can be finessed by declaring the gene a shared property of the transcripts). The Bio::DB::GFF schema also provides no control over feature types, but instead relies on the database administrator to maintain the proper list of types and part/subpart relationships.
These problems are solved by GadFly, the second schema supported by GBrowse. GadFly, which was originally developed for use with the Berkeley Drosophila Genome Project's annotation pipeline, represents features using a hierarchical coordinate scheme in which relative coordinates are translated into absolute coordinates as needed according to the current state of the genome assembly. GadFly is able to represent multiple levels of part/subpart relationships, and takes advantage of controlled vocabularies to describe feature types and gene functions.
GadFly uses a Perl API and a Bioperl-compliant object model for fetching and manipulating data. A strong emphasis has been placed on providing flexibility in the API to take advantage of the rich schema. For instance, it is possible to mix range-based queries with queries by gene function, protein domain, sequence similarity, and phylogeny.
Because of the need to support the richer schema, GadFly's performance is slower than that of an equivalent Bio:: DB::GFF database, and creating a feature load file will require more advanced planning on the part of the maintainers. However, GadFly is a good fit for MOD projects that prefer a robust, semantically rich schema, and it is straightforward to move feature data from one schema to the other. More information on GadFly, including an interactive schema diagram, can be found at http://www.fruitfly.org/developers/.
Plug-ins
GBrowse supports a plug-in architecture that allows third-party modules to extend GBrowse's capabilities. A plug-in is a Perl module that is placed in a specially designated plug-ins directory. When GBrowse initializes, it examines this directory and loads all plug-ins it finds. There are three types of plug-ins. Dumper plug-ins take the region of the genome currently under display and dump it out in text, HTML, or another format. Both the FASTA and the GFF dumping functions of GBrowse are implemented in this way. Finder plug-ins are responsible for searching the underlying Bio::DB::GFF or Gadfly databases for features of interest and returning a list of the features and/or genomic regions found. GBrowse then displays the search results to the user. A simple example of this type of plug-in is the OligoFinder plug-in, which accepts user input of an oligonucleotide of 15 bp or more in length, and returns the location(s) of the oligonucleotide on the genome. Lastly, there are Annotators, which add annotation tracks to the current view. The standard restriction-map generator is implemented in this way.
Many plug-ins have configuration settings. For example, the restriction map annotation plug-in maintains a list of restriction sites that the user can toggle on or off. To allow plug-ins to be configured by the end-user, each plug-in generates an HTML form that displays its settings. These settings are then saved in a persistent HTTP cookie from session to session, remembering the user's preferences. Plug-ins can also store low-level configuration information in files maintained by the administrator.
The plug-in mechanism is in principle very powerful. Because plug-ins are fully functional Perl programs, they can act as hooks into GBrowse to allow the browser to be used as a front end to similarity-searching programs, gene prediction engines and motif finders, and various types of visualization software.
Displaying Third-Party Annotations
GBrowse uses a simple scheme for fetching and displaying third-party annotations. Both uploaded and remotely located feature files are stored at the server side as a set of flat files. Uploaded files are stored on the server directly, whereas files located on remote Web or FTP sites are fetched and cached using a scheme in which the cached copy is freshened if its modification date is older than the modification date of the authoritative version on the remote site. This avoids the overhead of a network fetch if one is not needed.
Third-party feature files may use a mix of absolute and relative coordinate addressing; relative coordinates will be remapped into absolute coordinates before they can be displayed. At present, this remapping is deferred until just before the features are displayed on the screen. Although this ensures that the remapping is correct for the current state of the assembly, it has performance implications as the remapping is performed each time the features are needed. Subjectively, performance remains good for third-party feature files that contain up to several thousand annotated features, but a more sophisticated scheme will be needed to handle larger third-party feature sets.
Configuring GBrowse
To get GBrowse up and running, the database administrator will create a feature database, and then configure GBrowse to use it. Figure 6 shows the steps the administrator of a fictitious site named example.org would take to add a new track showing the positions of a set of targeted deletions. Assuming that the administrator is using the Bio::DB::GFF schema, the first step is to create a GFF-formatted file of the deletion endpoints (Fig. 6a). Each row of the file, named “deletions.gff” in this example, corresponds to one of the deletion features. It has columns indicating the sequence segment on which the feature is located (chromosome name in this example), its type, its start and end points, and the feature ID. The feature type is indicated by the combination of two fields historically known as the “source” and the “method,” which are typically used in GFF files to indicate the subtype and type of the feature, respectively. In this case we have chosen a source of “targeted” and a method of “deletion”. The feature name is indicated by the last two columns of the file, which indicates the name space (“Strain”) and the deletion name. Other columns correspond to attributes that are relevant to gene predictions and sequence alignments, such as the reading frame and alignment score. Empty fields are left blank or replaced with dots as in this example.
Figure 6.

Creating a new track of targeted deletions using GBrowse.
The next step is to load the data into the Bio::DB::GFF database. This is done by running an included script named “load_gff.pl” (Fig. 6b). If the database does not already exist, a command-line switch can be used to create and initialize it.
The last step is to configure GBrowse to recognize and display the new feature type. This is done by adding a new “stanza” to the GBrowse configuration file (Fig. 6c). The stanza is introduced by a pair of square brackets enclosing the internal identifier for the track, in this case “Knockout.” This is followed by a series of option = value pairs that describe what to show in the track. The “feature” option tells GBrowse to search the database for a feature of type “deletion:targeted,” where “deletion” and “targeted” are the method and source of this feature type. The “glyph” option instructs GBrowse to use the “span” glyph, which is rendered as a solid line with two vertical caps at the end, whereas the “fgcolor” option sets the color of the line to red. The “key” option sets the track legend, and the “citation” option (extending over several lines) provides a paragraph of information that describes the track in more detail. The “link” option provides a template for GBrowse's link generation. If the user clicks on a deletion glyph, he or she will be linked to http://example.org/cgi-bin/knockout?$name, where the special symbol “$name” is replaced at run time with the deletion name. Much more complex linking templates are possible, and for cases that do not lend themselves to template substitution rules, the GBrowse configuration file allows the “link” option to be replaced by the definition of a Perl subroutine. At run time, the subroutine will be passed a copy of the feature data, which it can inspect to construct the appropriate outgoing link. This mechanism is used by WormBase to construct outgoing links to the SwissProt-TREMBL database, which requires slightly different URLs to locate proteins in SwissProt and TREMBL (Bairoch and Apweiler 2000).
Changes made to the configuration file take effect immediately, and the knockout track will now appear among the tracks offered to the user. In addition to track-specific stanzas, the configuration file provides settings for such global options as the database adaptor to use and the stylesheet. If the administrator wishes to create more than one data source, he or she simply adds another configuration file, and the user will be offered a choice of data sources.
The process of adding a new track to a version of GBrowse running on top of the GadFly schema is similar, except that the database is loaded from GAME/XML format files using a script that comes with the GadFly distribution.
Extending GBrowse
GBrowse was designed with extensibility in mind. Hence there are multiple distinct layers at which software developers can add new code to extend the browser's capabilities. At the database layer, authors can implement new Bioperl adaptors, thereby allowing GBrowse to communicate with other database management systems; this is how one would adapt GBrowse to work with an existing Sybase or ACeDB database. At the data model layer, authors can implement “Aggregators”; these modules are responsible for building up Bioperl Sequence objects—which may contain arbitrarily complex part/subpart relationships—from their raw database representation, which is necessarily a flattened form. At the graphics rendering layer, authors can create new Glyph modules. Glyphs are responsible for generating graphical representations of sequence features. At the topmost application layer, software developers can create plug-ins that add new searching, annotating, and visualization capabilities to GBrowse.
As a concrete example of this extensibility, consider a bioinformaticist who has developed a splice-site prediction algorithm and wishes to integrate this into GBrowse so as to visualize the position of splice sites on the genome. She would extend GBrowse at two levels. At the graphical layer, the bioinformaticist would create a new “splice site glyph” that represents the splice site as an inverted L whose height corresponds to the predicted splice site's score and whose direction indicates whether the site is a donor or an acceptor. She would then create an Annotator plug-in that runs the prediction algorithm and creates a set of features to be fed into the splice-site glyph. Based on similar modules already in the code base, we estimate that the combined code for the glyph and the plug-in (not counting the actual prediction algorithm), would occupy roughly a page.
GBrowse Security
Security is a concern for any piece of server-side Web software because of the possibility of inadvertently passing untrusted information from the end-user to a system call that opens files or executes commands. GBrowse has been tested with, and passes, Perl's “taint” checking system, a set of checks that track untrusted data and raise an exception if such data are used in potentially dangerous system operations. Because of the performance impact, taint checking is turned off in the default GBrowse configuration, but the data provider has the option of reactivating it for an added level of security.
GBrowse Support
The GMOD Web site is hosted by SourceForge, which provides a polished user interface for user support, feature requests, and bug reports. From the GMOD main page, users can access a discussion forum devoted to GBrowse installation and configuration, a GBrowse mailing list, and a tracking system for bug reports and feature requests. These fora are monitored by GBrowse developers and other parties to the GMOD project, thereby providing a responsive environment for user questions and complaints. There are presently no plans to provide telephone support or a subscription-based user support service.
DISCUSSION
Along with genetic and physical map rendering tools, Web-based genome browsers are a class of application that the bioinformatics research community seems to be doomed to reinvent time and again. Genome browsers can be found at NCBI (Benson et al. 2002), UCSC (Kent et al. 2002), EnsEMBL (Hubbard et al. 2002), NCGR (http://www.ncgr.org/doc/cgs/CGS_User_Manual.pdf), The Sanger Centre (Rutherford et al. 2000), and at most of the model organism databases (at TAIR, http://www.arabidopsis.org/servlets/sv?action=closeup; at SGD, http://genome-www4.stanford.edu/cgi-bin/SGD/ORFMAP/ORFmap?chr=3&beg=&end=1; at WormBase, Stein et al. 2001; and at FlyBase, FlyBase 2002). Indeed, some of the model organism databases have invented the genome browser twice. For example, FlyBase developed two such browsers in the course of its history: GeneSeen, which uses a Java back end driven off flat files, and an earlier GadFly front end that uses Perl driven off a relational database. In addition to genome browsers produced by publicly funded efforts, there are many similar packages produced by private companies, including Celera (http://www.celera.com/genomics/academic/home.cfm?ppage=cds&cpage=default) and DoubleTwist (http://www.doubletwist.com/corporate/products/human_genome_database.shtml).
There are several reasons for this proliferation of browsers. First, there are legitimate differences in data models and user interface choices. Genome annotation groups wish to emphasize different aspects of the data set and to facilitate certain types of comparisons. It is impossible for one browser to meet all the various requirements that may be put upon it, and therefore there will always be a need for multiple such applications. Second are historical reasons: many of the annotation and curation groups listed earlier discovered they needed sequence-browsing software at around the same time, in a period when no packages existed; it is not surprising that development occurred on multiple parallel paths.
Now that multiple genome browsers have been implemented, however, the major factor hindering their dissemination is lack of portability. Most genome browsers are an integral part of a larger nonportable system and simply cannot be redistributed. Examples of this phenomenon include the NCBI, TIGR, and TAIR browsers. Others are part of a system that is portable as a whole, but not portable as individual modules. To use the genome browser requires that the prospective data provider adopt the entire data model, something that may not always be desirable. An example of this phenomenon is EnsEMBL, or any of the commercial genome database packages.
Presently, the closest fit to a genome browser that can be adapted as a component of a model organism database project is the UCSC human genome browser. However, several design decisions made to optimize the performance of the UCSC browser present obstacles to integrating the UCSC browser into a larger system. These decisions include the division of the feature data by chromosome into multiple stand-alone databases, the use of the C programming language for the middleware and user interface layers, and the use of the AutoSQL preprocessor package to marshal C structures into database tables. For these reasons, the UCSC browser is closely tied to the C programming language. However, data providers tend to use a scripting language such as Perl, Java, or Python. In addition, the UCSC browser offers a limited range of glyph shapes and attributes.
The Generic Genome Browser was designed from the bottom up for portability, extensibility, and modularity. It relies on no proprietary software, but only readily available open source software such as MySQL and the Bioperl libraries. It uses the Perl programming language throughout, a language that is familiar to the cohort of beginning to intermediate programmers that are most likely to be involved in setting up a new model organism database or other annotation project. It provides a choice of database back ends and is highly customizable.
GBrowse offers two complementary paths for integration into a larger model organism database project. The first path is to use it as a completely stand-alone product. Under this scheme, GBrowse and its database back end are separate from the rest of the project. Periodically the data provider dumps out the genome features and loads them into the GBrowse back end. Integration with the rest of the model organism project takes place at the level of the Web site, where the data provider uses link rules to forge outgoing links from the GBrowse page to other pages on the Web site. Similarly, incoming links use GBrowse's standard URL calling conventions to select the region of interest and a series of feature types to display. In practice, this scheme seems to work well. For example, the WormBase Web site maintains all genomic feature data in a GBrowse MySQL database, whereas all the other biological data on C. elegans resides in an ACeDB database. From the point of view of the end-user, however, the distinction between the two data sources is invisible.
The second path for integrating GBrowse with a model organism database project is at the database level. In this approach, the database schema for the GBrowse back end is incorporated into the larger project schema, allowing the same physical database to be used for the whole site. Not only is this an attractive solution from an administrative point of view, but it offers the ability to perform complex queries on the integrated data set. For example, integration with the Gene Ontology schema (another aspect of the GMOD project) would allow the database to be searched for physically adjacent genes that belong to common metabolic pathways. The Bio::DB::GFF schema was designed to accommodate this type of schema integration. In addition to having a relatively small number of tables, Bio::DB::GFF tables use a uniform prefix to reduce the chance of table name collision. A single join through the feature group table is all that is required to link foreign tables maintained by the model organism database with genomic coordinate information maintained in Bio:: DB::GFF.
Also by design, GBrowse is complementary to Apollo, the GMOD project's genome annotation editor. Unlike GBrowse, Apollo is a stand-alone Java application that runs on the end-user's machine. Once installed, Apollo provides rapid interactive zooming and scrolling, as well as the ability of authorized users to write back changes to the underlying database. In a typical MOD setting, Apollo would be used by curators to inspect and modify genomic annotations, whereas GBrowse would be accessed by end-users for Web-based access to the genome. However, Apollo and GBrowse integrate well both at the database level (by using the common GadFly schema) or at the file format level (by reading and writing GFF format feature files). This allows for configurations in which experienced end-users can use GBrowse to search for and select a genomic region of interest, and then launch Apollo to inspect the region with greater interactivity.
GBrowse and its underlying database schemata have been stable for roughly half a year, and the software should be considered suitable for production use. GBrowse has become the primary genome browser for the WormBase (http://www.wormbase.org) and FlyBase (http://www.flybase.org) Web sites, displacing the Web-based browsers that were there before. Companies and institutions presently using GBrowse in production systems include Texas A&M University (E. coli), Ingenium AG (mouse), Bristol-Myers Squibb (D. melanogaster), and the Medical College of Wisconsin (R. rattus). Should changes to the database schemata be necessary, the development team is committed to supporting the changes in an upwardly compatible manner, either by supporting older versions of the database in the adaptor layer, or by providing database conversion tools.
An important limitation on GBrowse at present is its restriction to a limited set of databases. MySQL was the first database management system targeted by this project because of its widespread use in the bioinformatics community, its open source status, and its excellent performance in read-mostly environments. A high priority of the project is to extend this support to other DBMSs and database schemata. An Oracle (Loney and Koch 2000) adaptor for Bio::DB::GFF is presently undergoing testing, and a PostgreSQL (Stinson 2001) is in the early stages. A PostgreSQL port of the GadFly schema is complete, but has not yet been tested with GBrowse. In addition, a beta release of a GenBank/EMBL adaptor layer is also available in recent versions of the browser. This layer acts as a transparent proxy on the remote sequence-retrieval facilities provided by the NCBI and EBI, allowing end-users to browse the feature tables of any entry that has been submitted to GenBank or EMBL.
Although preparing a new adaptor for each underlying database schema is effective for a limited number of such cases, this strategy cannot scale indefinitely and will ultimately limit our ability to improve the GBrowse feature set. For this reason, an important near-term goal of the project is to complete an adaptor layer for the Distributed Annotation System (DAS; Dowell et al. 2001). DAS is a standard protocol for electronically publishing genomic annotations that was developed and is actively promoted by several of the coauthors. The protocol has achieved modest penetration among genome databases; it is supported by EnsEMBL, the UCSC genome browser, WormBase, and FlyBase, among others, but has not been adopted by the large archival databases such as GenBank. With the DAS adaptor installed, MOD curators will be able to configure GBrowse to navigate and render genomic annotations served by any local or remote DAS-compliant data source and to superimpose these data sources on a single display. This will provide great flexibility at some cost in performance because of the additional network overhead. However, the performance when rendering large third-party feature sets is expected to improve dramatically because DAS indexing is able to select features that are contained within the genomic region of interest, rather than transferring the entire feature set as occurs with the current flat-file third-party annotation system.
As it happens, a site that is running GBrowse can readily become a DAS data source. This is because of the availability of DAS server software that runs on top of the Bio::DB::GFF and GadFly schemata (see http://www.biodas.org for availability).
In addition, we are looking to enhance GBrowse in a number of other respects. High on our priority list are:
A more flexible query interface, including the ability to search features by particular properties, such as submitting author, and to combine properties with Boolean operators. This will likely be accomplished using the plug-in architecture.
The ability to generate and mark up sequence alignments.
The ability to download FASTA files of derived features, such as spliced transcripts and translated peptides.
A facility that allows the end user to toggle from absolute to relative coordinates easily, so that researchers can view the region in clone coordinates or relative to a gene, STS marker, or other landmark; also the ability to flip the entire display left to right when the end-user is examining a minus strand gene or other feature.
Plug-ins that implement interfaces to gene and motif-finder programs, PCR design, in silico restriction digests, and other common sequence analysis utilities.
Internationalization of the help screens, navigation buttons, and label text.
Performance enhancements.
Although the GMOD project's ultimate goal is to create a full off-the-shelf model organism database, only a handful of components are released at present, and a group that needs to implement such a system now should look carefully at the available alternatives. For a project whose primary task is automated sequence annotation, the EnsEMBL system may well be the best choice because it provides a complete solution consisting of an annotation pipeline, a database to record the pipeline's output, and a Web-based system for browsing gene predictions, functional annotations, and supporting evidence. The main caveat is that EnsEMBL does not provide other components needed by a MOD, such as stock center functions or nomenclature management, and customizing the EnsEMBL code base to provide these features will require a significant level of software development expertise. Customized versions of EnsEMBL are also likely to be incompatible with future EnsEMBL development. On the other hand, for a project that is adding annotations to the genome of an organism that already has a MOD (e.g., mouse), it might be best to forego setting up one's own database entirely, and instead establish a cocuration arrangement in which the project forwards its annotations to the MOD for incorporation and display, either by using DAS or an ad hoc protocol.
As a stand-alone application, GBrowse is positioned between these two extremes. It is recommended for use by a group of modest size that is performing hand or automated annotation of genomic regions and either using the Apollo annotation editor, or which has the minimal amount of scripting skill needed to create a tab-delimited file representing the location and nature of its annotations. The software is particularly suitable for those wishing to place an experimental data set in its genomic context; examples of such data sets include microarray results, EST alignments, and the distribution of knockouts in an insertional mutagenesis study. In this case, GBrowse provides a lightweight way to display those annotations, search them, and share them with colleagues.
The GMOD project continues to progress, with new modules for managing genetic maps, comparative genomic analysis, and literature curation in early release form on the GMOD Web site, and software for managing nomenclature histories, ontologies, expression data, biological pathways, phenotypes, and stock center functionality slated for release over the next two years. Together with GBrowse and Apollo, these modules will one day form the core of a model organism database “constructor set” that will reduce the cost and time required to set up a new database, while simultaneously helping to standardize user interfaces and operating procedures among them.
METHODS
GBrowse requires the following software components:
- 1.
The Perl interpreter, version 5.6.0 or higher (http://www.perl.org).
- 2.
The Apache Web server, version 1.3 or higher (http://www.apache.org).
- 3.
The MySQL database management system, version 3.23.43 or higher (http://www.mysql.org).
- 4.
The following Perl modules, all of which are available at http://www.cpan.org: GD, DBI, DBD::mysql, Digest::MD5, Text::Shellwords.
- 5.
The Bioperl distribution, version 1.02 or higher (http://www.bioperl.org).
In theory, any Web server that supports the CGI protocol will support GBrowse, but only Apache has been tested at this time. Although not required, GBrowse can be used in conjunction with the mod_perl embedded Perl interpreter [http://perl.apache.org], in which case it benefits from a noticeable performance boost.
As described in Results, GBrowse supports two distinct relational database schemata, but both schemata depend, to differing extents, on MySQL-specific features.
WEB SITE REFERENCES
http://genome-www4.stanford.edu/cgi-bin/SGD/ORFMAP/ORFmap?chr=3&beg=&end=1; Chromosomal Features Map Display; Saccharomyces Genome Database (SGD).
http://stein.cshl.org/software/GD/; The GD Graphics Module; L. Stein.
http://www.arabidopsis.org/servlets/sv?action=closeup; SeqViewer Page; The Arabidopsis Information Resource (TAIR).
http://www.bioxml.org/Projects/game/game0.1.html; Genome Annotation Markup Elements (GAME).
http://www.celera.com/genomics/academic/home.cfm?ppage=cds&cpage=default; Celera Discovery System, Celera, Inc.
http://www.doubletwist.com/corporate/products/human_genome_database.shtml; The DoubleTwist Annotated Human Genome Database, DoubleTwist, Inc.
http://www.fruitfly.org/developers/; BDGP Developer's Resources.
http://www.gmod.org; GBrowse.
http://www.hgsc.bcm.tmc.edu/projects/bovine/; Bovine Genome Project, Human Genome Sequencing Center at Baylor College of Medicine.
http://www.labbook.com/products/xmlbsml.asp; LabBook, Inc., XML Standard–BSML.
http://www.ncgr.org/doc/cgs/CGS_User_Manual.pdf; Using the Comparative Genomics System, National Center for Genome Resources.
http://www.nhgri.nih.gov/HGP/HGP_goals/5yrplan.html; Understanding Our Genetic Inheritance; The First Five Years: Fiscal Years 1991–1995; National Human Genome Research Institute.
http://www.nps.ars.usda.gov/menu.htm?newsid=1696; USDA Holds First International Meeting on Comparative Insect Genomics; United States Department of Agriculture.
http://www.wormbase.org/db/seq/gbrowse; C. elegans genome features used in example.
Acknowledgments
We acknowledge the following individuals for their help in the development, documentation, and/or testing of the software described here: Tony Cox, Michael Zhang, Jim Kent, James Tisdall, Gudmundur Thorisson, and Richard Durbin. In addition, we thank Simon Twigger, Leonardo Marino-Ramirez, and Deborah Simon for their roles as beta testers of the software and for their thoughtful feedback. We thank Chia Jen Siao for her comments on the manuscript, Anita Matthes for her technical assistance during its preparation, and three anonymous reviewers for their constructive critiques. This work was supported by NIH grants HG00739 and P41HG02223, as well as a grant from the Howard Hughes Medical Institute.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL lstein@cshl.org; FAX (516) 367-8389.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.403602.
REFERENCES
- Bairoch A, Apweiler R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 2000;28:45–48. doi: 10.1093/nar/28.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennetzen JL, Chandler VL, Schnable P. National Science Foundation-sponsored workshop report. Maize genome sequencing project. Plant Physiol. 2001;127:1572–1578. doi: 10.1104/pp.127.4.1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson DA, Karsch-Mizrachi I, Lipman DJ, Ostell J, Rapp BA, Wheeler DL. GenBank. Nucleic Acids Res. 2002;30:17–20. doi: 10.1093/nar/30.1.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackwell JM. Parasite genome analysis. Progress in the Leishmania genome project. Trans R Soc Trop Med Hyg. 1997;91:107–110. doi: 10.1016/s0035-9203(97)90187-5. [DOI] [PubMed] [Google Scholar]
- Dowell RD, Jokerst RM, Day A, Eddy SR, Stein L. The Distributed Annotation System. BMC Bioinformatics. 2001;2:7. doi: 10.1186/1471-2105-2-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FlyBase. The FlyBase database of the Drosophila genome projects and community literature. Nucleic Acids Res. 2002;30:106–108. doi: 10.1093/nar/30.1.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner MJ. The genome of the malaria parasite. Curr Opin Genet Dev. 1999;9:704–708. doi: 10.1016/s0959-437x(99)00032-5. [DOI] [PubMed] [Google Scholar]
- Guttman A. Proceedings of the ACM SIGMOD Conference. 1984. R-Trees: A dynamic index structure for spatial searching; pp. 47–57. . Boston, MA. [Google Scholar]
- Harris NL. Genotator: A workbench for sequence annotation. Genome Res. 1997;7:754–762. doi: 10.1101/gr.7.7.754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman SL, Subramanian GM, Collins FH, Venter JC. Plasmodium, human and Anopheles genomics and malaria. Nature. 2002;415:702–709. doi: 10.1038/415702a. [DOI] [PubMed] [Google Scholar]
- Hubbard T, Barker D, Birney E, Cameron G, Chen Y, Clark L, Cox T, Cuff J, Curwen V, Down T, et al. The EnsEMBL genome database project. Nucleic Acids Res. 2002;30:38–41. doi: 10.1093/nar/30.1.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, Zahler AM. The intronerator: Exploring introns and alternative splicing in Caenorhabditis elegans. Nucleic Acids Res. 2000;28:91–93. doi: 10.1093/nar/28.1.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent, W.J., Sugnet, C.W., Furey, T.S., Roskin, K.M., Pringle, T.H., Zahler, A.M., and Hausler, D. 2002. The Human Genome Browser at UCSC. Genome Res. (in press). [DOI] [PMC free article] [PubMed]
- Kuspa A, Sucgang R, Shaulsky G. The promise of a protist: The Dictyostelium genome project. Funct Integr Genomics. 2001;1:279–293. doi: 10.1007/s101420000033. [DOI] [PubMed] [Google Scholar]
- Loney K, Koch G. Oracle 8i: The complete reference. Englewood Cliffs, NJ: McGraw-Hill Professional Publishing; 2000. [Google Scholar]
- Mural RJ, Parang M, Shah M, Snoddy J, Uberbacher EC. The Genome Channel: A browser to a uniform first-pass annotation of genomic DNA. Trends Genet. 1999;15:38–39. doi: 10.1016/s0168-9525(98)01624-2. [DOI] [PubMed] [Google Scholar]
- Ouelette BF. The GenBank sequence database. In: Baxevanis AD, Ouellette BF, editors. Bioinformatics: A practical guide to the analysis of genes and proteins. 2nd ed. New York: Wiley-Liss; 2001. pp. 16–45. [Google Scholar]
- Pearson WR. Flexible sequence similarity searching with the FASTA3 program package. Methods Mol Biol. 2000;132:185–219. doi: 10.1385/1-59259-192-2:185. [DOI] [PubMed] [Google Scholar]
- Pennisi E. Genomics. Rat genome off to an early start. Science. 2000;25:1267–1269. [PubMed] [Google Scholar]
- Reese MG, Hartzell G, Harris NL, Ohler U, Abril JF, Lewis SE. Genome annotation assessment in Drosophila melanogaster. Genome Res. 2000;10:483–501. doi: 10.1101/gr.10.4.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutherford K, Parkhill J, Crook J, Horsnell T, Rice P, Rajandream MA, Barrell B. Artemis: Sequence visualization and annotation. Bioinformatics. 2000;16:944–945. doi: 10.1093/bioinformatics/16.10.944. [DOI] [PubMed] [Google Scholar]
- Sasaki T, Burr B. International Rice Genome Sequencing Project: The effort to completely sequence the rice genome. Curr Opin Plant Biol. 2000;3:138–141. doi: 10.1016/s1369-5266(99)00047-3. [DOI] [PubMed] [Google Scholar]
- Stajich, J.E., Block, D., Boulez, K., Brenner., S.E., Chervitz, S.A., Dagdigian, C., Fuellen, G., Gibert, J.G.R., Korf, I., Lapp, H., et al. 2002. The Bioperl toolkit: Perl modules for the life sciences. Genome Res. (this issue). [DOI] [PMC free article] [PubMed]
- Stein L, Sternberg P, Durbin R, Thierry-Mieg J, Spieth J. WormBase: Network access to the genome and biology of Caenorhabditis elegans. Nucleic Acids Res. 2001;29:82–86. doi: 10.1093/nar/29.1.82. . (The browsing software described here has since been replaced with GBrowse.) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stinson B. PostgreSQL essential reference. New York: New Rider's; 2001. [Google Scholar]