

Abstract
Biotin is a necessary cofactor of numerous biotin-dependent carboxylases in a variety of microorganisms. The strict control of biotin biosynthesis in Escherichia coli is mediated by the bifunctional BirA protein, which acts both as a biotin–protein ligase and as a transcriptional repressor of the biotin operon. Little is known about regulation of biotin biosynthesis in other bacteria. Using comparative genomics and phylogenetic analysis, we describe the biotin biosynthetic pathway and the BirA regulon in most available bacterial genomes. Existence of an N-terminal DNA-binding domain in BirA strictly correlates with the presence of putative BirA-binding sites upstream of biotin operons. The predicted BirA-binding sites are well conserved among various eubacterial and archaeal genomes. The possible role of the hypothetical genes bioY and yhfS–yhfT, newly identified members of the BirA regulon, in the biotin metabolism is discussed. Based on analysis of co-occurrence of the biotin biosynthetic genes and bioY in complete genomes, we predict involvement of the transmembrane protein BioY in biotin transport. Various nonorthologous substitutes of the bioC-coupled gene bioH from E. coli, observed in several genomes, possibly represent the existence of different pathways for pimeloyl-CoA biosynthesis. Another interesting result of analysis of operon structures and BirA sites is that some biotin-dependent carboxylases from Rhodobacter capsulatus, actinomycetes, and archaea are possibly coregulated with BirA. BirA is the first example of a transcriptional regulator with a conserved binding signal in eubacteria and archaea.
Biotin (vitamin H) is an essential cofactor for a class of important metabolic enzymes, biotin carboxylases and decarboxylases (Perkins and Pero 2001). The biotin biosynthetic pathway is widespread among microorganisms. The well-studied systems of biotin biosynthesis from Escherichia coli, Bacillus subtilis, and Bacillus sphaericus differ in the first step of biosynthesis. B. subtilis and B. sphaericus use pimeloyl-CoA synthase encoded by the bioW gene to synthesize pimeloyl-CoA from pimelic acid. In addition, pimelic acid formation in B. subtilis has been proposed to use cytochrome P450 encoded by bioI (Stok and De Voss 2000). In E. coli, pimeloyl-CoA is synthesized from L-alanine and/or acetate via acetyl-CoA, instead of pimelic acid (Ifuku et al. 1994), and products of the bioC and bioH genes are required for pimeloyl-CoA synthesis in E. coli. The pathway from pimeloyl-CoA to biotin is similar in E. coli and bacilli and uses products of the bioF, bioD, bioA, and bioB genes (Fig. 1). Genes encoding biotin transporters have not been identified in bacteria until now, but E. coli can uptake biotin by active transport (Piffeteau and Gaudry 1985), and a gene for biotin transport, bioP, has been mapped on the E. coli chromosome (Eisenberg 1985).
Figure 1.

The biotin biosynthesis pathway in bacteria.
The operon organization of the biotin biosynthetic genes differs between E. coli and bacilli. E. coli has bioBFCD operon located divergently with the bioA gene and single bioH gene (DeMoll 1994). In contrast, B. subtilis has the single bioWAFDBI operon (Perkins et al. 1996). Two unlinked biotin biosynthetic operons, bioDAYB and bioXWF, were described in B. sphaericus (Gloeckler et al. 1990). The functions of two new biotin-related genes, bioX and bioY, are presently unknown; however, it has been proposed that BioX of B. sphaericus and BioC of E. coli may function as acyl carrier proteins involved in the pimeloyl-CoA synthesis (Lemoine et al. 1996). Recently, four biotin biosynthetic gene clusters, orf1–bioDA, orf2–bioFB, bioH–orf3, and bioFIIHIIC, were characterized in Gram-positive bacterium Kurthia sp. (Kiyasu et al. 2001). The authors of this study suggested that, in contrast to B. subtilis and B. sphaericus, Kurthia sp. produces pimeloyl-CoA by a pathway similar to that of E. coli.
The biotin operon of E. coli is negatively regulated by biotin and the bifunctional protein BirA (DeMoll 1994). The biotin–protein ligase BirA mediates biotinylation of acetyl-CoA carboxylase via a two-step reaction. Firstly, the adenylate of biotin is synthesized from substrates biotin and ATP and, at the second step, transferred to a unique lysine residue on carboxylase. When biotin is unclaimed, two generated BirA–biotinyl–5′-AMP monomers bind cooperatively to the bioO operator between the divergent bioA and bioBCDF operons and repress transcription in both directions. The BirA protein is composed of the N-terminal DNA-binding (D-b) domain containing a helix–turn–helix (HTH) structure, the central domain, and the C-terminal domain. The central catalytic domain contains the binding site for biotinyl–5′-AMP and also is required for transcriptional regulation (Kwon et al. 2000). The BirA protein of B. subtilis has a similar structure and also can act as a repressor of the bioWAFDBI operon (Bower et al. 1996). Recently, two new BirA-regulated operons of unknown function, yhfUST and yuiG, were detected in B. subtilis by expression microarray analysis (Lee et al. 2001). Imperfect palindromic sequences, which are partially similar to the bioO operator from E. coli, were found upstream of the BirA-regulated operons from B. subtilis, B. sphaericus, and Kurthia sp. (Gloeckler et al. 1990; Kiyasu et al. 2001; Lee et al. 2001).
The large number of complete genomes now available provides an opportunity to perform global comparison of whole metabolic pathways and regulons in a variety of bacteria. The comparative analysis of binding sites for transcriptional regulators in bacterial genomes is a powerful approach to functional annotation of genomes (for review, see Gelfand 1999). The general assumption in such studies is that true sites mostly occur upstream of orthologous genes, whereas false positives are scattered at random in the genome. In addition, analysis of gene clustering on the chromosome allows one to detect functionally coupled genes (Overbeek et al. 1999).
Here, we report the comparative study of the biotin regulon and metabolic pathway in all available prokaryotic genomes. It is shown that birA is the most widely distributed biotin-related gene in bacteria. However, only a fraction of BirA orthologs possess the N-terminal D-b domain with the HTH motif (D-b-BirA). Presence of D-b-BirA in a genome coincides with occurrence of potential BirA sites upstream of biotin-related genes. The BirA-mediated regulation was found in such diverse bacterial lineages as proteobacteria, low-GC Gram-positive bacteria, and archaea. At that, BirA is the only transcriptional regulator with the binding signal conserved in eubacteria and archaea. On the practical side, this analysis allowed us to predict new members of biotin regulons, to assign biotin-transport function to BioY, and to detect nonorthologous displacement of bioH in several lineages and individual genomes.
RESULTS AND DISCUSSION
Orthologs of birA and biotin biosynthetic genes (BBS) from E. coli and B. subtilis were identified in all available bacterial genomes by similarity search (Table 1). The biotin–protein ligase BirA is widely distributed in eubacteria and archaea. Only Buchnera sp., Borrelia burgdorferi, Aeropyrum pernix, thermoplasmas, and mycoplasmas have neither the BBS genes nor birA, which is consistent with the lack of biotin-dependent carboxylases in the genomes of these microorganisms. The BBS genes are less widespread than birA: among all complete genomes, Sinorhizobium meliloti, Rickettsia prowazekii, Deinococcus radiodurans, Thermotoga maritima, Treponema pallidum, most archaea, and Gram-positive pathogens from the Bacillus/Clostridium group lack the BBS genes, but have birA. Among archaeal genomes, only Methanococcus jannaschii has a cluster of the BBS genes. Phylogenetic analysis of the BBS proteins shows that this archaeal BBS gene cluster may be the result of possible horizontal gene transfer from bacilli. The detailed phylogenetic and positional analysis of the BBS genes is given below.
Table 1.
Operon Structure and Predicted BirA Sites for the Biotin Biosynthetic Genes in Prokaryotes
| Genome | AB | BirA | Biotin biosynthetic genes | Biotin transporters | Biotin-dependent caroxylases | BirA sites | Score | Pos | |
|---|---|---|---|---|---|---|---|---|---|
| D-b | BPL | ||||||||
| alpha-Proteobacteria | |||||||||
| Caulobacter crescentus | CO | 0 | + | bioB / bioA <> bioF–bioD / bioC | |||||
| Sinorhizobium meliloti | SM | 0 | + | bioC | cbiO–cbiQ–bioY–yhfT–yhfS | ||||
| Mesorhizobium loti | MLO | 0 | + | bioB–bioF–bioD–bioA–bioZ / bioC | bioY1 / bioY2–X | ||||
| Agrobacterium tumefaciens | AT | 0 | + | bioB–bioF–bioD–bioA–bioZ / bioC | cbiO–cbiQ–bioY | ||||
| Rhodopseudomonas palustris | RPA | 0 | + | bioB / bioF–bioD–bioA / bioC | bioY–X–X | ||||
| Bradyrhizobium japonicum | BJA | 0 | + | bioB / bioF–bioD–bioA / bioC | bioY–X–X | ||||
| Rhodobacter capsulatus # | RS | 0 | + | bioB–bioF–bioD–bioA–bioG / bioC | cbiO1–cbiQ1–bioY1 | madYZGB–birA– | |||
| cbiO2–cbiQ2–bioY2 | madAECDHKFLM | ||||||||
| M. magnetotacticum # | MMA | 0 | + | [bioB bioF] / bioD–bioA / bioC | |||||
| Brucella melitensis | BME | 0 | + | bioB–bioF–bioD–bioA–bioZ / bioC | bioY1 / bioY2-X | ||||
| Rickettsia prowazekii | RP | 0 | + | none | bioY | ||||
| beta-Proteobacteria | |||||||||
| Bordetella pertussis # | BP | 0 | + | bioA <> bioF / bioB | cbiO–cbiQ–bioY | ||||
| Burkholderia fungorum # | BU | 0 | + | bioA–bioF–bioD–bioB / bioC | |||||
| Burkholderia pseudomallei # | BPS | − | + | bioA–bioF–bioD–bioB / bioC | |||||
| Nitrosomonas europaea | NE | + | + | bioB–bioF–bioH–bioC–bioD / $ bioA | cTGTcttgC-(15)-GcTTgACAA | −246 | 5.99 | ||
| Neisseria meningitidis | NM | − | + | bioB / bioH–bioC2 /bioF–bioG–bioC1 / bioA–bioD | |||||
| Methylobacillus flagellatus # | MFL | + | + | $ bioB–bioF–bioH–bioC–bioD / bioA–X | aTGTAAAtg-(15)-GcTTgACAA | −68 | 7.10 | ||
| Ralstonia solanacearum | RSO | 0 | + | bioA–bioF–bioD / X–X–bioB / bioC | |||||
| Ralstonia eutropha # | REU | 0 | + | bioA–bioF–bioD–bioB / bioC | |||||
| gamma-Proteobacteria | |||||||||
| Escherichia coli | EC | + | + | bioA <$> bioB–bioF–bioC–bioD / bioH | TTGTAAACC-(16)-GGTTTACAA | −80 | 9.10 | ||
| Salmonella typhi | TY | + | + | bioA <$> bioB–bioF–bioC–bioD / bioH | TTGTAAACC-(16)-GGTTTACgA | −80 | 8.49 | ||
| Klebsiella pneumoniae # | KP | + | + | bioA <$> bioB–bioF–bioC–bioD / bioH | TTGTAAACC-(16)-GGTTTACAA | −207 | 9.10 | ||
| Yersinia pestis | YP | + | + | bioA <$> bioB–bioF–bioC–bioD / bioH | TTGTAAACC-(16)-GGTTgACAg | −88 | 8.87 | ||
| Vibrio cholerae | VC | + | + | bioA <$> bioB–bioF–bioC–bioD / bioH | aTGTAAACC-(15)-tGTTgACAg | −94 | 8.12 | ||
| Francisella tularensis # | FT | + | + | bioA <$> bioB–bioF–bioC–bioD | TTGTAAACC-(15)-aGTTgACAt | −90 | 8.44 | ||
| Legionella pneumophila # | LP | + | + | [bioA / [bioB–bioF–bioH–bioD / bioC | |||||
| Haemophilus influenzae | HI | − | + | bioA–bioF–bioG–bioC–bioD / bioB | |||||
| Haemophilus ducreyi # | DU | − | + | bioA–bioF–bioG–bioC–bioD / bioB | |||||
| Pasteurella multocida | VK | − | + | bioA–bioF–bioG–bioC–bioD / bioB | |||||
| A. actinomycetemcomitans # | AB | − | + | bioA–bioF–bioG–bioC–bioD / bioB | |||||
| Pseudomonas aeruginosa | PA | + | + | $ bioB–bioF–bioH–bioC–bioD / bioA | aTGTAgtCC-(14)-GGTTgACAg | −130 | 7.43 | ||
| Pseudomonas putida | Ppu | + | + | $ bioB–bioF–bioH–bioC–bioD / bioA | TTGTAAACC-(15)-aGTTgACAg | −125 | 8.47 | ||
| Pseudomonas fluorescencs | PU | + | + | $ bioB–bioF–bioH–bioC–bioD / bioA | aTGTAAACC-(15)-GGTTgACAg | −128 | 8.71 | ||
| Shewanella putrefaciens # | SH | + | + | bioA <$> bioB–bioF–bioC–bioD / bioH | TgGTAAACC-(15)-cGTTgACAg | −90 | 7.78 | ||
| Thermochromatium tepidum # | CTE | + | + | $ bioB–bioF–bioH–bioC]/ [X–bioA | TTGTAAACC-(15)-aGTTgACAA | −112 | 8.60 | ||
| Xylella fastidiosa | XFA | − | + | bioB / bioF–bioH / bioD / bioC / bioA | |||||
| Acinetobacter calcoaceticus # | AC | 0 | + | bioB / bioH–bioA–bioF–bioC–bioD | |||||
| Buchnera sp. | BUC | 0 | 0 | bioA <> bioB–bioD | |||||
| epsilon-Proteobacteria | |||||||||
| Helicobacter pylori | HX | 0 | + | bioA / bioD / X–bioF / bioC / bioB–X | |||||
| Campylobacter jejuni | CJ | 0 | + | bioA <> bioF–bioG–bioC / X–bioD / X–bioB–X | |||||
| Magnetococcus # | MCO | + | + | $ bioF–bioH–bioC1–bioB–X–bioD / bioA / bioC2 | aaGTAAACC-(16)-aGTTgACtA | −46 | 7.44 | ||
| Bacillus/Clostridium group | |||||||||
| Bacillus subtilis | BS | + | + | $ bioW–bioA–bioF–bioD–bioB–biol | ATTGTTAAC-(15)-GTTAACAAT | −127 | 8.84 | ||
| $bioY1 | tTTGTTAAC-(15)-GTTgACAAT | −89 | 8.54 | ||||||
| $bioY2–yhfT–yhfS | tATGTaAAC-(15)-GTTgACATa | −88 | 8.32 | ||||||
| Bacillus sphaericus # | BW | ? | ? | $ bioD–bioA–bioY–bioB | tgTGTTAAC-(16)-GTTAACtAa | −52 | 7.86 | ||
| $ bioX–bioW–bioF | tgTGTTAAC-(15)-GTTAACtca | −67 | 7.49 | ||||||
| Bacillus halodurans | HD | + | + | $ bioB | ATTGTTAAC-(15)-GTTtACAAT | −58 | 8.72 | ||
| $ bioD–bioA | tATGTTAAC-(15)-GTTAACATa | −42 | 8.64 | ||||||
| $ bioF–bioH–bioC | tATGTTAAC-(15)-GTTAACAAT | −68 | 8.74 | ||||||
| $ bioY | tATGTcAAC-(15)-GTTgACAAa | −89 | 8.24 | ||||||
| tATGTTAAC-(15)-GTaAACATT | −35 | 8.08 | |||||||
| Bacillus stearothermophilus # | BE | + | + | $ bioY1–bioD–bioA / [bioB | AATGTaAAC-(15)-GTTgACAAa | −86 | 8.42 | ||
| $bioF | AATGTaAAC-(16)-GTTtACATa | −46 | 8.50 | ||||||
| $ bioY2 | tTTGTTAAC-(15)-GTTtACATa | −33 | 8.52 | ||||||
| Bacillus cereus | ZC | + | + | $ bioA–bioD–bioF–bioH–bioC–bioB | AATGTTAAC-(15)-GTTAACATT | −140 | 8.84 | ||
| $ bioY1 | AATGTTAAC-(16)-GTTAACATT | −32 | 8.84 | ||||||
| $ bioY2–yhfT–yhfS | tTTGTaAAC-(15)-GTTgACAAa | −110 | 8.32 | ||||||
| Clostridium acetobutylicum | CA | + | + | $ bioY1–bioD–bioA | ATTGTTAAC-(16)-GTTAACAAT | −44 | 8.84 | ||
| 0 | + | (D–b–birA) <$> bioY–bioB | ATTGTaAAC-(16)-GTTtACAAT | −44 | 8.60 | ||||
| $ bioY2–X | ATTGTaAAC-(16)-GTTtACAAT | −19 | 8.60 | ||||||
| Clostridium botulinum # | CB | + | + | [bioY–bioB–bioD | $ bioY | ATTGTTAAC-(16)-GTTgACAAT | −81 | 8.64 | |
| Clostridium difficile # | DF | + | + | $ bioB | tAaGTaAAC-(16)-GTTgACAAa | −44 | 7.91 | ||
| ATaGTaAAC-(16)-GTTgACcAa | −108 | 7.17 | |||||||
| $ bioY–yhfS–yhfT | AATGTaAAC-(16)-GTTgACAAa | −113 | 8.42 | ||||||
| Clostridium perfringens | CP | + | + | $ bioY–bioB–bioD | ATTGTaAAC-(16)-GTTgACAAT | −127 | 8.52 | ||
| $ (D–b–birA) | ATTGTagAC-(16)-GTTgtCAAT | −133 | 6.82 | ||||||
| Enterococcus faecalis | EF | + | + | none | $ bioY | ATTGTTAAC-(16)-GTTAACAAT | −49 | 8.84 | |
| Heliobacillus mobilis # | HMO | + | + | [bioD / [bioA | $ bioY | ATTGTcAAC-(16)-GTTgACAAT | −110 | 8.44 | |
| Kurthia sp. # | Kur | ? | ? | $ bioY–bioD–bioA | tATGTTAAC-(14)-GTTgACATa | −101 | 8.44 | ||
| $ orf2–bioF–bioB | tATGTTAAC-(14)-GTTAACATa | −55 | 8.64 | ||||||
| $ bioF–bioH–bioC / bioH | tATGTTAAC-(14)-GTTAACATa | −50 | 8.64 | ||||||
| Listeria innocua | LI | + | + | none | $ bioY | AATGTTAAC-(15)-GTTtACATT | −178 | 8.72 | |
| Lactococcus lactis | LL | + | + | none | (D–b–birA)–bioY <$> yhfT–yhfS | AcaGTTAAC-(16)-GgTtACtgT | −98 | 6.48 | |
| 0 | + | bioY | |||||||
| Staphylococcus aureus | SAX | + | + | $ bioD–bioA–bioB–bioF–bioW–bioX | AATGTaAAC-(15)-GTTtACATT | −56 | 8.60 | ||
| $ bioY | ATTGTaAAC-(15)-GTTtACAAT | −74 | 8.60 | ||||||
| $ yhfT–yhfS | AATGTTAAC-(15)-GTTtACATT | −49 | 8.72 | ||||||
| Streptococcus pneumoniae | PN | + | + | none | $ bioY | tTTGTTAcC-(16)-GTTgACATc | −86 | 7.41 | |
| Steptococcus pyogenes | ST | + | + | none | $ bioY | AcaGTTAcC-(16)-GTTgACAAa | −72 | 7.14 | |
| $ yhfS–yhfT | AAaGTcAcC-(16)-GgTtACAgT | −270 | 6.56 | ||||||
| Streptococcus equi # | SEQ | + | + | none? | $ bioY | gTTGTcAAC-(15)-GgTAgCAAT | 1 | 6.66 | |
| $ yhfS–yhfT | AAaGTTAAC-(16)-GgTtACAAT | −567 | 7.75 | ||||||
| Actinobacteriae | |||||||||
| Corynebacterium glutamicum # | CGL | 0 | + | bioB / bioA–bioD | bioY–cbiO–cbiQ | birA <> pccB1–pccB2 | |||
| Corynebacterium diphtheriae # | DI | 0 | + | bioB1 / bioA–bioD / bioW–bioF / bioB2 | bioY–cbiO–cbiQ | birA <> pccB | |||
| Mycobacterium tuberculosis | MT | 0 | + | bioB / bioA–bioF–bioD | birA <> pccB–X–X–X–pccA | ||||
| Streptomyces coelicolor # | SX | 0 | + | bioF <> bioB–bioA–bioD | bioY | birA <> pccB–X–X–pccA | |||
| Thermomonospora fusca # | TFU | 0 | + | none? | bioY–cbiO–cbiQ | birA–ppc <> pccB–pccA | |||
| CFB/Green sulfur bacteria group | |||||||||
| Bacteroides fragilis # | BX | 0 | + | bioA–bioF–bio(GC)–bioD | |||||
| Cytophaga hutchinsonii # | CHU | 0 | + | bioB / bioF–bioD–bioA | |||||
| Porphyromonas gingivalis # | PG | 0 | + | bioB–bioA] / X–bioD / bioG–bioC / bioF] | |||||
| D-b | BPL | ||||||||
| Cyanobacteria | |||||||||
| Nostoc sp. | NPU | 0 | + | bioB / bioD / bioF / bioA | bioY–lspA | ||||
| Synechocystis sp. | CY | 0 | + | bioB–bioY–IspA / bioD / bioF / bioA | |||||
| Prochlorococcus marinus | CK | 0 | + | X–X–bioB / bioF–X–bioC–bioD–bioA | bioY–IspA | ||||
| Synechococcus sp. | SN | 0 | + | X–X–bioB / bioF–X–bioC–bioD–bioA | bioY–IspA | ||||
| Others | |||||||||
| Aquifex aeolicus | AA | 0 | + | X–X–bioB / bioW–X–X / X–X–bioD / bioA / bioC | |||||
| Chlamydia trachomatis | QT | 0 | + | bioB–bioF–bioD–bioA–bioW | bioY | ||||
| Chlorobium tepidum | CL | + | + | $ birA–bioB–bioF–bioG–bioC–bioD–bioA–fadD | TTGTcAACC-(14)-GGTTTACAA | −143 | 9.00 | ||
| Chloroflexus aurantiacus # | CAU | 0 | + | none | bioY | ||||
| Deinococcus radiodurans | DR | − | + | none | bioY–cbiO–cbiQ | ||||
| Fusobacterium nucleatum # | FN | 0 | + | bioB–bioD–bioA / bioF–bioG–bioC | |||||
| Thermotoga maritima | TM | 0 | + | none | fabH–fabZ–fabK–bioY–fabD | ||||
| Thermus thermophilus # | TQ | + | + | $ bioB | TcGTAAACt-(15)-GGTTTACgA | 21 | 7.48 | ||
| $ bioY | acGTcAACC-(15)-GGTTgACgA | 615 | 7.52 | ||||||
| Treponema pallidum | TP | 0 | + | none | bioY–cbiO–HTP1 | ||||
| Archaea | |||||||||
| Archaeoglobus fulgidus | AG | + | + | none | $ bioY–cbiO–HTP2 | cTcGTTAAC-(15)-GTTAACgAT | −22 | 6.39 | |
| $ pycA | gcTGTaAAt-(16)-GaTAACAAT | −173 | 6.03 | ||||||
| Halobacterium sp. | HSL | + | + | none | $ bioY–cbiO–HTP3 | gTcGTaAAC-(16)-GTTgACgAc | −50 | 5.70 | |
| 0 | + | pccB–pccA–X–$(D–b–birA) | tAaaTaAAC-(14)-GTTgAgtTa | −118 | 5.80 | ||||
| M. thermoautotrophicum | TH | 0 | + | none | bioY | pycA–birA | |||
| Methanococcus jannaschii | MJ | 0 | + | bioB1 / bioB2 <> bioW–bioF–bioD–bioA | |||||
| Methanosarcina barkeri # | MBA | + | + | none? | $ bioY–cbiO–cbiO–cbiQ | AATGTaAAC-(16)-GTTAACAAT | −275 | 8.72 | |
| $ pycB–pycA–(D–b–birA) | gTaGTTcAC-(16)-GTTAACAgg | −365 | 5.81 | ||||||
| Methanosarcina mazei | MMZ | + | + | none | $ bioY–cbiO–cbiO–cbiQ | AATGTaAAC-(16)-GTTAACAAT | −269 | 8.72 | |
| $ pycB–pycA–(D–b–birA) | AAcGgcAgC-(15)-GTTAACAAT | −349 | 6.11 | ||||||
| Pyrococcus abyssii | PO | + | + | none | bioY <$> (D–b–birA) | tTcGTTAAC-(16)-GTTAACcAa | −43 | 6.96 | |
| 0 | + | ||||||||
| Pyrococcus furiosus | PF | + | + | none | bioY > (D–b–birA) | gTgGTTAAC-(16)-GTTACgAa | −55 | 6.49 | |
| 0 | + | ||||||||
| Pyrococcus horikoshii | PH | 0 | + | none | bioY | ||||
| Sulfolobus solfataricus | STO | 0 | + | none | |||||
The genome abbreviations are given in column AB. Unfinished genomes are marked by #. The names of taxonomic groups are given in bold. The signs + and 0 in the columns “BirA D–b” and “BirA BPL” denote the existence or absence of the N-terminal regulatory domain (D-b) and C-terminal catalytic domain (BPL) of BirA, respectively; − denotes N-terminal BirA domain not similar to the known regulatory BirA domain. Other columns show the operon structure and regulation of the biotin-related genes. Genes forming one candidate operon (with spacer <100 bp) are separated by dashes. Different loci are separated by slashes. The direction of transcription in divergons is shown by angle brackets. Predicted BirA sites are denoted by $. The contig ends are shown by square brackets. Bio(GC) is the fusion of the bioG and bioC genes. HTP1, HTP2, and HTP3 are nonhomologous hypothetical transmembrane proteins clustered with bioY–cbiO. The other genes of unknown function are denoted by X. The birA genes are shown only if they are colocalized with other biotin-related genes. The positions of the site are given relative to annotated translation starts. The site scores are computed using positional nucleotide weight matrices of two types, proteobacterial and nonproteobacterial, as described in Methods. The BirA sites of the proteobacterial type are given in bold.
BirA Regulon
To analyze possible transcriptional regulation of the BBS genes, we started with identification of the N-terminal regulatory domains in the detected BirA proteins. Using multiple alignment, we compiled the list of 46 sequences of the BirA N-terminal domains that have the same length as the known regulatory domain of E. coli BirA. To determine the significance of the possible helix–turn–helix (HTH) regulatory motif in each of the collected sequences, the HTH motif prediction program (Dodd and Egan 1990) was used (Fig. 2). After that, eight sequences without HTH motifs were removed, and 38 BirA proteins with the predicted DNA-binding regulatory domains (D-b-BirA) were retained (Table 1). We also retained the BirA protein from Bacillus cereus, although it was predicted to contain no HTH motif. This looks like a false-negative prediction. Indeed, not only is BirA highly conserved among bacilli, but the B. cereus genome has several strong BirA sites upstream of biotin-related operons. To support the selection of D-b-BirA, the phylogenetic tree of 50 BirA N-terminal domains was constructed (Fig. 3). It shows that each sequence without a potential HTH motif is highly diverged from the D-b-BirA sequences and looks like an outgroup in this tree.
Figure 2.

Multiple alignment of the BirA N-terminal domains and identification of the HTH motif. The known secondary structure of the Escherichia coli BirA is shown in the first row. The α2 and α3 helices form the helix–turn–helix (HTH) structure. The score and the probability of the candidate HTH motif are given. A score of <2.5 is not significant. Non-HTH proteins are boxed, except BirA from Bacillus cereus, which is a false-negative prediction (see text). The genome abbreviations are listed in Table 1.
Figure 3.

Maximum likelihood tree of the N-terminal domains of BirA. Domains containing the regulatory HTH motif are shown in solid lines. Other N-terminal domains of BirA (without HTH) are shown as outgroups by broken lines. The proteobacterial and nonproteobacterial subtrees are separated by arrowtail signs. The genome abbreviations are listed in Table 1.
D-b-BirA is widely distributed in the Bacillus/Clostridium group, gamma-proteobacteria, and archaea. In addition, it was found in Nitrosomonas europaea, Methylobacillus flagellatus, Magnetococcus sp., and Thermus thermophilus. The N-terminal domains of BirA from the Pasteurellaceae family of gamma-proteobacteria possibly have lost their regulatory function. The genomes of Clostridium acetobutylicum, Lactococcus lactis, Halobacterium sp., Pyrococcus abyssii, and Pyrococcus furiosus have two BirA paralogs, with and without the N-terminal regulatory domain. The phylogenetic analysis of the catalytic BirA domains shows that paralogous BirA in the first three genomes could result from a recent duplication. In P. abyssii and P. furiosus, BirA without the N-terminal regulatory domain is close to the other archaeal BirA, whereas the second BirA (D-b-BirA) has a weakly conserved catalytic domain and a well-conserved N-terminal regulatory domain.
Based on the phylogenetic analysis of the D-b domains, all D-b-BirAs were divided into two major groups, proteobacterial and nonproteobacterial (Fig. 3). Consistent with this, two different recognition rules (profiles) for the BirA sites were constructed using the sets of upstream regions of the BBS genes from various genomes. The BirA profile for proteobacteria (with consensus 5′-tTGTaAACC-N14 … 16-GGTTtACAa-3′, where strongly conserved positions are shown in capitals) is more strict than that for other bacteria (5′wwTGTtAAC-N14 … 16-GTTaACAww-3′, where ‘w’ stands for A or T). The constructed profiles were used to detect new candidate members of the BirA regulons in the genomes containing D-b-BirA. Proteobacteria possess only one strong BirA site per genome occurring upstream of the BBS operon. However, most Gram-positive bacteria and some archaea have multiple BirA sites located upstream of BBS genes and new genes of the BirA regulon (Table 1). For a control, we checked the genomes without D-b-BirA for the existence of BirA sites upstream of the BBS operons, and found none.
After comparison of the BirA regulons from numerous bacteria, we predicted several new biotin-regulated genes. A gene of unknown function, bioY (so named by Gloeckler et al. 1990), is widely distributed in bacteria and often clusters with genes of biotin metabolism. The homologs of BioY form a unique protein family (InterPro entry IPR003784), and have no significant similarity to any gene of known function. Analysis of the BirA sites showed that bioY is always under regulation of the biotin repressor in genomes containing regulatory D-b-BirA. The existence of the BirA-regulated bioY in several complete genomes that have no BBS genes indicates that bioY is probably not involved in biotin biosynthesis. On the other hand, proteins of the BioY family have six candidate transmembrane segments, an arrangement typical for prokaryotic transporters. The phylogenetic tree of the BioY protein family consists of several branches, and within each branch most members are positionally linked to BBS genes, or have upstream candidate BirA-binding sites, or both (Fig. 4A). Taken together, these observations strongly imply that all BioY paralogs are transporters of biotin or some biotin precursor.
Figure 4.

Maximum likelihood trees of the predicted biotin-related transporter BioY (A), the hypothetical long-chain-fatty acid-CoA ligase YhfT (B), and the hypothetical acetyl-CoA-acetyltransferase YhfS (C). Genes predicted to be regulated by BirA are boxed and shown in bold. The co-occurrence of the bioY, yhfS, and yhfT genes in one genome is shown by thick lines. Background colors signify: (black) single bioY gene; (blue) bioY from the biotin biosynthetic operon; (red) bioY in one operon with cbiO–cbiQ; (yellow) bioY in one operon with lspA; (magenta) bioY in the fadH–fabZ–fabK–bioY–fabD operon; (green) bioY positionally linked to the yhfS-yhfT gene pair. The bioY genes positionally linked to birA are shown by broken lines. The genome abbreviations are listed in Table 1.
Another gene pair of unknown function, yhfS–yhfT, has been detected in several bacteria from the Bacillus/Clostridium group and in S. meliloti. Except for the latter genome, the yhfS–yhfT genes are always under predicted regulation by BirA. YhfT and YhfS are homologous to numerous long-chain fatty acid-CoA ligases and acetyl-CoA-acetyltransferases, respectively. Each of them forms a separate branch on the phylogenetic tree for the corresponding protein family (Fig. 4B,C). One of the bioY paralogs from B. subtilis, yhfU, belongs to the yhfUST operon, and transcription of this operon is repressed by BirA (Lee et al. 2001). In addition, yhfU and yhfS–yhfT are clustered in the genomes of B. cereus, Lactococcus lactis, Clostridium difficile, and S. meliloti; whereas Streptococcus pyogenes, Streptococcus equi, and Staphylococcus aureus have separate BirA-regulated yhfST and yhfU operons. Surprisingly, all YhfU paralogs except one from C. difficile form a separate branch in the phylogenetic tree of the BioY family (Fig. 4A). Again, occurrence of the positionally linked yhfU–yhfS–yhfT genes in complete genomes without BBS genes rules out their involvement in the first steps of biotin biosynthesis. A plausible hypothesis is that the YhfS–YhfT proteins are involved in fatty acid metabolism, the pathway that requires biotin at one of the early steps (cf. clustering of bioY with fatty acid biosynthetic genes in T. maritima; see below).
Positional Analysis of Biotin Genes
To reveal new biotin-related genes, we analyzed putative operon structures and chromosomal clustering of the BBS, birA, and bioY genes. In some eubacterial and archaeal genomes, bioY is clustered with a hypothetical two-component ABC cassette that encodes ATPase and permease components from the CbiO and CbiQ families, respectively (Table 1; Fig. 4A). The cbiN–cbiO–cbiQ operon of Salmonella typhimurium encodes the permease, ATPase, and the second permease components, respectively, of a putative cobalt transporter (Roth et al. 1993). Analysis of the phylogenetic trees for the CbiO and CbiQ protein families shows the existence of separate tree branches for the bioY-linked CbiO and CbiQ components of putative ABC transporters from S. meliloti, R. capsulatus, Agrobacterium tumefaciens, Bordetella pertussis, Thermomonospora fusca, two corynebacteria, and D. radiodurans (data not shown). The bioY genes from T. pallidum, Halobacterium sp., and Archaeoglobus fulgidus form possible operons with cbiO homologs and hypothetical transmembrane proteins (with six predicted TMS) that are not similar to any known protein. Both Methanosarcina genomes have BirA-regulated bioY–cbiO1–cbiO2–cbiQ operons encoding two paralogous ATPase components from the CbiO family. Computational approaches alone cannot explain the possible functional link between the predicted biotin transporter BioY and the putative ABC transporter CbiO–CbiQ, but the obtained data seem to be sufficiently strong to warrant experimental analysis.
Another interesting finding is that bioY from T. maritima was found in one operon with genes involved in fatty acid biosynthesis (Table 1). One logical explanation of this linkage is that fatty acid biosynthesis requires biotin as a coenzyme for a hypothetical biotin carboxylase. In addition, positional linkage of the bioY gene with a hypothetical signal peptidase lspA was observed in all cyanobacteria; the functional meaning of this observation is unclear.
Some differences in the gene organization and BirA-mediated regulation of the bioY genes were observed in three Pyrococcus genomes. Strong BirA sites in the common regulatory regions of divergently transcribed bioY and birA genes were predicted in the genomes of P. abyssii and P. furiosus. Besides the regulatory birA gene, these two genomes also contain the second birA gene, encoding BirA without the regulatory domain. In contrast, Pyrococcus horikoshii has no regulatory birA gene, and BirA sites were not found in this genome.
We predicted possible coregulation of various biotin-dependent carboxylases and BirA in some genomes (Table 1). The pycA and pycB genes encoding the biotin-dependent pyruvate carboxylase were found in one candidate operon with birA in two Methanosarcina genomes. These Methanosarcina operons and the single pycA gene from A. fulgidus are preceded by weak BirA sites. The genes encoding subunits of putative propionyl-CoA carboxylase (pccA and pccB) are clustered on the chromosome with the birA gene in all actinobacteria and Halobacterium sp. Finally, in R. capsulatus, birA is located within a long gene cluster encoding components of the malonate decarboxylase Na+ pump. The BirA-regulated gene clusters from C. acetobutylicum, L. lactis, and some archaea contain the birA gene itself; therefore, the biotin repressors from these bacteria can be autoregulated.
The bioC–bioH gene pair is required for the synthesis of pimeloyl-CoA in E. coli. The bioC gene is widely distributed in bacteria, whereas bioH was not found in many bioC-containing bacterial genomes. Instead, we predict several nonorthologous gene displacements of bioH in some of these genomes. It was recently shown that the bioZ gene from the bioABFDZ operon of Mesorhizobium loti can complement bioH of E. coli (Sullivan et al. 2001). The orthologs of bioZ with the same gene organization were found in A. tumefaciens and Brucella melitensis.
Using comparative analysis, we have detected displacement of bioH by another gene, named here bioG, in some proteobacteria (including all Pasteurellaceae), the CFB group of bacteria, and Fusobacterium nucleatum (Table 1). The bioG gene always forms an operon with bioC and other BBS genes in these genomes; furthermore, in Bacteroides fragilis there is a single gene encoding a fused protein BioC–BioG. Interestingly, all gamma-proteobacteria except Pasteurellaceae possess the bioC–bioH gene pair, whereas all Pasteurellaceae have bioC–bioG. Neisseria meningitidis has both bioC–bioH and bioC–bioG gene pairs, and the latter likely has been acquired from Haemophilus influenzae or a closely related bacterium, as the respective genes are highly similar. The phylogenetic tree of the BioC family has a separate branch for the proteins associated with BioG (Fig. 5).
Figure 5.

Maximum likelihood tree of BioC. The proteins predicted to be associated with (blue) BioH, (red) BioG, (yellow) BioZ, and (green) BioK. The genome abbreviations are listed in Table 1.
Another bioC-linked gene, named bioK, was found in two cyanobacteria, Synechococcus sp. and Prochlorococcus marinus. The genomes of these bacteria contain the bioFKCDA operon and the bioB gene. Two other cyanobacteria, Synechocystis sp. and Nostoc sp., have all biotin biosynthetic genes except bioC and bioK. Therefore, they possibly use a different pathway for pimeloyl-CoA synthesis.
Using similarity search, we detected that BioC possesses an S-adenosylmethionine binding motif (InterPro entry IPR000379) and belongs to the methyltransferase superfamily. BioK and BioG are not similar to any known protein. The BioZ protein is similar to the 3-oxoacyl-[acyl-carrier-protein] synthase FabH involved in fatty acid biosynthesis in bacteria. Another BioC-linked protein, BioH, possesses the active-site serine of a wide variety of enzymes including esterases, lipases, and peptidases (InterPro entry IPR000379) and is similar to arylesterase EstE from Pseudomonas fluorescens (26% identity). All bioK and bioG genes, as well as most bioH genes, are located immediately upstream of the bioC gene in the biotin operon.
The observed diversity of enzymes for the first step of biotin biosynthesis can reflect either frequent nonorthologous gene displacements, or possible use of different substrates for biotin biosynthesis. In contrast, B. subtilis, S. aureus, Corynebacterium diphtheriae, Aquifex aeolicus, and M. jannaschii possess pimeloyl-CoA synthase encoded by the bioW gene and can use pimelate as a biotin precursor (Table 1).
It remains unclear why the comparative analysis of regulation and operon structures failed to identify missing BBS genes in the complete genomes of Clostridium perfringens and C. acetobutylicum. The former has no the bioF and bioA counterparts, whereas the latter lacks only bioF. However, these bacteria possess the predicted biotin transporter BioY. It would be interesting to check if these bacteria can synthesize biotin de novo, and if they can, to search for genes missing in their incomplete BBS pathways.
Conclusions
The biotin–protein ligase BirA is a ubiquitous enzyme in bacteria. In addition, BirA can act as a repressor of transcription when it has the N-terminal DNA-binding domain. Using a global analysis of BirA proteins and DNA-binding sites in available bacterial genomes, we have found that the BirA regulon is widely distributed in eubacteria and archaea. A correlation exists between the presence of D-b-BirA and finding of the BirA sites in bacterial genomes. Conservation of the BirA binding sites across large phylogenetic distances allows us to suggest that D-b-BirA is the first example of an ancient DNA-binding transcriptional factor common to eubacteria and archaea. It is unlikely that numerous BirA regulons in various archaea result from mass gene transfer from bacteria, as this scenario would involve many similar, but independent events (although some cases of horizontal transfer are very clear). In contrast, analysis of regulatory systems for biosynthesis of riboflavin and thiamin showed that they are operated by conserved RNA elements, the RFN element (Vitreschak et al. 2002) and the Thi-box (Miranda-Rios et al. 2001), respectively. These unique regulatory elements are widely distributed in eubacteria and, in addition, several Thi-boxes have been found in archaeal genomes (Vitreschak et al. 2002). Thus, it seems very likely that, in general, the regulatory systems for vitamin biosynthesis are ancient.
Comparative analysis of the biotin regulon in complete genomes resulted in new functional assignments for the bioY, yhfS, and yhfT genes. The first of them, bioY, widely distributed in eubacteria and archaea, is a member of the BirA regulon in all genomes containing D-b-BirA, and it has been predicted to encode a transporter for biotin or biotin-related compounds. Proteins YhfS and YhfT, associated with BioY, can be involved in the metabolic pathway that requires biotin as a coenzyme. The systematic comparison of putative operon structures revealed the conserved gene string bioY–cbiO–cbiQ in some bacterial genomes. Such functional linkage between the putative ABC transporter CbiO–CbiQ and the biotin transporter BioY is enigmatic.
Positional analysis resulted in dissection of novel interesting examples of coregulation of biotin-related genes. Positional linkage between birA and genes encoding biotin-dependent carboxylases was found in Actinobacteria and some archaea, and a fraction of these genes were predicted to be regulated by the biotin repressor. Several genomes have divergently transcribed birA and bioY genes with predicted BirA sites in their common regulatory region. Another example of coregulation of bioY with genes of fatty acid biosynthesis in T. maritima can be easily explained, as biotin is a required cofactor of carboxylase, the latter being involved in the first step of fatty acid biosynthesis.
The enzymes mediating the first step of the biotin biosynthetic pathway are diverse. BioW and BioC represent two major types of enzymes involved in the synthesis of pimeloyl-CoA, a biotin precursor. Moreover, another type of pimeloyl-CoA synthetase, namely, PauA, was found recently in Pseudomonas mendocina (Binieda et al. 1999). In contrast to BioW, PauA belongs to the newly recognized superfamily of acyl-CoA synthetases (Sanchez et al. 2000) and is involved in catabolism rather than biosynthesis. The most interesting observation is that various bacteria have different BioC-associated proteins (BioH, BioG, BioK, or BioZ). It can be explained either by utilization of different sources for biotin biosynthesis or by nonorthologous displacements of the BioC-linked proteins.
This report once again shows the power of comparative genomics for prediction of regulatory sites and functional annotation of genomes, especially when experimental data are limited. In particular, this approach is a powerful tool for prediction of missing transport genes, shown by this study and in the analysis of riboflavin (Vitreschak et al. 2002) and thiamin (A. Vitreschak, D. Rodionov, A. Mironov, and M. Gelfand, in prep.) regulons.
METHODS
Complete and partial bacterial genomes were downloaded from GenBank (Benson et al. 2000). Preliminary sequence data were also obtained from the Web sites of the Institute for Genomic Research (http://www.tigr.org), the University of Oklahoma's Advanced Center for Genome Technology (http://www.genome.ou.edu/), the Wellcome Trust Sanger Institute (http://www.sanger.ac.uk/), the DOE Joint Genome Institute (http://jgi.doe.gov), and the ERGO database (Overbeek et al. 2000; http://ergo.integratedgenomics.com/ERGO/). The gene identifiers from the ERGO database and GenBank are used throughout.
The existence of BirA with an N-terminal DNA-binding domain (D-b-BirA) is a prerequisite to the comparative analysis of the BirA regulons in bacteria. Therefore, the bacterial genomes containing D-b-BirA were selected and divided into two major groups, proteobacterial and nonproteobactertial including archaeal, according to the phylogenetic tree of the DNA-binding domains of D-b-BirA (Fig. 3). Two training sets were composed; each of them included the upstream regions of the biotin biosynthetic genes (operons) from one of the above genomic groups.
For construction of the BirA profiles, we used the ”inverted repeat” option in the SignalX program (Mironov et al. 2000) with a 14–16-bp spacer between two 9-bp units of the inverted repeat. The positional nucleotide weights in the profile were defined as
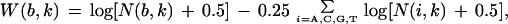 |
where N(b,k) is the count of nucleotide b in position k (Mironov et al. 1999). The score of a candidate site was calculated as the sum of the respective positional nucleotide weights:
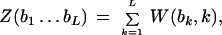 |
where L is the length of the site. All genomes containing D-b-BirA were scanned using the constructed profiles, and the genes with candidate regulatory sites in the upstream regions were selected.
Protein alignment was performed using the Smith–Waterman algorithm implemented in the GenomeExplorer program (Mironov et al. 2000). Orthologous proteins were defined by the best-bidirectional-hits criterion (Tatusov et al. 2000). Distant homologs were identified using PSI-BLAST (Altschul et al. 1997). Multiple sequence alignments were constructed using CLUSTALX (Thompson et al. 1997). Phylogenetic trees were created by the maximum likelihood method implemented in PHYLIP (Felsenstein 1981) and drawn using the GeneMaster program (A.A. Mironov, unpubl.). Prediction of potential transmembrane segments in protein sequences was done using TMpred (http://www.ch.embnet.org/software/TMPRED_form.html). Helix–turn–helix (HTH) DNA-binding motifs were analyzed using the weight matrix method (Dodd and Egan 1990; http://npsa-pbil.ibcp.fr/). The significance of a candidate HTH motif in a given sequence was estimated using the HTH score and probability reported by the above program. In addition, the InterPro database (Apweiler et al. 2000; http://www.ebi.ac.uk/interpro/) was used to verify the protein functional and structural annotation.
WEB SITE REFERENCES
http://ergo.integratedgenomics.com/ERGO/; ERGO database.
http://jgi.doe.gov; DOE Joint Genome Institute.
http://npsa-pbil.ibcp.fr; Network Protein Sequence Analysis server.
http://www.ch.embnet.org/software/TMPRED_form.html; TMpred Server.
http://www.ebi.ac.uk/interpro/; InterPro database.
http://www.genome.ou.edu; University of Oklahoma's Advanced Center for Genome Technology.
http://www.sanger.ac.uk; Wellcome Trust Sanger Institute.
http://www.tigr.org/; Institute for Genomic Research.
Acknowledgments
The authors are grateful to Andrei Osterman, Olga Vassieva, Sveta Gerdes, and Alexandra Rachmaninova for helpful discussions. This study was partially supported by grants from INTAS (99-1476) and HHMI (55000309). It is a part of the “missing genes” project of Integrated Genomics.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL rodionov@genetika.ru; FAX 7-095-3150501.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.314502.
REFERENCES
- Altschul S, Madden T, Schaffer A, Zhang J, Zhang Z, Miller W, Lipman D. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apweiler R, Attwood TK, Bairoch A, Bateman A, Birney E, Biswas M, Bucher P, Cerutti L, Corpet F, Croning MD, et al. InterPro—An integrated documentation resource for protein families, domains and functional sites. Bioinformatics. 2000;16:1145–1150. doi: 10.1093/bioinformatics/16.12.1145. [DOI] [PubMed] [Google Scholar]
- Benson DA, Karsch-Mizrachi I, Lipman DJ, Ostell J, Rapp BA, Wheeler DL. GenBank. Nucleic Acids Res. 2000;28:15–18. doi: 10.1093/nar/28.1.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binieda A, Fuhrmann M, Lehner B, Rey-Berthod C, Frutiger-Hughes S, Hughes G, Shaw NM. Purification, characterization, DNA sequence and cloning of a pimeloyl-CoA synthetase from Pseudomonas mendocina 35. Biochem J. 1999;340:793–801. [PMC free article] [PubMed] [Google Scholar]
- Bower S, Perkins JB, Yocum RR, Howitt CL, Rahaim P, Pero J. Cloning, sequencing, and characterization of the Bacillus subtilis biotin biosynthetic operon. J Bacteriol. 1996;178:4122–4130. doi: 10.1128/jb.178.14.4122-4130.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMoll E. Biosynthesis of biotin and lipoic acid. In: In: Neidhardt FC, editor. Escherichia coli and Salmonella. Cellular and molecular biology. Washington, DC.: American Society for Microbiology; 1994. pp. 704–709. [Google Scholar]
- Dodd IB, Egan JB. Improved detection of helix–turn–helix DNA-binding motifs in protein sequences. Nucleic Acids Res. 1990;18:5019–5026. doi: 10.1093/nar/18.17.5019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg MA. Regulation of the biotin operon in E. coli. Ann NY Acad Sci. 1985;447:335–349. doi: 10.1111/j.1749-6632.1985.tb18449.x. [DOI] [PubMed] [Google Scholar]
- Felsenstein J. Evolutionary trees from DNA sequences: A maximum likelihood approach. J Mol Evol. 1981;17:368–376. doi: 10.1007/BF01734359. [DOI] [PubMed] [Google Scholar]
- Gelfand MS. Recognition of regulatory sites by genomic comparison. Res Microbiol. 1999;150:755–771. doi: 10.1016/s0923-2508(99)00117-5. [DOI] [PubMed] [Google Scholar]
- Gloeckler R, Ohsawa I, Speck D, Ledoux C, Bernard S, Zinsius M, Villeval D, Kisou T, Kamogawa K, Lemoine Y. Cloning and characterization of the Bacillus sphaericus genes controlling the bioconversion of pimelate into dethiobiotin. Gene. 1990;87:63–70. doi: 10.1016/0378-1119(90)90496-e. [DOI] [PubMed] [Google Scholar]
- Ifuku O, Miyaoka H, Koga N, Kishimoto J, Haze S, Wachi Y, Kajiwara M. Origin of carbon atoms of biotin. 13C-NMR studies on biotin biosynthesis in Escherichia coli. Eur J Biochem. 1994;220:585–591. doi: 10.1111/j.1432-1033.1994.tb18659.x. [DOI] [PubMed] [Google Scholar]
- Kiyasu T, Nagahashi Y, Hoshino T. Cloning and characterization of biotin biosynthetic genes of Kurthia sp. Gene. 2001;265:103–113. doi: 10.1016/s0378-1119(01)00354-7. [DOI] [PubMed] [Google Scholar]
- Kwon K, Streaker ED, Ruparelia S, Beckett D. Multiple disordered loops function in corepressor-induced dimerization of the biotin repressor. J Mol Biol. 2000;304:821–833. doi: 10.1006/jmbi.2000.4249. [DOI] [PubMed] [Google Scholar]
- Lee JM, Zhang S, Saha S, Santa Anna S, Jiang C, Perkins J. RNA expression analysis using an antisense Bacillus subtilis genome array. J Bacteriol. 2001;183:7371–7380. doi: 10.1128/JB.183.24.7371-7380.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemoine Y, Wach A, Jeltsch JM. To be free or not: The fate of pimelate in Bacillus sphaericus and in Escherichia coli. Mol Microbiol. 1996;19:645–647. doi: 10.1046/j.1365-2958.1996.t01-4-442924.x. [DOI] [PubMed] [Google Scholar]
- Miranda-Rios J, Navarro M, Soberon M. A conserved RNA structure (Thi box) is involved in regulation of thiamin biosynthetic gene expression in bacteria. Proc Natl Acad Sci. 2001;98:9736–9741. doi: 10.1073/pnas.161168098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mironov AA, Koonin EV, Roytberg MA, Gelfand MS. Computer analysis of transcription regulatory patterns in completely sequenced bacterial genomes. Nucleic Acids Res. 1999;27:2981–2989. doi: 10.1093/nar/27.14.2981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mironov AA, Vinokurova NP, Gelfand MS. GenomeExplorer: Software for analysis of complete bacterial genomes. Mol Biol. 2000;34:222–231. [Google Scholar]
- Overbeek R, Fonstein M, D'Souza M, Pusch GD, Maltsev N. The use of gene clusters to infer functional coupling. Proc Natl Acad Sci. 1999;96:2896–2901. doi: 10.1073/pnas.96.6.2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overbeek R, Larsen N, Pusch GD, D'Souza M, Selkov E, Jr, Kyrpides N, Fonstein M, Maltsev N, Selkov E. WIT: Integrated system for high-throughput genome sequence analysis and metabolic reconstruction. Nucleic Acids Res. 2000;28:123–125. doi: 10.1093/nar/28.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins JB, Pero JG. Vitamin biosynthesis. In: Sonenshein AL, et al., editors. Bacillus subtilis and its relatives: From genes to cells. Washington, DC.: American Society for Microbiology; 2001. pp. 279–293. [Google Scholar]
- Perkins JB, Bower S, Howitt CL, Yocum RR, Pero J. Identification and characterization of transcripts from the biotin biosynthetic operon of Bacillus subtilis. J Bacteriol. 1996;178:6361–6365. doi: 10.1128/jb.178.21.6361-6365.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piffeteau A, Gaudry M. Biotin uptake: Influx, efflux and countertransport in Escherichia coli K12. Biochim Biophys Acta. 1985;816:77–82. doi: 10.1016/0005-2736(85)90395-5. [DOI] [PubMed] [Google Scholar]
- Roth JR, Lawrence JG, Rubenfield M, Kieffer-Higgins S, Church GM. Characterization of the cobalamin (vitamin B12) biosynthetic genes of Salmonella typhimurium. J Bacteriol. 1993;175:3303–3316. doi: 10.1128/jb.175.11.3303-3316.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez LB, Galperin MY, Muller M. Acetyl-CoA synthetase from the amitochondriate eukaryote Giardia lamblia belongs to the newly recognized superfamily of acyl-CoA synthetases (nucleoside diphosphate-forming) J Biol Chem. 2000;275:5794–5803. doi: 10.1074/jbc.275.8.5794. [DOI] [PubMed] [Google Scholar]
- Stok JE, De Voss J. Expression, purification, and characterization of BioI: A carbon—carbon bond cleaving cytochrome P450 involved in biotin biosynthesis in Bacillus subtilis. Arch Biochem Biophys. 2000;384:351–360. doi: 10.1006/abbi.2000.2067. [DOI] [PubMed] [Google Scholar]
- Sullivan JT, Brown SD, Yocum RR, Ronson CW. The bio operon on the acquired symbiosis island of Mesorhizobium sp. strain R7A includes a novel gene involved in pimeloyl-CoA synthesis. Microbiology. 2001;147:1315–1322. doi: 10.1099/00221287-147-5-1315. [DOI] [PubMed] [Google Scholar]
- Tatusov RL, Galperin MY, Natale DA, Koonin EV. The COG database: A tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000;28:33–36. doi: 10.1093/nar/28.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The CLUSTAL-X windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;25:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitreschak AG, Rodionov DA, Mironov AA, Gelfand MS. Regulation of riboflavin biosynthesis and transport genes in bacteria by transcriptional and translational attenuation. Nucleic Acids Res. 2002;30:3141–3151. doi: 10.1093/nar/gkf433. [DOI] [PMC free article] [PubMed] [Google Scholar]