

Abstract
It is expected that one of the merits of comparative genomics lies in the transfer of structural and functional information from one genome to another. This is based on the observation that, although the number of chromosomal rearrangements that occur in genomes is extensive, different species still exhibit a certain degree of conservation regarding gene content and gene order. It is in this respect that we have developed a new software tool for the Automatic Detection of Homologous Regions (ADHoRe). ADHoRe was primarily developed to find large regions of microcolinearity, taking into account different types of microrearrangements such as tandem duplications, gene loss and translocations, and inversions. Such rearrangements often complicate the detection of colinearity, in particular when comparing more anciently diverged species. Application of ADHoRe to the complete genome of Arabidopsis and a large collection of concatenated rice BACs yields more than 20 regions showing statistically significant microcolinearity between both plant species. These regions comprise from 4 up to 11 conserved homologous gene pairs. We predict the number of homologous regions and the extent of microcolinearity to increase significantly once better annotations of the rice genome become available.
Comparative genome analysis has demonstrated that across different plant species, which diverged from a common ancestor but currently tend to vary largely in genome sizes, gene content and order are often conserved. Especially, comparative genetic mapping in the grasses revealed a high degree of conservation of markers within large chromosomal segments (for reviews, see Gale and Devos 1998; Keller and Feuillet 2000). Because, in general, different plant species use homologous genes for similar functions, these observations have great potential. Comparative genome mapping experiments can be a powerful and efficient tool to transfer biological information from a well-studied reference genome to related plant species. However, there are some serious drawbacks when using comparative genetic maps based on recombinational mapping of DNA markers. First, when the marker density is low, small exceptions to colinearity will not be observed, and second, the fact that most genes are organized in multigene families makes it difficult to determine whether real orthologous loci are being compared. Consequently, one can imagine that many experiments suffer from a bias toward promoting colinear regions and miss exceptions to colinearity (Bennetzen 2000a).
The various sequencing efforts over the last few years, such as the complete genome sequence of the model plant Arabidopsis thaliana (Arabidopsis Genome Initiative 2000), the YAC and BAC insert libraries of several grass genomes (Panstruga et al. 1998; Feuillet and Keller 1999) and the International Rice Genome Sequencing Project (Sasaki and Burr 2000), make it possible to investigate whether the degree of colinearity found in comparative genetic mapping experiments is also observed at the gene level. The existence of colinearity between model species and other plant species, even in a limited number of small regions, could provide the opportunity to use these model systems to identify candidate genes in other plants. Comparative sequence analysis at the submegabase level indicates that microcolinearity is abundant between closely related plant species, although exceptions do appear (Chen and Bennetzen 1996; Kilian et al. 1997; Tikhonov et al. 1999; Tarchini et al. 2000). A high degree of conservation of gene content and order between orthologous loci of rice, maize, and sorghum has been reported (Chen et al. 1997). These grass species diverged from a common ancestor ∼50 million years ago. Also, within related dicots, microcolinearity can be observed. For example, conserved gene content and order have been demonstrated between tomato and Arabidopsis, which diverged ∼112 million years ago (Ku et al. 2000), between Arabidopsis and soybean (Grant et al. 2000), and between tomato, Arabidopsis and Capsella (Rossberg et al. 2001). All of these comparative studies revealed that rearrangements, such as inversions, deletions, insertions, and tandem duplications, are an important mechanism responsible for breaking up colinearity, and consequently, make it hard to detect the remnants of colinearity. In addition, these rearrangement processes appear to be more active in some plant lineages than in others (Devos et al. 1993; Devos and Gale 1997; Schmidt 2000).
When comparing more anciently diverged plant species, such as monocots and dicots, more rearrangements are expected to have occurred and, consequently, gene content and order to be less conserved. Recent DNA sequence analysis seems to confirm this assumption and several lines of evidence result in a plastic model in which the modern plant genome is characterized by a series of nested duplications in addition to the species-specific levels of rearrangements (Arabidopsis Genome Initiative 2000; Vision et al. 2000; Wendel 2000). Whether these currently observed large-scale gene duplications are the result of polyploidization, successive hyperploidizations, or a large number of iteration events (entire genome duplication, entire chromosome duplication, and generic duplications of unspecific DNA regions within the same or between two chromosomes, respectively) is still highly debated. Nevertheless, all of the different actors identified so far in playing a role in the evolution of plant nuclear genomes make the picture rather complicated. Consequently, solid conclusions about genetic colinearity between Arabidopsis and rice, both expected to have a great value as a model system for dicots and monocots, respectively, are still missing, although several examples showing traces of microcolinearity have been reported (Devos et al. 1999; Van Dodeweerd et al. 1999; Liu et al. 2001; Mayer et al. 2001).
To carefully study genome evolution using the massive amount of sequence data that becomes available, we have developed a flexible tool, called ADHoRe (Automatic Detection of Homologous Regions), that detects genomic regions with statistically significant conserved gene content and order. Particularly, ADHoRe was developed to find large regions of colinearity, taking into account phenomena such as gene loss, inversions, and tandem duplications. This general concept makes it possible to use ADHoRe for analysis within one genome, that is, to look for paralogous regions with duplicated genes (Raes et al. 2002), or for comparisons between genomes of different organisms, that is, to look for synteny.
RESULTS
In this study, we have applied a new tool to estimate the frequency and significance of microcolinearity between distantly related plant species such as Arabidopsis and rice. Therefore, publicly available rice genomic sequences (as a series of BACs) from seven different chromosomes were used to compare with the complete Arabidopsis genome sequence. For both plant species, gene annotation was retrieved from public resources (see Methods). Important to note is that no prior information of macrocolinearity was incorporated into this analysis.
In total, using ADHoRe, we detected 105 cases of microcolinearity between Arabidopsis and rice before removing nonsignificant colinear regions, from which 75 are between individual rice BACs and a segment of the Arabidopsis genome and 30 are between overlapping rice clones and an Arabidopsis genomic segment. Applying the default 99% cut-off level, which retains all colinear regions that have a probability to be generated by chance of <1%, 24 segments showing conserved gene content and order between Arabidopsis and rice remain (listed in Table 1). Of these statistically significant regions, 18 (69%) show colinearity between an individual rice BAC and an Arabidopsis genomic segment, whereas 8 (31%) show colinearity between Arabidopsis and overlapping rice BACs. The distributions of the number of conserved genes within these homologous regions between Arabidopsis and rice for the different significance levels are shown in Figure 1. As expected for these classes of colinear regions characterized by a small number of conserved genes and a large number of nonhomologous intervening genes, the probability that they were generated by chance is the highest. Consequently, applying more stringent conditions reduces the number of these colinear regions. For all significance levels, most of the statistically significant colinear segments are characterized by four conserved genes (referred to as anchor points hereafter).
Table 1.
Overview of the Rice Data Set Useda
| Chromosome | Sequenced (%) | Total data set | Annotated genes | Gene densityb | Overlapping BACs | Genes | ||
|---|---|---|---|---|---|---|---|---|
| MB | BACs | BACs | MB | |||||
| 1 | 100.0 | 50.68 | 370 | 10,300 | 4.92 | 266 | 34.97 | 6237 |
| 2 | 44.7 | 18.40 | 154 | 3692 | 4.98 | 2 | 0.30 | 38 |
| 4 | 92.4 | 18.90 | 143 | 3766 | 5.02 | 75 | 10.76 | 2064 |
| 6 | 63.0 | 21.57 | 159 | 4410 | 4.89 | 6 | 0.94 | 163 |
| 7 | 75.5 | 20.33 | 164 | 4149 | 4.90 | 7 | 0.86 | 168 |
| 8 | 46.2 | 16.85 | 139 | 3398 | 4.96 | 24 | 3.55 | 615 |
| 10 | 95.6 | 19.28 | 145 | 3806 | 5.07 | 73 | 10.83 | 1892 |
| Total | 166.01 | 1274 | 33,521 | 4.95 | 453 | 62.00 | 11,177 | |
Status on January 14, 2002; source TIGR (http://www.tigr.org).
Genes/kb.
Figure 1.

Distribution of the number of conserved genes within colinear regions of Arabidopsis and rice. The black, gray, and white histograms show the distribution of the blocks emerged by maximally 100%, 5%, and 1% chance, or 0%, 95%, and 99% significance levels, respectively. We propose to use the 99% significance level (i.e., maximally 1% probability to be generated by chance) as default setting.
The largest homologous segment between Arabidopsis and rice that ADHoRe could detect contained 11 conserved genes and is shown in Figure 2A. Detailed analysis showed that within this rice region on chromosome 1 (326.8 kb), originally 64 genes have been predicted, resulting in a gene density of one gene per 5.1 kb. The homologous Arabidopsis segment on chromosome 3 shows a gene density of one gene per 3.4 kb. However, validating the automatic rice gene prediction using Expressed Sequence Tag (EST) information and comparisons with putative homologs (see Methods) shows that only ∼32 genes are present, resulting in a gene density of one gene per 10 kb. As a result, the number of nonhomologous intervening genes between the anchor points drastically decreases, and consequently, the biological significance or quality of the colinear region to be homologous increases (see Methods). An analogous approach was applied to determine whether all nonhomologous intervening Arabidopsis genes were real genes. If not, removing genes in the Arabidopsis genome could also result in a higher degree of conservation within a colinear area. However, no indications were found that some of these intervening nonhomologous Arabidopsis genes were falsely predicted.
Figure 2.

Examples of colinearity found between overlapping rice BACs and segments of the Arabidopsis genome. (A) Colinear segment between rice BACs (P0005H10, P0414E03, and P0529H11) and part of the Arabidopsis chromosome 3. Arrows indicate genes present on the genomic segment (black line), black bands connecting Arabidopsis and rice genes indicate anchor points (homologs), whereas gray bands indicate a tandem duplication. Genes probably erroneously predicted in rice are indicated in red (see text for details). LTRs are represented as hatched boxes. White boxes indicate gene products with similarity to proteins encoded by transposable elements. (gag) Retrotransposon gag protein; (rve) integrase core domain; (rvt) reverse transcriptase (RNA-dependent DNA polymerase); (rvp) retroviral aspartyl protease; (MUDR) MuDR family transposase. (B) Colinear segment between rice BACs (P0506B12 and P0031D11) and a segment of Arabidopsis chromosome 3.
Careful analysis of the long stretch of genomic sequence within the rice BAC clone P0414E03, characterized by a low gene density and no conservation with Arabidopsis, showed that multiple transposable elements have been integrated into this particular region (Fig. 2A). Analysis of putative genes and ORFs revealed high similarities with proteins encoded by transposable elements (e.g., gag protein, reverse transcriptase, integrases, RNaseH). In addition, different sets of long repetitive elements were discovered, which allowed us to reconstruct a number of distinct transposable elements involved in plant gene and genome evolution (Grandbastien 1992; Vicient et al. 2001). On the basis of organization of the proteins encoded in these transposons, three gypsy-like LTR-retrotransposons (Bennetzen 2000b) and one Mutator (Lisch et al. 2001) transposable element could be identified, together with other transposon-like remnants. In the homologous Arabidopsis genome segment, no retrotransposable elements were detected. Figure 2B shows another colinear region between rice chromosome 1 and Arabidopsis chromosome 3, characterized by eight anchor points. Removing dubiously predicted rice genes results in a gene density of one gene per 7.7 kb (or 42 genes on the stretch of 305.1-kb rice genomic sequence). The probability of this colinear region to be generated by chance is <1%. Several rearrangements can be clearly observed; since the divergence of rice and Arabidopsis, two genes have undergone tandem duplications in Arabidopsis, whereas other genes have been inverted in Arabidopsis or in rice. A more drastic rearrangement event is shown in Figure 3. This colinear region between rice chromosome 1 and Arabidopsis chromosome 5 is characterized by five pairs of homologs (anchor points). Within the rice genomic fragment, a gypsy-like LTR-retrotransposon has been inserted, resulting in a much longer rice segment (96.8 kb) compared with the homologous Arabidopsis segment (39.8 kb). Next to the local gene inversions observed in a number of colinear regions, this example shows a more complex inversion event. Genes 03 and 06 located on rice BAC B1088C09 are part of a segment colinear with Arabidopsis chromosome 5, although their gene order and orientation are not conserved compared with the other anchor points. Therefore, a chromosomal segment encoding these two genes (or their Arabidopsis orthologs) seems to have been inverted after both species diverged from each other. However, reconstructing the history leading to this configuration requires an additional inversion event. Because for gene 06, in contrast to all other genes conserved within this homologous region, the orientation compared with the homologous Arabidopsis gene is different (see twisted black band in Fig. 3), one extra gene inversion is required to explain the current gene organization between these two genomic fragments. Finally, gene 06 experienced a tandem duplication resulting in gene 07, or vice versa.
Figure 3.

Colinearity between an individual rice BAC and a segment of the Arabidopsis chromosome 5. Interpretation is as in Fig. 2.
DISCUSSION
It is estimated that rice and Arabidopsis have diverged ∼200 million years ago (Yang et al. 1999; Wikström et al. 2001). Nevertheless, applying our newly developed tool to detect homologous regions between both plants revealed numerous examples of significant microcolinearity. On the other hand, of the total set of colinear regions present between rice and Arabidopsis, probably only a subset can be considered as genuine orthologous regions that originated from a common ancestral region. The major cause of this phenomenon is the fact that many genes are organized in multigene families, and consequently, the discrimination between paralogous and orthologous gene sequences is extremely difficult. Therefore, we incorporated a routine in the ADHoRe algorithm to determine whether a colinear region could have been generated by chance out of homologous gene couples. In other words, it was tested whether a particular colinear region is a homologous region or purely consists of homologous gene couples organized in a colinear way by chance. Analysis of a number of colinear regions characterized by a high probability to be generated by chance showed that low overall-similarity signals, such as similarities between DNA-binding sites, or badly conserved gene content and order were detected (data not shown).
Combining numerous rice BACs resulted in a set of long genomic rice stretches that could be investigated for colinearity with Arabidopsis. Although only a small fraction of the final rice genome sequence was used in this study (∼38%, for which 62 MB was organized in overlapping BACs), already >20 regions between rice and Arabidopsis were found with biologically relevant colinearity, consisting of 4 up to 11 conserved genes. Because a large number of short colinear regions are found between individual rice BAC clones and an Arabidopsis genome segment, a major fraction of these regions were removed because they could represent colinear regions generated by chance. However, with more rice genomic sequence data becoming freely accessible very fast, we expect that concatenation of additional BACs will generate longer colinear stretches with Arabidopsis. Therefore, a number of colinear regions currently not retained in our final results could become statistically significant when analyzed over longer distances. Consequently, the real number of rice regions showing real microcolinearity with Arabidopsis will most probably be higher than presented here. Preliminary results on the draft sequence of the rice genome show that larger colinear segments may exist between Arabidopsis and rice (Goff et al. 2002). However, as the annotation of the draft sequence is not yet publicly available, a comparison with the results described here remains difficult.
Detailed analysis of some colinear regions indicates that the quality of the rice annotation used in this comparison is not outstanding. Although the RiceGAAS system (Sakata et al. 2002) tries to benefit from combining a number of different gene prediction programs, a large number of errors still seem to be present. The crude quality assessment performed here to determine whether a predicted gene is a real gene (i.e., sensitivity) revealed that a major fraction of the protein-encoding genes were falsely predicted. Consequently, the initial gene density determined by the gene prediction system decreased drastically when removing unreliable predicted genes. In addition, a number of genes were split (one gene predicted as two separate genes) and some exons or complete genes were missing, which could be demonstrated by incorporating EST information. Especially the large number of ORFs predicted as genes poses a problem, because a small number of these ORFs actually are confirmed by EST information, but the major fraction was not. All of these annotation inaccuracies will definitely have their repercussions on the correct interpretation of the rice genome sequence, in a way similar to that faced in annotating the Arabidopsis genome sequence (Pavy et al. 1999). Therefore, further improvement and retraining of rice gene prediction programs, together with newly developed extrinsic gene prediction methods seems inevitable for fully exploiting the rice genome sequence (Rouzé et al. 1999; Bennetzen 2002).
Next to the incorrectly predicted protein-encoding genes, a subset of these erroneously predicted genes seems to correspond with transposable elements. Although detailed analyses can unambiguously identify these elements, the presence of these elements annotated as protein-encoding genes is a major problem when performing genome-wide analyses such as described here. Although in the Arabidopsis genome 2109 Class I transposable elements have been described already (Arabidopsis Genome Initiative 2000), an additional screening reveals that within the Arabidopsis proteome nearly 600 predicted protein-encoding genes are present with high similarity to some retrotransposable elements (data not shown). Furthermore, it should be noted that the largest fraction of these genes resembling retrotransposable elements has been identified on chromosomes 1, 2, and 4. Because chromosomes 2 and 4 have been sequenced and analyzed first within the Arabidopsis sequencing project, an imperfect annotation protocol for transposons at that moment could be an explanation for this observation. For ∼36% of these detected genes, an EST matches the structural annotation, which could explain why these genes have been allocated as protein-encoding genes in the automatic annotation protocols. Nevertheless, additional efforts seem most likely to increase the quality of the current annotation on a full-genome level toward transposable elements in both rice and Arabidopsis (Le et al. 2000).
Although transposable elements integrate and retrotransposons amplify within plant genomes, when correctly annotated, they should not interfere with the presented algorithm to detect homologous regions. Consequently, this level of complexity generated by transposable elements can be masked in our method, if all transposable elements are defined as such and not as protein-encoding genes in the genomic sequence. Analysis of multiple colinear regions showed that the number of retrotransposable elements in rice was considerably higher than in the homologous Arabidopsis segments, although the actual number of retrotransposable elements in Arabidopsis is probably higher than described so far (Arabidopsis Genome Initiative 2000). Accumulation of retrotransposons in plant genomes clearly seems to be dependent of both the evolutionary lineage and the efficiency of mechanisms repressing this activity (Bennetzen and Kellog 1997; Fedoroff 2000).
It is clear that all sorts of rearrangements have occurred since rice and Arabidopsis diverged from each other ∼200 million years ago. Detailed analysis of colinearity between Arabidopsis and rice identified tandem duplications and gene loss, as well as gene and block inversions, although the frequency of these detectable events is rather low. In other words, it is not possible to trace all rearrangements that are responsible for the nonhomologous genes present in colinear regions. The main driving force responsible for degrading colinearity is seemingly a complex evolutionary mechanism, consisting of species-specific levels of large and small rearrangements (due to duplications, inversions, insertions, and deletions), transposon activity, and perhaps other unknown mechanisms. Ideally, the continuous improvement of data sets, methods, and additional genome sequences from intervening species will give us further insight into these mechanisms and their frequencies within different species.
Finally, the question remains whether, after detecting colinearity between genomes, the functions of the genes in one genome may be transferred to the homologous genes of the other genome. One major problem lies in the fact that a particular region of a chromosome can be duplicated in rice as well as in Arabidopsis. Even more drastically, complete genome duplication events may have occurred in both Arabidopsis (e.g., Arabidopsis Genome Initiative 2000; Vision et al. 2000) and rice (e.g., Goff et al. 2002; Yu et al. 2002). Because after such a duplication event, all genes are present in duplicate, one copy may degenerate through loss-of-function mutations, or both duplicates may remain redundant, experience subfunctionalization, or diverge in function through positive Darwinian selection (e.g., Ohno 1970; Force et al. 1999; Hughes 1999; Van de Peer et al. 2001). This results in a situation in which one genomic segment of one species maps with two or more different segments in the other genome, or vice versa. Transferring functional annotations from one genome to the other genome, thus, has to be done with caution, as genes belonging to paralogous regions may have considerably diverged in function.
METHODS
The ADHoRe Algorithm
Detection of Homologous Genes
To detect chromosomal locations of colinear genes, one has to look for regions that can be paired up because they contain sets of similar genes. Therefore, a data set containing all gene products, their absolute or relative position on a genomic sequence, and their orientation is required. The whole procedure is controlled by two parameters as follows: the gap size G, which describes the maximal number of intervening, nonhomologous genes tolerated between two homologous genes within a colinear segment, and Q, the quality of the colinear regions (see below). Figure 4 presents a flowchart of the algorithm. For all gene products on two genomic fragments for which gene colinearity is to be detected, initially an all-against-all sequence similarity search is performed, using BLASTP (Altschul et al. 1990). In a second step, all of these results are converted into sequence identity scores (over a given alignable region) between query and hit sequences. Two protein sequences with >30% sequence identity over an alignable region of 150 amino acids are considered as being homologous. For matching sequences with an alignable region smaller than 150 amino acids, the Homology-derived Secondary Structure of Proteins (HSSP) identity cut-off curve is used to determine whether the two sequences are homologous (Rost 1999). With this procedure, all pairs of homologous proteins between both genomic fragments are determined.
Figure 4.

Flowchart of the ADHoRe strategy used to define colinear regions between two genomic fragments. White boxes represent data items, gray boxes represent routines, and arrows indicate the dataflow.
The information on homologous genes is then stored in a matrix of (m · n) elements (m and n being the total number of genes on each genomic fragment), each non-zero element (x, y) being a pair of homologous genes (x and y denote the coordinates of these genes). Figure 5 shows such a small hypothetical matrix, in which gray elements indicate gene pairs having the same orientation, whereas black elements indicate homologous pairs of genes having an opposite orientation. In the matrix, colinear regions are represented as diagonal lines, whereas tandem duplications are manifested as purely horizontal or vertical lines; inversions can be detected by looking at the organization of the elements, and block duplications followed by gene loss form gaps in diagonal regions. To detect colinear regions, it is obvious that one has to find more or less diagonal series of elements in the matrix. This way of presenting the information reduces the problem to a clustering problem. When the matrix is constructed, it is subjected to a number of procedures that, in the end, returns all colinear regions present between both genomic fragments. In general, these procedures can be subdivided into three steps, preprocessing of the data, the actual clustering of homologous genes or blocks of genes, and postprocessing.
Figure 5.

Matrix representation of homologous genes. Arrows indicate the orientation of the genes on the two genomic fragments compared. Homologous genes with the same orientation are colored in gray; homologous genes with an opposite orientation are in black.
Preprocessing of the Data
As discussed above, during the preprocessing step, the two genomic fragments are compared, and homologous gene pairs are determined using BLAST and HSSP, after which, these are stored in a matrix. The orientation of the two genes determines the value in the matrix, whereas nonhomologous pairs are represented as empty elements in the matrix.
The next step during the preprocessing is the removal of irrelevant data points, which we designate negative filtering. During this step, all elements that cannot belong to a cluster because they are too far away from other elements in the matrix, are removed. The last step in the preprocessing is to remap tandem duplicated blocks. Because we are looking for diagonal regions in the matrix, purely horizontal or vertical regions due to tandem duplications are remapped. This is done by collapsing all tandem duplications of a gene with the same orientation and within a distance G. This way, it is easier to detect diagonal regions, as they are no longer interrupted by horizontal or vertical elements. At the end of the preprocessing, the elements in the matrix are separated according to their orientation, yielding the two orientation classes (see Figs. 4 and 5). This separation is made to facilitate the clustering and is based on the observation that colinear regions consist primarily of elements with the same orientation class. At the end of the process, both orientation classes are again combined, enabling the reconstruction of duplicated regions that have been subjected to small gene inversions.
Clustering of Genes and Blocks of Genes
A colinear region is defined in the matrix representation as a number of points showing diagonal proximity. Therefore, a special distance function was used, yielding a shorter distance for points that are in diagonally closer proximity than points that are in horizontal or vertical proximity. The formula for this function is:
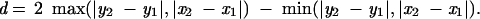 |
Because the triangle inequality does not hold for this function, it cannot be regarded as a real distance function, but rather as a diagonal pseudo distance (DPD) function. Figure 6 shows the result of applying such a distance function on a hypothetical example.
Figure 6.

Graphical representation of the DPD function. Every rectangle represents a cell of the matrix. The central dot corresponds with an element of a cluster. Because the DPD distance to element a is 2 and the DPD distance to element b is 5, a is in closer proximity to the central dot under investigation than b. According to the orientation class, a specific region of the environment is masked (which corresponds to an infinite distance).
The actual clustering step is conceived as an iterative process, gradually increasing the gap size until the final gap size—one of the parameters of the algorithm—has been reached. During each iteration, the gap size represents the maximal distance between two points in a cluster. In each iteration, new clusters can be formed and existing clusters can be extended. The algorithm details of the clustering step are depicted in Figure 7. Starting with the elements of either one of the two orientation classes (a set of singletons, i.e., elements not yet clustered), the DPD function is used to cluster the elements according to the initial gap size. By default, the initial gap size is set to 3 and is then increased in 10 exponential steps until the final gap size G has been reached. This results in a set of clusters and a set of singletons.
Figure 7.

Flowchart of the ADHoRe core algorithm. Dark gray boxes represent the different steps in the clustering process, white boxes the data items, and the light gray boxes the actions performed during each iteration step. Arrows indicate the dataflow.
Subsequently, the second parameter of the algorithm comes into play. This parameter determines to which extent the elements of a cluster fit on a diagonal line. This quality is estimated by calculating the coefficient of determination (r2) by linear regression through the points in the clusters. Only clusters with a sufficiently high quality (higher than the cut-off Q, set by the second parameter) will be kept; the constituting elements of the other clusters are reassigned the status of singletons. Within each iteration, the remaining data set after applying the DPD clustering and the quality filtering is a collection of retained clusters and a collection of singletons (from the orientation class being analyzed) not yet clustered, or initially clustered, but rejected by the quality filtering (Fig. 7).
In the next step, which also uses the DPD function, it is tested whether the clusters can be enriched with singletons from the same orientation class without badly affecting the cluster's diagonal properties. Therefore, three conditions must be fulfilled. First, the candidate singleton must be within a distance smaller than or equal to the current gap size in the iteration. Second, the candidate singleton must be positioned within the 99% confidence interval of the cluster. This confidence interval is computed by considering the best-fit line y = ax + b through all of the points in the cluster using the least-squares fit method. Usually, the points in the cluster show a certain degree of deviation from this line. This deviation can be explained by two factors: (1) the error on the calculation of the constants a and b of the regression line, and (2) the error caused by the deviation of the point xi, yi from this line. Assuming a normal distribution of this deviation, we can calculate a confidence interval that indicates the maximum deviation a candidate singleton can have from the best-fit line. Finally, if a singleton lies within these boundaries, it is also checked whether adding this singleton to the cluster will not decrease the r2 value (see above) below the specified r2 cutoff. If all criteria are met, the singleton is then added to the cluster. If not, the original configuration of both cluster and singleton is restored.
The last step of the core algorithm aims at joining clusters. For each cluster within a distance smaller than g (g being the gap size in the current iteration) of another existing cluster, it is tested whether it can be merged with that cluster, again without badly affecting the cluster's diagonal properties. To determine whether two clusters can be joined, we first check whether the distance between the diagonal lines through the central points of both clusters is not larger than g (using DPD). Next, we check whether the distance between the endpoints of both clusters is small enough. If the clusters have overlapping x or y coordinates, we consider the distance between them to be 0. In this case, we have to check whether from both clusters at least one point lies in the confidence interval of the other or whether all points of one cluster lie in the interval of the other. This is to avoid grouping of closely, in-parallel-aligned clusters. Finally, we check whether the r2 value of the resulting merged cluster does not drop below the specified r2 cutoff.
The resulting new data set again consists of a number of clusters and a number of singletons, which are used as input for the next iteration during the process (Fig. 4). During the next iteration, the gap size is increased and new clusters are made or existing clusters extended, until the final gap size has been reached. The result is a set of clusters for each orientation class.
Postprocessing
When all clusters have been compiled as described above, the fraction of colinear regions (clusters) that are not significant needs to be removed. The goal of this procedure is to determine the fraction of colinear regions that could have occurred purely by chance, and therefore are not biologically significant. This is implemented as a statistical test, sampling a large number of reshuffled data sets and calculating the probability that a colinear region, characterized by a number of conserved genes and an average gap size, can be found by chance. Using a default significance level of 99%, all regions with a probability to be generated by chance smaller than 1% are retained. The second step during postprocessing is to combine the results for the two sets of clusters with different orientations. First, we try to enrich clusters from one orientation class with singletons from the other orientation class. This step is similar to the third step in the clustering algorithm, in which clusters are extended without badly affecting the quality. Second, it is tested whether clusters from the two different orientation classes can be merged. By combining the results of both orientation classes, it is possible to reconstruct larger colinear regions that might have been subjected to one or more inversion events.
The Rice Data Set
For rice chromosomes 1, 2, 4, 6, 7, 8, and 10 (a set of chromosomes for which a large fraction of the chromosome was already sequenced), the public data of the different centers was collected (status January 14, 2002). All BAC sequences for which map position information was available and that were linked to one chromosome only were downloaded from the different consortia websites, for which an overview can be found at http://www.tigr.org/tdb/e2k1/osa1/BACmapping/description.shtml.
Concatenation of Rice BACs
To obtain large stretches of genomic rice sequence to compare with Arabidopsis, we used a simple strategy to build rice contigs. Initially, for all BAC clones, the BAC extremities were compared with BAC ends of neighboring BACs using BLASTN (Altschul et al. 1990). These BAC ends were defined as the first and the last 20% of the genomic BAC sequence. For each BAC, the 25 closest neighboring BACs were scanned, given their putative map position. Two BACs were considered overlapping when an alignable region >300 bp showed >95% sequence identity. Next, all pairs of overlapping BACs were used to build larger stretches of adjacent overlapping BAC sequences (pair A-B and pair B-C producing stretch A-B-C, etc.). In the case in which one BAC overlapped with multiple other BACs, preferentially the BAC resulting in the longest stretch was selected. Note that these BAC stretches were not physically assembled into a contig sequence, but that this information was only used to locate and order the BACs relative to each other. This procedure divided the initial data set into two large fractions, a set of overlapping BACs (in total, 453 BACs, or 37% of the total size of the original data set) and a set of remaining individual BACs.
Annotation
For all rice BACs, gene annotation was performed using RiceGAAS (Sakata et al. 2002). This system combines a total of 14 analysis programs and automatically generates gene annotation for all rice BACs present in GenBank. For all BACs retained in the data set, the predicted coding sequence and corresponding protein sequences were retrieved from the RiceGAAS website (http://ricegaas.dna.affrc.go.jp/). An overview of the number of BACs and proteins used can be found in Table 2. Finally, using the two sets of BAC clones (overlapping and individual BACs) and their corresponding gene annotation, gene lists were made and used as input for the ADHoRe algorithm. Parameters used for the ADHoRe algorithm were G = 20 for the maximum gap size and Q = 0.8 to denote the quality of the cluster. In total, 1000 reshuffled data sets were used to calculate the probability that a colinear region, characterized by a number of conserved genes and an average gap size, could have been generated by chance.
Table 2.
Overview of the Colinear Regions Detected Between Arabidopsis and Rice (99% significance level)
| Ricea | Arabidopsisb | Anchor points | BAC typec | Clone name | Arabidopsis ORFd | Qe | Pchance (%)f |
|---|---|---|---|---|---|---|---|
| 1 | 3 | 11 | O | P0529H11, P0005H10, P0414E03 | At3g54100 | 0.988 | 0.00 |
| 1 | 4 | 10 | O | P0481E12, P0046E05 | At4g18870 | 0.880 | 0.25 |
| 1 | 2 | 10 | O | P0439E11, P0031D02, B1088C09, P0485B12 | At2g30300 | 0.964 | 0.03 |
| 1 | 3 | 8 | O | P0506B12, P0031D11 | At3g55180 | 0.973 | 0.99 |
| 1 | 2 | 7 | O | P0506B12, P0031D11 | At2g39400 | 0.989 | 0.20 |
| 1 | 1 | 6 | O | P0480C01, B1131B07 | At1g34060 | 0.889 | 0.89 |
| 1 | 3 | 5 | O | P0454H12, OJ1529_G03 | At3g08670 | 0.986 | 1.00 |
| 4 | 5 | 5 | I | OSJNBa0038O10 | At5g23280 | 0.984 | 0.45 |
| 4 | 2 | 5 | I | OSJNBa0042L16 | At2g23380 | 0.977 | 0.80 |
| 6 | 3 | 5 | I | P0698A06 | At3g14230 | 0.926 | 0.64 |
| 1 | 2 | 5 | O | P0518C01 | At5g59480 | 0.945 | 0.57 |
| 4 | 5 | 4 | I | OSJNBa0088H09 | At5g06340 | 0.969 | 0.63 |
| 8 | 5 | 4 | I | P0543D10 | At5g43420 | 0.919 | 0.44 |
| 8 | 5 | 4 | I | P0705A05 | At5g43420 | 0.929 | 0.63 |
| 8 | 5 | 4 | I | P0690C12 | At4g08100 | 0.929 | 0.63 |
| 4 | 2 | 4 | I | OSJNBa0084K20 | At2g43230 | 1.000 | 0.44 |
| 10 | 1 | 4 | I | OSJNBa0026O12 | At1g03900 | 0.963 | 0.30 |
| 4 | 3 | 4 | I | OSJNBa0033G16 | At3g11630 | 0.985 | 0.63 |
| 10 | 1 | 4 | I | OSJNBb0044B19 | At1g03900 | 0.987 | 0.25 |
| 4 | 3 | 4 | I | OJ1661_E06 | At3g11630 | 0.985 | 0.63 |
| 6 | 5 | 4 | I | P0468G03 | At5g57140 | 0.999 | 0.89 |
| 4 | 3 | 4 | I | OSJNBa0088H09 | At3g52470 | 0.995 | 0.63 |
| 8 | 4 | 4 | I | OJ1005_B05 | At4g22730 | 0.983 | 0.20 |
| 2 | 1 | 4 | I | OJ1288_G09 | At1g78080 | 0.980 | 0.63 |
Rice chromosome.
Arabidopsis chromosome.
O = overlapping BACs, I = individual BAC clone.
Gene indicating the position of the homologous Arabidopsis segment.
Score obtained by quality filtering (see text for details).
Probability to be generated by chance.
For the genomic rice regions showing homology with an Arabidopsis genomic segment, which were analyzed in detail, the quality of the annotation retrieved from RiceGAAS was estimated. Therefore, for each predicted gene, we checked for the existence of a rice EST and for homology of the corresponding protein with any other protein present in the public protein databases. All predicted genes not confirmed by an EST and not showing similarity with any other protein were not considered as genes. Although these criteria are not biologically correct (i.e., these genes could be rice specific, not confirmed by ESTs and occur as a unique gene, not part of a multigene family in the rice genome), they were used here to determine rather crudely the quality of the annotation system. The same criteria applied to the total set of predicted genes in Arabidopsis shows that only 0.31% (79/25439) of the genes are selected. Thus, on the basis of the ratios found in the Arabidopsis genome, we expect that from the complete set of rice genes we remove in this way, <0.3% might be real genes. For all analyzed rice segments, on average, 45% of the predicted genes were removed.
Annotation of Transposable Elements
Initially, the genomic BAC sequence was screened for repetitive elements using REPuter (Kurtz and Schleiermacher 1999). In addition, predicted genes and ORFs were screened against a collection of protein families and domains using PFAM (Bateman et al. 2002) to determine similarities with proteins encoded in transposable elements. Artemis was used for sequence visualization and annotation (Rutherford et al. 2000).
Arabidopsis Data Set
Genomic sequences and gene annotation for the complete Arabidopsis genome was downloaded from the TIGR Arabidopsis thaliana Database (version August 2001, http://www.tigr.org/tdb/e2k1/ath1/) and processed with in-house Perl scripts.
WEB SITE REFERENCES
http://ricegaas.dna.affrc.go.jp/; RiceGAAS, Rice Genome Automated Annotation System.
http://www.psb.rug.ac.be/; Department homepage.
http://www.sanger.ac.uk/Software/Pfam/; PFAM, a collection of protein families and domains.
http://www.tigr.org/; The Institute for Genomic Research.
Acknowledgments
We thank Stephane Rombauts and Pierre Rouzé for helpful discussions, and Martine De Cock for help in preparing the manuscript. K.V. and C.S. thank the Vlaams Instituut voor de Bevordering van het Wetenschappelijk-Technologisch Onderzoek in de Industrie for a predoctoral fellowship.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL yvdp@gengenp.rug.ac.be; FAX 32-9-264-5349.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.400202.
REFERENCES
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Arabidopsis Genome Initiative. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature. 2000;408:796–815. doi: 10.1038/35048692. [DOI] [PubMed] [Google Scholar]
- Bateman A, Birney E, Cerruti L, Durbin R, Etwiller L, Eddy SR, Griffiths-Jones S, Howe KL, Marshall M, Sonnhammer EL. The Pfam protein families database. Nucleic Acids Res. 2002;30:276–280. doi: 10.1093/nar/30.1.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennetzen JL. Comparative sequence analysis of plant nuclear genomes: Microcolinearity and its many exceptions. Plant Cell. 2000a;12:1021–1029. doi: 10.1105/tpc.12.7.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ————— Transposable element contributions to plant gene and genome evolution. Plant Mol Biol. 2000b;42:251–269. [PubMed] [Google Scholar]
- ————— The rice genome. Opening the door to comparative plant biology. Science. 2002;296:60–63. doi: 10.1126/science.1071402. [DOI] [PubMed] [Google Scholar]
- Bennetzen JL, Kellog E. Do plants have a one-way ticket to genomic obesity? Plant Cell. 1997;7:1509–1514. doi: 10.1105/tpc.9.9.1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Bennetzen JL. Sequence composition and organization in the Sh2/A1-homologous region of rice. Plant Mol Biol. 1996;32:999–1001. doi: 10.1007/BF00041383. [DOI] [PubMed] [Google Scholar]
- Chen M, SanMiguel P, de Oliveira AC, Woo SS, Zhang H, Wing R, Bennetzen JL. Microcolinearity in sh2-homologous regions of the maize, rice, and sorghum genomes. Proc Natl Acad Sci. 1997;94:3431–3435. doi: 10.1073/pnas.94.7.3431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devos KM, Gale MD. Comparative genetics in the grasses. Plant Mol Biol. 1997;35:3–15. [PubMed] [Google Scholar]
- Devos KM, Atkinson MD, Chinoy CN, Harcourt RL, Koebner RMD, Liu CJ, Masojc P, Xie DX, Gale MD. Chromosomal rearrangements in the rye genome relative to that of wheat. Theor Appl Genet. 1993;85:673–680. doi: 10.1007/BF00225004. [DOI] [PubMed] [Google Scholar]
- Devos KM, Beales J, Nagamura Y, Sasaki T. Arabidopsis-rice: Will colinearity allow gene prediction across the eudicot-monocot divide? Genome Res. 1999;9:825–829. doi: 10.1101/gr.9.9.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedoroff N. Transposons and genome evolution in plants. Proc Natl Acad Sci. 2000;97:7002–7007. doi: 10.1073/pnas.97.13.7002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feuillet C, Keller B. High gene density is conserved at syntenic loci of small and large grass genomes. Proc Natl Acad Sci. 1999;96:8265–8270. doi: 10.1073/pnas.96.14.8265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Force A, Lynch M, Pickett FB, Amores A, Yan YL, Postlethwait J. Preservation of duplicate genes by complementary, degenerative mutations. Genetics. 1999;151:1531–1545. doi: 10.1093/genetics/151.4.1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gale MD, Devos KM. Plant comparative genetics after 10 years. Science. 1998;282:656–659. doi: 10.1126/science.282.5389.656. [DOI] [PubMed] [Google Scholar]
- Goff S, Ricke D, Lan TH, Presting G, Wang R, Dunn M, Glazebrook J, Sessions A, Oeller P, Varma H, et al. A draft sequence of the rice genome (Oryza sativa L. ssp. japonica) Science. 2002;296:92–100. doi: 10.1126/science.1068275. [DOI] [PubMed] [Google Scholar]
- Grandbastien M. Retroelements in higher plants. Trends Genet. 1992;8:103–108. doi: 10.1016/0168-9525(92)90198-d. [DOI] [PubMed] [Google Scholar]
- Grant D, Cregan P, Shoemaker RC. Genome organization in dicots: Genome duplication in Arabidopsis and synteny between soybean and Arabidopsis. Proc Natl Acad Sci. 2000;97:4168–4173. doi: 10.1073/pnas.070430597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes AL. Adaptive evolution of genes and genomes. New York, NY: Oxford University Press; 1999. [Google Scholar]
- Keller B, Feuillet C. Colinearity and gene density in grass genomes. Trends Plant Sci. 2000;5:246–251. doi: 10.1016/s1360-1385(00)01629-0. [DOI] [PubMed] [Google Scholar]
- Kilian A, Chen J, Han F, Steffenson B, Kleinhofs A. Towards map-based cloning of the barley stem rust resistance genes Rpg1 and rpg4 using rice as an intergenomic cloning vehicle. Plant Mol Biol. 1997;35:187–195. [PubMed] [Google Scholar]
- Ku HM, Vision T, Liu J, Tanksley SD. Comparing sequenced segments of the tomato and Arabidopsis genomes: Large-scale duplication followed by selective gene loss creates a network of synteny. Proc Natl Acad Sci. 2000;97:9121–9126. doi: 10.1073/pnas.160271297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtz S, Schleiermacher C. REPuter: Fast computation of maximal repeats in complete genomes. Bioinformatics. 1999;15:426–427. doi: 10.1093/bioinformatics/15.5.426. [DOI] [PubMed] [Google Scholar]
- Le QH, Wright S, Yu Z, Bureau T. Transposon diversity in Arabidopsis thaliana. Proc Natl Acad Sci. 2000;97:7376–7381. doi: 10.1073/pnas.97.13.7376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisch DR, Freeling M, Langham RJ, Choy MY. Mutator transposase is widespread in the grasses. Plant Physiol. 2001;125:1293–1303. doi: 10.1104/pp.125.3.1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Sachidanandam R, Stein L. Comparative genomics between rice and Arabidopsis shows scant collinearity in gene order. Genome Res. 2001;11:2020–2026. doi: 10.1101/gr.194501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer K, Murphy G, Tarchini R, Wambutt R, Volckaert G, Pohl T, Dusterhoft A, Stiekema W, Entian KD, Terryn N, et al. Conservation of microstructure between a sequenced region of the genome of rice and multiple segments of the genome of Arabidopsis thaliana. Genome Res. 2001;11:1167–1174. doi: 10.1101/gr.161701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno S. Evolution by gene duplication. New York, NY: Springer-Verlag; 1970. [Google Scholar]
- Panstruga R, Buschges R, Piffanelli P, Schulze-Lefert P. A contiguous 60 kb genomic stretch from barley reveals molecular evidence for gene islands in a monocot genome. Nucleic Acids Res. 1998;26:1056–1062. doi: 10.1093/nar/26.4.1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavy N, Rombauts S, Déhais P, Mathé C, Ramana DV, Leroy P, Rouzé P. Evaluation of gene prediction software using a genomic data set: Application to Arabidopsis thaliana sequences. Bioinformatics. 1999;15:887–899. doi: 10.1093/bioinformatics/15.11.887. [DOI] [PubMed] [Google Scholar]
- Raes J, Vandepoele K, Simillion C, Saeys Y, Van de Peer T. Investigating ancient duplication events in the Arabidopsis genome. In: Meyer A, Van de Peer Y, editors. Genome evolution. Dordrecht, The Netherlands: Kluwer Academic Publishers; 2002. (in press). [PubMed] [Google Scholar]
- Rossberg M, Theres K, Acarkan A, Herrero R, Schmitt T, Schumacher K, Schmitz G, Schmidt R. Comparative sequence analysis reveals extensive microcolinearity in the lateral suppressor regions of the tomato, Arabidopsis, and Capsella genomes. Plant Cell. 2001;13:979–988. doi: 10.1105/tpc.13.4.979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rost B. Twilight zone of protein sequence alignments. Protein Eng. 1999;12:85–94. doi: 10.1093/protein/12.2.85. [DOI] [PubMed] [Google Scholar]
- Rouzé P, Pavy N, Rombauts S. Genome annotation: Which tools do we have for it? Curr Opin Plant Biol. 1999;2:90–95. doi: 10.1016/S1369-5266(99)80019-3. [DOI] [PubMed] [Google Scholar]
- Rutherford K, Parkhill J, Crook J, Horsnell T, Rice P, Rajandream M, Barrell B. Artemis: Sequence visualization and annotation. Bioinformatics. 2000;16:944–945. doi: 10.1093/bioinformatics/16.10.944. [DOI] [PubMed] [Google Scholar]
- Sakata K, Nagamura Y, Numa H, Antonio B, Nagasaki H, Idonuma A, Watanabe W, Shimizu Y, Horiuchi I, Matsumoto T, et al. RiceGAAS: An automated annotation system and database for rice genome sequence. Nucleic Acids Res. 2002;30:98–102. doi: 10.1093/nar/30.1.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki T, Burr B. International Rice Genome Sequencing Project: The effort to completely sequence the rice genome. Curr Opin Plant Biol. 2000;3:138–141. doi: 10.1016/s1369-5266(99)00047-3. [DOI] [PubMed] [Google Scholar]
- Schmidt R. Synteny: Recent advances and future prospects. Curr Opin Plant Biol. 2000;3:97–102. doi: 10.1016/s1369-5266(99)00048-5. [DOI] [PubMed] [Google Scholar]
- Tarchini R, Biddle P, Wineland R, Tingey S, Rafalski A. The complete sequence of 340 kb of DNA around the rice Adh1-adh2 region reveals interrupted colinearity with maize chromosome 4. Plant Cell. 2000;12:381–391. doi: 10.1105/tpc.12.3.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tikhonov AP, SanMiguel PJ, Nakajima Y, Gorenstein NM, Bennetzen JL, Avramova Z. Colinearity and its exceptions in orthologous adh regions of maize and sorghum. Proc Natl Acad Sci. 1999;96:7409–7414. doi: 10.1073/pnas.96.13.7409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van de Peer Y, Taylor JS, Braasch I, Meyer A. The ghost of selection past: Rates of evolution and functional divergence of anciently duplicated genes. J Mol Evol. 2001;53:436–446. doi: 10.1007/s002390010233. [DOI] [PubMed] [Google Scholar]
- van Dodeweerd AM, Hall CR, Bent EG, Johnson SJ, Bevan MW, Bancroft I. Identification and analysis of homoeologous segments of the genomes of rice and Arabidopsis thaliana. Genome. 1999;42:887–892. [PubMed] [Google Scholar]
- Vicient CM, Jaaskelainen MJ, Kalendar R, Schulman AH. Active retrotransposons are a common feature of grass genomes. Plant Physiol. 2001;125:1283–1292. doi: 10.1104/pp.125.3.1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vision TJ, Brown DG, Tanksley SD. The origins of genomic duplications in Arabidopsis. Science. 2000;290:2114–2117. doi: 10.1126/science.290.5499.2114. [DOI] [PubMed] [Google Scholar]
- Wendel JF. Genome evolution in polyploids. Plant Mol Biol. 2000;42:225–249. [PubMed] [Google Scholar]
- Wikström N, Savolainen V, Chase MW. Evolution of the angiosperms: Calibrating the family tree. Proc R Soc Lond B Biol Sci. 2001;268:2211–2220. doi: 10.1098/rspb.2001.1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YW, Lai KN, Tai PY, Li WH. Rates of nucleotide substitution in angiosperm mitochondrial DNA sequences and dates of divergence between Brassica and other angiosperm lineages. J Mol Evol. 1999;48:597–604. doi: 10.1007/pl00006502. [DOI] [PubMed] [Google Scholar]
- Yu J, Hu S, Wang J, Wong GK, Li S, Liu B, Deng Y, Dai L, Zhou Y, Zhang X, et al. A draft sequence of the rice genome (Oryza sativa L. ssp. indica) Science. 2002;296:79–92. doi: 10.1126/science.1068037. [DOI] [PubMed] [Google Scholar]