

Abstract
This paper reports an analysis of the encoded proteins (the proteome) of the genomes of human, fly, worm, yeast, and representatives of bacteria and archaea in terms of the three-dimensional structures of their globular domains together with a general sequence-based study. We show that 39% of the human proteome can be assigned to known structures. We estimate that for 77% of the proteome, there is some functional annotation, but only 26% of the proteome can be assigned to standard sequence motifs that characterize function. Of the human protein sequences, 13% are transmembrane proteins, but only 3% of the residues in the proteome form membrane-spanning regions. There are substantial differences in the composition of globular domains of transmembrane proteins between the proteomes we have analyzed. Commonly occurring structural superfamilies are identified within the proteome. The frequencies of these superfamilies enable us to estimate that 98% of the human proteome evolved by domain duplication, with four of the 10 most duplicated superfamilies specific for multicellular organisms. The zinc-finger superfamily is massively duplicated in human compared to fly and worm, and occurrence of domains in repeats is more common in metazoa than in single cellular organisms. Structural superfamilies over- and underrepresented in human disease genes have been identified. Data and results can be downloaded and analyzed via web-based applications at http://www.sbg.bio.ic.ac.uk.
[Supplemental material is available online at http://www.genome.org.]
The interpretation and exploitation of the wealth of biological knowledge that can be derived from the human genome (Lander et al. 2001; Venter et al. 2001) requires an analysis of the three-dimensional structures and the functions of the encoded proteins (the proteome). Comparison of this analysis with those of other eukaryotic and prokaryotic proteomes will identify which structural and functional features are common and which confer species specificity. In this paper, we present an integrated analysis of the proteomes of human and 13 other species considering the folds of globular domains, the presence of transmembrane proteins, and the extent to which the proteomes can be functionally annotated. This integrated approach enables us to consider the relationship between these different aspects of annotation and thereby enhance previous analyses of the human and other proteomes (e.g., Koonin et al. 2000; Frishman et al. 2001; Iliopoulos et al. 2001), including the seminal papers reporting the human genome sequence (Lander et al. 2001; Venter et al. 2001).
A widely used first step in a bioinformatics-based functional annotation is to identify known sequence motifs and domains from manually curated databases such as PFAM/INTERPRO (Bateman et al. 2000) and PANTHER (Venter et al. 2001). This strategy was used in the original analyses of the human proteome (Lander et al. 2001; Venter et al. 2001). These annotations tend to be reliable, as these libraries have been carefully constructed to avoid false positives whilst maintaining a high coverage. In the absence of a match to these characterized motifs/domains, suggestion for a functional annotation comes from a homology to a previously functionally annotated sequence. However, transfer of function via an identified homology is problematic and the extent of the difficulty has been recently quantified (e.g., Devos and Valencia 2000; Wilson et al. 2000; Todd et al. 2001). Below 30% pair-wise sequence identity, two proteins often may have quite different functions even if their structures are similar. Because of this problem, global bioinformatics analyses of genomes generally do not use functional transfer from distant homologies for annotation. However, specific analyses by human experts still extensively employ this strategy, particularly as any suggestion of function can be refined from additional information or from further experiments.
A powerful source of additional information is available when the three-dimensional coordinates of the protein are known. The structure often provides information about the residues forming ligand-binding regions that can assist in evaluating the function and specificity of a protein. For example, recently we have shown that spatial clustering of invariant residues can assist in assessing the validity of function transfer in this twilight zone (Aloy et al. 2001). At higher levels of identity, knowledge of structure can assist in analyzing ligand specificity and the effect of point mutations.
A valuable tool in exploiting three-dimensional information is the databases of protein structure in which domains with similar three-dimensional architecture are grouped together. Here, we use the structural classification of proteins (SCOP) (Conte et al. 2000). In SCOP, protein domains of known structure that are likely to be homologs are grouped by an expert into a common superfamily based on their structural similarity together with functional and evolutionary considerations. SCOP is widely regarded as an accurate assessment of which domains are homologs. However, SCOP remains subjective and one cannot exclude the possibility that two domains placed within the same superfamily only share a common fold as a result of convergent evolution and therefore are not homologous.
The above considerations have led us to focus our analysis on the following three objectives: (i) to estimate the extent to which the known proteomes can be annotated in terms of structure and function and how reliable we consider these annotations to be; (ii) to place the occurrence of particular SCOP structural superfamilies in terms of their biological and species-specific contexts; and (iii) to derive evolutionary insights from frequency-based analysis of homologous SCOP domains.
Strategy for Structural and Functional Annotations
Protein sequences from the human genome and from 13 other species were analyzed (for details, see Methods). The main strategy was to use the sensitive protein sequence similarity search program PSI-BLAST (Altschul et al. 1997) to scan each protein sequence against a database composed of a nonredundant set of sequences augmented by the sequences of each SCOP domain and, to ensure up-to-date coverage, each protein entry of the PDB (Berman et al. 2000).
Functional annotation was considered as sequence matches to the PFAM domain library (excluding families of unknown function), which is now part of the more general INTERPRO database. In the absence of a match to these characterized motifs/domains, we need to evaluate functional annotation via transfer from homology. To represent this approach computationally, we simply considered a functional annotation if a homolog contains some textual description of function (see legend to Fig. 1A). Thus, the total of the proteome that can be functionally annotated is the sections that are assigned to a PFAM domain or, if no assignment to PFAM, the sections that are homologous to a protein with a text functional description.
Figure 1.

Structural and functional annotation of the proteomes. (A) Coverage for each species is reported as the fraction of the residues in the proteome that are annotated. This allows for partial coverage of any sequence. Structural annotation is a homology to a known structure. Functional annotation is when there is no structural annotation but there is a homology to an entry from SwissProt or PIR that has a description other than those that contain any of the following words: “hypothetical”, “probable”, “putative”, “predicted”. Any homology denotes a sequence similarity to a structurally or functionally unannotated protein, such as one described as hypothetical. Nonglobular denotes remaining sequence regions that were predicted as transmembrane, signal peptide, coiled-coils, or low-complexity. Remaining residues are classified as orphans. (B) Structural and functional annotations that cover the entire protein sequence. For structural annotation, we required that >95% of the sequence was structurally annotated and there was no unannotated segment of >30 residues. Functional annotation is evaluated after assigning structures and requires the same constraints. Finally, any homolog (including those of unknown function) is assigned to the remainder (with the same sequence length constraints).
Status of Structural and Functional Annotations
Figure 1A shows the structural annotation status of the proteomes expressed as the fraction of the total residues in each proteome. We use the residue fraction to include situations when only part of a protein sequence is annotated, as one cannot quantify this as a fraction of domains because one does not know the number of domains in unannotated regions. Thirty-nine percent of the human proteome can be structurally annotated from either having a known protein structure or via a PSI-BLAST detectable homology to a known structure. This percentage is higher than that for yeast, fly, and worm and is comparable to the coverage of many bacteria and archaea. A further 38% of the human genome falls into the category of functional annotation without structure. Because nearly every protein structure has some functional annotation, the total functional annotation of the human proteome is 77%. The remainder are (i) either homologous to another protein of unknown function, or (ii) orphan regions without any detectable homology, or (iii) an unannotated, nonglobular region (a region of low complexity, coiled-coil, or a transmembrane segment).
We also consider how many protein sequences can be fully annotated. To allow for gaps, we require that >95% of a particular sequence be covered without gaps of >30 residues (Fig. 1B). The fraction of the human protein sequences that are fully annotated in terms of structure is only 15%. A further 14% of the human protein sequences are fully annotated in terms of function but not structure. The fraction of fully covered annotated sequences for human is much higher than for worm, fly, and yeast. Another 8% of the human sequences are fully covered by hypothetical sequences or sequences of unknown function.
The accuracy of the above analysis is dependent on the quality of the gene prediction. For the eukaryotic genomes analyzed, particularly for the human proteome, this is problematic and it is anticipated that several new genes will be identified and some present assignments modified. The human proteome we analyzed is based on gene predictions that are confirmed by matches to expressed sequence tags (ESTs) or homologs in other species (see http://www.ensembl.org). This use of homology would contribute to the high level of structural and functional annotation and, if additional genes were identified, the values for coverage probably would be somewhat lower. We can obtain an upper estimate of the magnitude of this problem by noting that the human genome has 6% by residue of orphans. In worm, this figure is 17%, and it is considered that most genes have been identified in this genome (Reboul et al. 2001). Similar figures for orphans are found in yeast and fly. If we then assume that the true figure for orphan proteins in the human genome is 17%, then any other section of the annotation as shown in the bar charts (e.g., of structural coverage) should be reduced by 83/94 (i.e., 0.88). Thus, the structural coverage is reduced from 39% to 34%. In practice, the true value is expected to lie between these two extremes.
However, even for prokaryotes, errors in gene prediction can affect our survey. For example, the proteome of the archaea Aeropyrum pernix contains the largest fraction of orphan regions. This result may be because the gene prediction in A. pernix produced many very short questionable open reading frames (ORFs) (Skovgaard et al. 2001).
Reliability of Annotation
The reliability of the structural annotation from homology model-building depends on the level of sequence identity between the protein of known structure with that of unknown structure for which one wants to build a model (Sanchez and Sali 1998; Bates and Sternberg 1999) (Fig. 2A). Only 2% of the residues in the human proteome are from domains for which there is an actual crystal structure or which share >97% sequence identity with an experimental structure. However, 11% are within the identity range of 97% to 40%, and homology models are likely to be of sufficient accuracy to place residues reasonably accurately. Below 30%, modeling is likely to reveal only general features of the fold.
Figure 2.

Reliability of annotation. (A) Reliability of structural annotation. Homologies are dissected into sequence similarity bands. The >97% identity effectively reports a match to an experimentally determined structure or to one that differs in only a few residues. Structures based on these annotations are accurate. The next band down to 40% denotes annotations for which models can be constructed that are expected to be reasonably accurate (Sanchez and Sali 1998; Bates and Sternberg 1999). Between 40% and 30% sequence identity automated modeling is difficult. Below 30% identity, the sequence alignment suggested by the annotation is expected to have many errors and the structural annotation primarily provides an indication of the 3-D fold. (B) Reliability of functional annotation. Functional annotation is distinguished between reliable (≥30% sequence identity) and “fuzzy” (<30% sequence identity). The fractions are cumulative, that is, regions that are assigned to a PFAM domain and a structure are counted first, then we count regions for which we have a PFAM domain but no structural assignment.
Figure 2B provides an assessment of the reliability of functional annotation. We consider that a match to a PFAM domain (excluding domains of unknown function) constitutes a reliable functional annotation. For the human proteome, 26% of the residues can be assigned to PFAM domains, this includes 19% for which we have a structural assignment, which often will assist in functional annotation. Next, we identify those proteins where the closest homolog that has a text functional description (see legend to Fig. 1A) shares at least 30% sequence identity. This cutoff was chosen because studies have shown that below this value, homologs often have diverged to radically different functions (Devos and Valencia 2000; Wilson et al. 2000; Todd et al. 2001). A total of 41% of the proteome could potentially be functionally annotated based on a homology to a protein with at least 30% sequence identity. This 41% contains 15% without any match to PFAM but with an assigned structure that could help to refine the proposed annotation. A further 8% of the proteome is below the 30% identity cut off for functional annotation. Of this fraction, 50% (4% of the total proteome) has a structural homologue that is of great value in assessing the validity of functional transfer. However the remaining 4% of the proteome with functional assignment below the 30% cutoff is without any structural information, and annotations for these sequence regions must be considered highly tentative.
SCOP Superfamilies
Table 1 reports the commonly occurring SCOP superfamilies in human, fly, worm, yeast, and average values for archaea and bacteria. Complete tables can be downloaded from our web site, http://www.sbg.bio.ic.ac.uk.
Table 1.
Commonly Occurring SCOP Superfamilies in the Proteomes
| SCOP superfamily | Human | Fly | Worm | Yeast | Archaea | Bacteria | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N | R | N | R | N | R | N | R | N | R | N | R | |
| Classic zinc finger, C2H2 | 5092 | 1 | 1096 | 1 | 190 | 10 | 74 | 9 | – | 269 | – | – |
| Immunoglobulin* | 1214 | 2 | 483 | 3 | 457 | 2 | 8 | 91 | 1 | 135 | 4 | 94 |
| EGF/laminin | 1192 | 3 | 320 | 4 | 413 | 4 | – | – | – | – | – | – |
| P-loop containing nucleotide triphosphate hydrolases* | 847 | 4 | 575 | 2 | 516 | 1 | 408 | 1 | 126 | 1 | 168 | 1 |
| Fibronectin type III* | 842 | 5 | 247 | 7 | 222 | 8 | 1 | 301 | – | – | 1 | 237 |
| Cadherin | 608 | 6 | 222 | 10 | 135 | 21 | – | – | 3 | 72 | – | – |
| RNA-binding domain | 587 | 7 | 282 | 5 | 199 | 9 | 128 | 3 | – | – | – | 420 |
| Protein kinase-like (PK-like)* | 557 | 8 | 271 | 6 | 434 | 3 | 142 | 2 | 3 | 72 | 5 | 82 |
| Hemeodomain-like | 334 | 9 | 144 | 18 | 145 | 17 | 32 | 20 | 1 | 221 | 17 | 16 |
| Spectrin repeat | 327 | 10 | 227 | 9 | 150 | 13 | – | – | – | – | – | – |
| PH domain-like* | 327 | 10 | 140 | 19 | 100 | 31 | 23 | 29 | – | – | – | – |
| SH3 domain | 304 | 12 | 105 | 23 | 70 | 37 | 29 | 23 | – | – | – | 454 |
| EF-hand* | 284 | 13 | 163 | 14 | 120 | 26 | 23 | 29 | – | – | – | 420 |
| Ankyrin repeat | 278 | 14 | 120 | 21 | 128 | 24 | 31 | 22 | – | – | 1 | 342 |
| Complement control module/SCR domain | 277 | 15 | 57 | 38 | 52 | 43 | – | – | – | – | – | – |
| PDZ domain-like | 265 | 16 | 103 | 24 | 89 | 32 | 6 | 120 | 1 | 169 | 6 | 64 |
| Ligand-binding domain of low-density lipoprotein receptor | 247 | 17 | 196 | 12 | 143 | 18 | 3 | 194 | – | – | – | – |
| Tetratricopeptide repeat (TPR)* | 215 | 18 | 171 | 13 | 115 | 27 | 98 | 5 | 4 | 48 | 16 | 19 |
| RING-finger domain, C3HC4 | 207 | 19 | 108 | 22 | 122 | 25 | 33 | 19 | – | – | – | – |
| Trp-Asp repeat (WD-repeat) | 193 | 20 | 198 | 11 | 142 | 19 | 114 | 4 | 2 | 121 | 3 | 157 |
| C2 domain (Calcium/lipid-binding* domain, CaLB) | 186 | 21 | 68 | 32 | 89 | 32 | 32 | 20 | – | – | – | – |
| NAD(P)-binding Rossmann-fold domains* | 177 | 22 | 150 | 16 | 130 | 23 | 88 | 7 | 27 | 3 | 72 | 2 |
| ARM repeat* | 177 | 22 | 137 | 20 | 105 | 28 | 80 | 8 | 1 | 221 | – | – |
| SH2 domain* | 161 | 24 | 59 | 37 | 72 | 35 | 8 | 91 | – | – | – | – |
| Thioredoxin-like* | 152 | 25 | 148 | 17 | 148 | 14 | 50 | 12 | 8 | 21 | 18 | 13 |
| C-type lectin-like* | 149 | 26 | 40 | 53 | 310 | 5 | – | – | – | – | – | 454 |
| Glucocorticoid receptor-like (DNA-binding domain)* | 143 | 27 | 69 | 31 | 281 | 6 | 14 | 59 | – | – | – | – |
| ConA-like lectins/glucanases* | 136 | 28 | 66 | 34 | 105 | 28 | 8 | 91 | 1 | 169 | 3 | 157 |
| Actin-like ATPase domain* | 135 | 29 | 65 | 35 | 38 | 56 | 58 | 10 | 2 | 97 | 12 | 26 |
| No. distinct proteins in proteome | 28,913 | 13,922 | 16,323 | 6,237 | 2,176 | 2,789 | ||||||
| No. distinct superfamilies in proteome | 546 | 518 | 482 | 434 | 328 | 499 | ||||||
R, the rank of a superfamily within a proteome.
N, the frequency of domains within this superfamily.
Denotes that our analysis showed that several PFAM (Bateman et al. 2000) families (and hence several INTERPRO families) are included within the single SCOP superfamily. The number of distinct proteins and the number of domains per superfamily (N) for archaea and bacteria are averages, whereas the number of distinct superfamilies are totals over the species (seven for bacteria and three for archaea).
First, we consider the commonly occurring superfamilies in the human proteome. The most common domain in human is the C2H2 classic zinc finger, which occurs four times more often than the next most common domain, the immunoglobulin. The P-loop SCOP superfamily involved in nucleotide triphosphate hydrolysis is the fourth most common in human and second in fly, but the most common in the other analyzed proteomes. In general, the commonly occurring superfamilies in the human proteome reflect the eukaryotic and multicellular organization. Commonly observed superfamilies involved in or part of cell-surface receptors, protein-protein, or cell–cell interaction, signaling, or cytoskeleton structure are represented by superfamilies such as: immunoglobulin, EGF/laminin, fibronectin, cadherin, protein kinase, homeo-domain, tetratricopeptide repeat, spectrin repeat, PH-domain, and SH3-domain.
In general, the fly and worm have similar ranking of the common superfamilies to those in human reflecting the multicellular organization. There are, however, some differences. The c-type lectins are at rank 26 with 149 domains in human but at rank five with 310 domains in worm. C-type lectins have a wide spectrum of functions associated with carbohydrate binding and occur membrane bound and soluble. The high occurrence of c-type lectins has previously been noted but not explained by Koonin and coworkers (Koonin et al. 2000). We also are unable to explain this observation. Similarly, the most common DNA binding domain in worm is the glucocorticoid receptor which is at rank six in worm (281 domains) but only at rank 27 (143 domains) in human and at rank 31 in the fly (69 domains). In contrast to the rank order, the domain frequencies of the top superfamilies in human are generally much higher than the corresponding frequencies in fly and worm, whereas the frequencies in fly and worm are often similar. The human proteome is roughly double the size of that of fly or worm, but for several of the most common superfamilies in human (the first eight ranks), we observe a scaling factor of more than two. At lower ranks, the ratio is generally around two. The first superfamily that occurs with roughly the same frequency in human, fly, and worm is the thioredoxin-like domain (152, 148, and 148 domains, respectively). Proceeding down the rank order of occurrence in human, the first superfamily with a lower frequency of domains in human than in other multicellular eukaryotes is the c-type lectin (see above).
There are, however, major differences in rank order for the single-celled organisms. Several of the superfamilies in Table 1 have similar ranks in human, fly, and worm, whereas the rank in yeast often differs markedly (e.g., the immunoglobulin). Domains of superfamilies found in cell–cell interaction proteins and cell-surface proteins such as the fibronectin and cadherin are not found or only occur infrequently in the proteomes of the single cellular organisms. In bacteria, and especially in archaea, the top ranks are mainly occupied with superfamilies associated with enzymes. The most common DNA-binding domain in bacteria and archaea is the winged helix-turn-helix motif.
The abundance of several superfamilies in metazoans that are absent or have relatively low domain frequencies in yeast leads us to conclusions different than those recently published for the Schizosaccharomyces pombe genome (Wood et al. 2002). Results by Wood et al. show many new protein sequences in yeast (S. pombe and Saccharomyces cerevisiae) compared to prokaryotes, but only a few new sequences in metazoans (i.e., those proteins found in metazoans only). We find 84 SCOP superfamilies present in metazoa and yeast that we do not find in any of the processed prokaryota, and we find 113 new superfamilies in metazoa that we do not find in yeast (data not shown). Our analysis is based on structural domains rather than closely related full-length sequences, which allows us to find members of even diverse superfamilies. Our results suggest that in invention and expansion on the level of structural domains there may well be a bigger step from single-cellular eukaryotes to multicellular organisms than implied by Wood et al. (2002).
Proteins forming a particular SCOP superfamily are identified on the basis of both their similar structure and function. In contrast, PFAM, INTERPRO, and PANTHER are primarily sequence- and function-based families. Because homologies can be recognized from structural conservation that are undetectable by sequence-based methods, one SCOP superfamily can include several PFAM, INTERPRO, or PANTHER families. In addition, SCOP is a structural domain database whereas PFAM identifies a single sequence motif that can be repeated to form a structural domain. For example, PFAM describes each of the β-sheet motifs of a WD-repeat by itself whereas SCOP considers the entire barrel of seven of these motifs as a domain. Thus, there are several differences between the rank of commonly occurring SCOP domains compared to the results from sequence-based analysis (Lander et al. 2001; Venter et al. 2001).
Our results are in broad agreement with similar analyses by others (Frishman et al. 2001; Iliopoulos et al. 2001; Lander et al. 2001; Venter et al. 2001), in particular with results from others describing the distribution of SCOP folds and superfamilies in different genomes. Differences in methodology, different confidence cutoffs, and different sequence databases used for the analysis do not allow a direct comparison of domain frequencies and annotation coverage in proteomes. However, the relative rank order for folds and superfamilies within a proteome are suitable for a comparison between different work. Recent work from Gough et al. (2001) using hidden Markov models for SCOP superfamilies shows generally similar ranks for the top 10 superfamilies in the processed genomes. The zinc finger is the most abundant superfamily in human followed by the immunoglobulin. Although results from the HMM superfamily analysis by Gough et al. (2001) on a more recent version of the human genome (based on ENSEMBL-4.28.1, see http://stash.mrc-lmb.cam.ac.uk/SUPERFAMILY/) gives different total numbers compared to our work, the general trend (i.e., ranks of superfamilies) is stable even for the different interpretations of the human genome. It should be noted that we have focussed our analysis on the globular parts of the proteomes, and we do not have PSI-BLAST homology assignments for the membrane all-alpha SCOP superfamily. However, we have BLAST assignments for close homologs of this superfamily. Therefore, this superfamily is found at far higher rank (i.e., further down the list) in our results compared to Gough et al. (2001), who constructed special HMMs for this superfamily.
Our superfamily rankings are more different than those in PartsList (Qian et al. 2001b), that reports the EGF/laminin superfamily at rank one for Caenorhabditis elegans (rank four in our analysis) and the P-loop at rank eight, compared to rank one in our results. The HMM superfamily analysis of the worm from Gough et al. (2001) ranks the P-loop at position two, following the membrane all-α superfamily.
Wolf et al. (1999) assigned SCOP-1.35 folds than several prokaryotes, yeast, and C. elegans using an automated processing pipeline similar to ours. Folds of coiled-coiled domains and immunoglobulins and those domains mainly found in viruses were omitted from their analysis. The top-ranking SCOP folds for archaea are similar to the ranks from our analysis, but there is more variation in ranks for bacteria, possibly because of differences in the set of bacterial genomes that we have chosen. As shown by Wolf et al. (1999), we also find more agreement between archaea and bacterial folds compared to eukaryotic folds. The fold analysis by Wolf et al. (1999) has been refined (Koonin et al. 2000) by including the IMPALA program into their processing pipeline.
SCOP Superfamilies Specific for Phylogenetic Branches
Table 2 presents SCOP superfamilies that just occur with one species or set of related species but not in any of the other organisms analyzed. In addition, each member of the superfamily was run against the nonredundant sequence database using PSI-BLAST (with the parameters described in the method) to identify other species not included in those from the genomes analyzed here. In Table 2, we exclude any superfamily that occurs less than four times in a particular branch (human, fly, worm, yeast, bacteria, archaea) to prevent erroneous inferences because of the inherent difficulties of automated annotation. This information identifies biological function specific for one branch of life.
Table 2.
Superfamilies Unique for One of the Processed Proteomes or Group of Proteomes
| SCOP superfamily | N | R | Functional description |
|---|---|---|---|
| Human | |||
| MHC antigen-recognition domain | 57 | 62 | Immune system |
| Interleukin 8-like chemokines | 48 | 71 | Immune system, growth factors |
| 4-helical cytokines | 47 | 75 | Immune system, diverse range of interferons and interleukins |
| Crystallins/protein S/yeast-killer toxin | 20 | 144 | Eye lens component |
| Serum albumin | 19 | 150 | Major blood plasma component |
| Colipase-like | 11 | 202 | Enzyme regulation for pancreatic lipases, development |
| RNase A-like | 8 | 237 | Different ribonucleases found in pancreas, eosinophil granules, and involved in angeogenesis |
| PKD domain | 7 | 260 | Possibly involved in extracellular protein-protein interaction |
| Defensin-like | 7 | 260 | Small antibacterial, fungal, and viral protein |
| Uteroglobin-like | 5 | 294 | Binding of phospholipids, progesterone, inhibits phospholipase A2 (involved in metabolism of biomembranes) |
| Midkine | 4 | 328 | Growth factors |
| Fly | |||
| Insect pheromone/odorant-binding proteins | 26 | 81 | Hormone related, sex recognition |
| Scorpion toxin-like | 6 | 220 | Drosomycin and defensin, antibiotic, antifungicide |
| Worm | |||
| Plant lectins/antimicrobial peptides | 4 | 234 | Antimicrobial peptides, pathogen response, antifungicides Homologous to plant proteins |
| Osmotin, thaumatin-like protein | 4 | 234 | Same description as for lectins above |
| Yeast | |||
| Zn2/Cys6 DNA-binding domain | 53 | 11 | Transcription factors |
| DNA-binding domain of Mlu1-box binding protein MP1 | 4 | 155 | Transcription factors |
| Bacteria | |||
| TetR/NARL DNA-binding domain | 112 | 19 | Transcription factors |
| IIA domain of mannitol-specific and ntr phosphotransferase EII | 28 | 99 | Carbohydrate transport system: part of phosphoenolpyruvate-dependent sugar phosphotransferase system (PTS) |
| Prokaryotic DNA-binding protein | 18 | 157 | Bacterial histone-like proteins |
| Zn2+ DD-carboxypeptidase, N-terminal domain | 17 | 165 | Found in enzymes involved in bacterial cell wall degradation, possibly peptidoglycan-binding domain |
| Glucose permease domain IIB | 17 | 165 | Part of PTS |
| Regulatory protein AraC | 14 | 182 | Part of the transcription regulation of the arabinose operon |
| LexA/signal peptidase | 11 | 211 | 1. Transcriptional regulation of SOS repair genes, protease domain of the LexA protein 2. Cleaves the N-terminal signal peptides of secreted or periplasmic proteins. |
| Histidine-containing phosphocarrier proteins (HPr) | 11 | 211 | Part of PTS |
| Periplasmic chaperone C domain | 11 | 211 | Assembly of extracellular periplasmic macromolecular structures |
| Duplicated hybrid motif | 10 | 224 | Part of PTS |
| Aspartate receptor, ligand-binding domain | 10 | 224 | Found in different membrane integral sensor and chemotaxis proteins, often associated with kinase domains |
The functional description is taken from PFAM/InterPro and SwissProt homologs. N and R are the same as in Table 1. For human, fly, worm, and yeast, the superfamilies with N >3 and for bacteria N >9 are listed.
Human Branch
The three most frequent domains are implicated with immunity, in particular the major histocompatibility complex (MHC) antigen-recognition domain, interleukin 8–like chemokines, and the 4-helical cytokines. Analysis of our results that include the complete sequence database showed that in addition to mammals the interleukin 8–like superfamily is also found in sequences from birds and fish, and the MHC antigen-recognition domain is also found in amphibia. Several of the other domains specific to the mammalian branch are also involved in immunity—MHC class II-associated invariant chain ectoplasmic trimerization domain and p8-MTCP1 (mature T-cell proliferation). The mammalian defensin is involved in defense against a wide range of microorganisms, whereas the defensin-like superfamily is also found as neurotoxin in some Cnidaria such as anemonae. At third in frequency in the human branch is serum albumin that is a major protein component of blood. Many of these superfamilies potentially specific for human rather than the other species for which we have annotated the genomes were also found in viruses, amphibia, reptiles, fish, and birds when considering the complete sequence database. These include the following frequently occurring domain families: colipase-like for enzyme regulation (particularly required by pancreatic lipases) and involved in development; RNaseA-like with different ribonucleases involved in endonuclease function in pancreas, blood (eosinophil granules), and in angiogenesis; the PKD domain, which is possibly involved in extracellular protein-protein interaction. The RNAase A-like was also found in Aspergillus.
Fly
Insect pheromone/odorant-binding proteins are the most common SCOP superfamily (which occurs 26 times). The next most common are the scorpion toxin-like domains that occur as parts of the fungicide drosomycin, and the antibacterial defensin. Thus, the insect form of immunity/defense leads to a commonly occurring branch-specific SCOP superfamily. However, in addition to arthropods, the scorpion-like toxin and the antibacterial defensin are also found in plants.
Worm
Two superfamilies occur with a frequency four (the osmotin, thaumatin-like proteins and the plant lectins/antimicrobial peptides). Both are involved in pathogen response. However, further examination of protein of the complete sequence database showed that both SCOP superfamilies occur in plant genomes with close homologs.
Yeast (S. cerevisiae)
This is dominated by the Zn-Cys DNA-binding domain of transcription factors. This family is also found in the recently sequenced genome of the yeast S. pombe (Wood et al. 2002).
Bacteria
Given the smaller size of bacterial genomes, we have pooled the superfamilies and their frequencies from the seven organisms we have annotated (i.e., the reported frequencies are the sums of domains in superfamilies from all seven bacterial proteomes, and not averages). Here, we discuss the higher ranking superfamilies. The most frequent domain is a transcription factor, the tetR/NARL DNA-binding domain (also found in some archaea and algae). This is followed by the dimerization domain of the AraC protein that is involved in the transcription regulation of that operon. Third is the superfamily of the DNA-bending protein. Other potentially specific superfamilies are involved in transport (especially the phosphate transferase system, possibly also present in fungi). There is one superfamily involved in the phosphate transferase system, the duplicated hybrid motif, that is also found in mouse (but not human) as has been previously noted (Nakamura et al. 1994). In addition, there are superfamilies specific for the cell wall synthesis, with one superfamily, the Zn2+ DD-carboxypeptidase, that is also found in plants.
Archaea
There are only three species of archaea in our set of organisms, and we did not find any frequently occurring archaea-specific SCOP superfamilies.
The general conclusion from this analysis is that three general classes of biological activity lead to commonly occurring branch-specific superfamilies. These functions are defense (e.g., immunity), transcriptional regulation, and hormone-related signaling.
Gene Duplication
The presence of multiple copies of any particular SCOP domains within the proteome is the result of domain duplication and divergence during evolution, both within and between proteins. The extent of this duplication can be quantified:
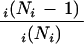 |
where Ni is the number of occurrence of domains in SCOP superfamily type i(Teichmann et al. 1998). This can be estimated from the frequencies of the SCOP superfamilies in a proteome, using these domains as a sample of the entire proteome. Note that the value is for domain duplication and is not necessarily a value for the fraction of the proteome residues that arose from duplication. Figure 3 shows that we estimate that 98% of the human proteome arose via duplication. There are 28,913 different peptide sequences in the data set of human proteome, and we found 23,573 SCOP domains within these sequences, which belong to only 546 different SCOP superfamilies with 23,027 duplication events. The figure shows that as the number of proteins in the genome increases, there is an increase in the extent of gene duplication from the 55% observed in Mycoplasma genitalium as the smallest genome. There is a very rapid increase in the extent of domain duplication in the bacteria and archaea until the size of smallest eukaryote included in our analysis (yeast) is reached. However, one does not observe a marked difference in the extent of duplication between the largest prokaryote (Escherichia coli, 4257 peptide sequences) and the smallest eukaryote (yeast, 6237 peptide sequences) despite the major differences in the organization of their genes, in terms of the presence of introns/exons and of chromosomes. Importantly, because several PFAM families are homologs, when the same estimate is made using PFAM one obtains a lower estimate of the extent of gene duplication in each species.
Figure 3.

Extent of domain duplication in different proteomes. The extent of duplication is estimated from the frequencies of observing domains in the different SCOP superfamilies and is shown as the fraction of total assigned domains for each proteome. The size of the human proteome is estimated at the number of protein sequences in the ENSEMBL dataset (∼29,000). Comparable results from frequencies of PFAM families (Bateman et al. 2000) are reported.
This estimate of domain duplication relies on two assumptions. First is that the duplication frequency of structurally characterized domains (i.e., SCOP) is a representative sample of all proteins in the genomes. This has been analyzed for proteins in the M. genitalium genome by Teichmann et al. (1998) who concluded that the SCOP superfamilies are representative for the proteins in the genome. However, a study by Gerstein (1998) on eight microbial genomes suggested that there are several differences, including in the lengths of the sequences, between the proteins in the PDB and those in the genomes. Nevertheless, the trend of increasing domain duplication, as the size of the proteome increases, is the same for SCOP and PFAM based analysis, suggesting that any bias from using just SCOP is not marked. The second assumption is that all the proteins have been identified in the genome, and one has to estimate the effect of uncharacterized proteins. However, the worm, where gene prediction is more accurate than in human, and therefore even rare, and orphan protein families are more likely to be identified (Reboul et al. 2001), yields a value for domain duplication of 95%, which is probably a lower estimate of the extent for humans.
The values for domain duplication are without a time scale and substantial further work is required to estimate the extent of duplications since divergence of the different phylogenetic branches. Recently, Qian et al. (2001a) have developed an evolutionary model and estimated the extent of fold acquisition within a species. Here, we consider the extent of duplication in the different species of the 10 most frequently occurring SCOP superfamilies found in the human proteome (Fig. 4). Taking the frequency in humans as 100%, Figure 4A shows that all of these 10 SCOP superfamilies have been expanded in human compared to all other species. The greatest expansion from worm and fly to human is for the classic zinc finger. This suggests the major increase in importance of transcriptional regulation in humans via zinc fingers compared to fly and worm. In contrast, the smallest extent of expansion from prokaryotes to human is for the P-loop that has a central role in housekeeping metabolism. This smaller rate of expansion is also observed for another housekeeping superfamily, the RNA-binding domain found at rank three in yeast. The protein kinase-like superfamily has a markedly bigger expansion in worm than in fly, and corresponds to 80% to the expansion in human. This may account for the expansion of certain types of signaling in worm. Note that three of the shown superfamilies are not found in yeast (EGF/laminin, cadherin, and the spectrin repeat), and one, the fibronectin, is only found once.
Figure 4.

Expansion of SCOP superfamilies. The 10 most abundant human superfamilies are shown. (A) Superfamily expansion relative to the human proteome. The expansion of a superfamily relative to the human proteome is plotted as the number of domains in superfamily X in proteome Y divided by the number of domains in superfamily X in human (times 100), so that all superfamilies are 100% in human. (B) Relative superfamily expansion. Number of domains in a superfamily normalized by the number of domains in all superfamilies for a proteome (multiplied by 100). (C) Average repetitiveness of superfamilies. For each superfamily, the number of domains divided by the number of sequences this superfamily is found in is plotted.
These results can be contrasted to analysis of the top superfamilies in bacteria. Of the top 10, seven are expanded in bacteria between 150 and 350% relative to human (data not shown). The two superfamilies that are reduced in bacteria compared to human are the periplasmic binding protein-like II (extra-cellular receptor domains in human and mainly extra-cellular solute binding domains in bacteria) with 70% and the thiolase-like domain (84%). In human, we do not find any CheY-like transcription factors at all.
Figure 4B shows the relative domain frequencies (number of observed domains in a superfamily normalized by the total number of domains in the proteome) of the top 10 human superfamilies for the processed genomes. The 5092 zinc-finger domains that were identified for human comprise more than 20% of the identified domains. Zinc-finger domains have an average length of just 27 residues, and together this corresponds to only 1.5% of the residues in the human proteome. Compared to the majority of the top 10 human superfamilies, the P-loop decreases its relative abundance from prokaryotes to human. Although the domain fraction comprised by P-loops is much lower than for the zinc-finger, because of its average length of 217 residues in human, the P-loop accounts for 2% of all residues. In yeast and worm, the protein kinase-like superfamily seems to have more importance than in fly and human. In addition the RNA-binding domain involved in a range of functions is more abundant in yeast than in the metazoan proteomes where this superfamily accounts for roughly the same fraction of domains. The worm proteome contains relatively more EGF/laminins compared to fly. In general, the relative abundance of the top 10 superfamilies in human, except for the zinc finger, is similar between the metazoan proteomes. Plotting the top 10 superfamilies for yeast shows a similar trend (data not shown); there are only slight changes in the relative domain abundance for most superfamilies between the eukaryotic proteomes. These results imply that in general, the most popular superfamilies in a particular proteome do not comprise a substantially different fraction of the domain repertoire in other proteomes. Given an increasing number of domains for larger proteomes, it may not be a change in relative domain abundance that leads to specialization.
In general, domains of superfamilies found at a high rank are often found in repeats. Here, we define a repeat as at least two domains of the same superfamily that are found within the same peptide sequence irrespective of the sequence distance between these domains. Indeed, the zinc finger is the most repeated domain in human. The average number of repeats for the zinc finger is seven (maximum 36), four (maximum 17), two (maximum five), and two (maximum five) per zinc finger containing sequence for human, fly, worm, and yeast, respectively. In fly and worm, the most repeated domain is the cadherin with on average 12 repeats in fly and eight in worm. The most repeated superfamily in yeast is the KH domain (probably involved in RNA binding) with four repeats on average, and in prokaryotes this is the thiolase-like superfamily (found in proteins of degradative pathways such as fatty acid β-oxidation) with two repeats on average.
Considering only the existence (and not the frequency) of a superfamily in a sequence to exclude the effect of repeats overall just slightly changes the order of the top ranks of superfamilies. The domain-based top 10 ranks in human are still present in the top 22 list that excludes repeats (except for the spectrin repeat at rank 43). The immunoglobulin, the EGF/laminin, and the fibronectin are still within the top 10 (data not shown). Figure 4C plots the average number of repeats within a protein for each of these 10 SCOP superfamilies in human. The most notable feature is that the fly has far more duplicated copies per protein for cadherins (cell surface) and spectrin repeats (cytoskeleton) compared to human. Both worm and fly have more repeated copies per protein of fibronectin and immunoglobulin than human. Overall, seven of the 10 superfamilies are repeated on average at least twice per sequence. The most abundant superfamilies in yeast and especially in bacteria are not as frequently found in repeats as the most popular superfamilies in metazoa (data not shown).
In general, this implies that repetitiveness on the domain level may play an important role in the divergence of the metazoan branch from single cellular eukaryotes. As mentioned above, several of the popular superfamilies in human are associated with cell-surface functions such as cell adhesion, for which long proteins with regular structure may be required.
We also considered the number of different domain-domain associations for the commonly occurring SCOP superfamilies. An association is taken when two different SCOP superfamilies occur within the same sequence (including self association). For a detailed analysis of pairs of adjacent domains and their phylogenetic distributions, see Apic et al. (2001). Figure 5A plots the number of partners for the 10 most common superfamilies in human, Figure 5B for those in yeast, and Figure 5C for bacteria (note, that for better scaling of the plots, in 5B and 5C only, superfamilies are shown that are not already plotted in 5A). The general trend is that the number of different associations is roughly similar for the three multicellular eukaryotes. An interesting feature is that there tends to be somewhat more domain pairings in fly compared to worm. Although the protein kinase-like superfamily is more popular in worm than in fly, and also more than in human when normalized by the number of domains in the proteome as in Figure 4B, the worm has fewer partners for this superfamily. In addition, the most popular partner for the protein kinase-like superfamily in human and fly is the SH3 domain with 43 occurrences in human and 14 in fly (partner data not shown); in worm there are only seven such cooccurrences. The most popular protein kinase-like partner in worm is the adenylyl and guanylyl cyclase catalytic domain with a frequency of 24, and five in human. In all three metazoan proteomes, the SH2 domain is a frequent partner for the protein-kinase like superfamily. The number of partners for EGF/laminin domains decreases from worm to fly, but in human there are more partners for this superfamily than in worm. A frequent domain partner for EGF/laminin domains in worm is the c-type lectin (found 22 times) that has been mentioned above (see SCOP Superfamilies section), which is not a partner for EGF/laminin domains in the fly but is found as an EGF/laminin partner 25 times in human. The immunoglobulin superfamily has more cooccurrences in fly than in worm and human. In fly, this superfamily combines with di-copper center-containing domains that are also found in human (but not as a partner of immunoglobulins). Also the hemocyanin N-terminal domain, absent in human and worm, is found in combination with immunoglobulins. In fly, the hemocyanin N-terminal domain, the di-copper center-containing domain and the immunoglobulin are in fact found together in sequences that belong to the invertebrate copper-containing oxygen transport proteins and larval storage proteins (InterPro family IPR000896). In human, a popular partner for immunoglobulins is the MHC antigen-recognition domain, which is not found at all in fly and worm.
Figure 5.



SCOP superfamily partners. The plots show the number of different SCOP superfamilies that are found together in the same sequence with a given superfamily, including the superfamily itself and irrespective of the order or sequence space between domains. This implies that at least two domains have to be identified in a sequence. Superfamily partners for the 10 most abundant superfamilies in human (A), in yeast (B) and bacteria (C) are plotted. Only those superfamilies not found within the first 10 ranks in human are shown in B (P-loop, protein kinase-like, tetratricopeptide repeat, and the classic zinc finger) and C (P-loop rank one in bacteria).
Figure 5B shows the top 10 superfamilies in yeast. Only the tetratricopeptide repeat, a domain probably involved in a wide range of protein-protein interactions, expands its domain partner repertoire in a step from yeast and worm to fly and to human. The other superfamilies have similar frequencies in the three metazoans.
Figure 5C shows that all the popular superfamilies in bacteria have markedly fewer cooccurrence partners in archaea, although seven of these superfamilies are also found in the top 10 superfamilies in archaea (data not shown). In worm, five of the popular bacterial superfamilies have an increased number of partners compared to yeast, fly, and human, possibly reflecting a closer phylogenetic relationship between worm and bacteria.
The plots in Figure 5 only show the number of different superfamily partners. However, even if the number of partners is similar, the actual frequencies and composition of these partnerships often shows great variation. Hegyi and Gerstein (2001) demonstrated that there is less functional conservation in multidomain than in single-domain proteins except if they have exactly the same domain combination, so that a superfamily can have different functional contexts. This observation from Hegyi and Gerstein suggests a higher degree of functional variation for a superfamily in different proteomes even if the number of domain partners is similar. For example, we find 15 partners for the c-type lectin in human and worm, but some of the frequently found partners are different. In worm, we find many spermadhesin and integrin A domains together with c-type lectins, whereas the integrin A is not found at all as a partner for c-type lectins in human, although the overall integrin domain frequency in human is more than twice as high than in worm. In human, we find more complement control modules (SRC domain) and immunoglobulins in combination with c-type lectins (the immunoglobulin is not found at all in the list of lectin partners in worm). In addition, it has been shown that in many cases of adjacent domains, the domain order is an important functional aspect (Apic et al. 2001; Bashton and Chothia 2002).
In summary, our analysis suggests that for most superfamilies, as the organism increases in complexity, specialization and diversity does not arise from an increasing number of domain combinations, rather from refinement and diversification of the superfamily repertoire itself and probably by changing the repertoire of domain partners.
The web site mentioned in Methods provides a link to an application that allows generic ranking of selected proteomes according to selected properties such as domain frequencies, superfamily partners, or domain repetitiveness of superfamilies. The results can be displayed as a table and as a plot similar to those shown in this paper.
SCOP Superfamilies in Disease Genes
The Online Mendelian Inheritance in Man (OMIM) database (Antonarakis and McKusick 2000) identifies genes that have been associated with human disease. Human proteins were associated with OMIM identifiers via the genelink table from Ensembl. Six thousand, six hundred fifty-six different OMIM entries are linked to 5856 human proteins, so that a human protein can be associated with several OMIM entries. We then evaluated the frequency of each SCOP superfamily in the proteome assigned to disease genes versus the nondisease genes. Seven thousand, six hundred twenty-one SCOP domains in 481 different superfamilies could be assigned to disease genes.
This analysis directly associates SCOP superfamilies with disease and nondisease genes. However, the cause of disease state could be the result of one (or a combination) of effects not directly involving the protein, for example alteration of regulation or deletion of the entire gene. In addition, any point mutation or deletion within a protein may not be within a particular SCOP domain. However, for many genes in OMIM, the location of the alteration (e.g., point mutation) is not known. Thus, to analyze the entire OMIM database, one can only perform a high level view of the distribution of SCOP superfamilies between disease and nondisease genes. A more focused analysis would consider only those genes where the location of the alteration has been identified (for a review of computational analysis of disease genes, see Sreekumar et al. 2001).
The overall frequencies of SCOP superfamilies in the two sets of genes are significantly different at >99.9% confidence. Table 3 reports the SCOP superfamilies that are significantly over- and underrepresented in the disease genes at >95% confidence as confirmed by a χ2 test. We have performed the analysis of the superfamilies in disease genes on the protein sequence level rather than on the domain level, so that we count only one domain per superfamily per protein sequence. The aim of our analysis is to describe general trends for superfamilies and their biological function in association with disease, and therefore we exclude superfamilies with low sequence frequencies but significantly high domain frequency as a result of repeats that confuse a trend analysis. An example for this is the extracellular domain of the cation-dependent mannose 6-phosphate receptor with 15 domains in disease proteins (one domain in the small mannose 6-phosphate receptor and 14 repeated domains in the big receptor) and only two domains in nondisease proteins. This receptor plays an important role in targeting lysosomal enzymes to the lysosome.
Table 3.
Over- and Under-represented SCOP Superfamilies in OMIM Disease Genes
| SCOP superfamily | R | ND | NnD | f | Description |
|---|---|---|---|---|---|
| Interleukin 8-like Chemokines (V) | 62 | 36 | 12 | 3.00 | Mainly small inducible cytokines (single domain proteins), immunoregulatory and inflammatory processes, homeostasis, development. Secreted proteins, activity via GPCRs. |
| Nuclear receptor ligand-binding domain (M) | 56 | 40 | 15 | 2.67 | Growth factor inducible intracellular steroid/thyroid receptors coupled with a DNA-binding domain (mostly glucocorticoid-receptor–like) such as estrogen receptor (breast cancer associated). Transcription factors and enhancers. |
| Cystine-knot cytokines (E) | 49 | 42 | 17 | 2.47 | Growth factors belonging to TGF-β, cell determination, differentiation and growth. Neurotrophins, differentiation, and function of neurones. |
| Periplasmic binding protein-like I | 96 | 21 | 9 | 2.33 | Glutamate receptors, ionotropic (ion channels) and metabotropic (GPCRs with activity via a second messenger), also receptors for atrial natriuretic clearance peptides, involved in regulation of blood pressure. |
| Serpins (M) | 76 | 26 | 12 | 2.17 | Serine protease inhibitors of the blood-clotting cascade. |
| 4-helical cytokines (V) | 66 | 32 | 15 | 2.13 | Different interferons and interleukins (extracellular single-domain proteins), regulatory in differentiation and proliferation, antiviral, immune, and inflammatory response. |
| Winged helix DNA-binding domain | 21 | 70 | 57 | 1.23 | Associated with at least 25 disease entries. Transcription factors (activation and repression). Dominated by forkhead family members, important in embryogenesis of the nervous system in mammals, associated with different leukemia; ETS family of oncogene products; histones (chromatin remodelling), and others. |
| Helix-loop-helix DNA-binding domain (E) | 28 | 54 | 45 | 1.20 | Transcriptional control for cell-type determination during development, also transcriptional control of histone acetyltransferases (preparing chromatin for transcription). |
| Glucocorticoid receptor-like (DNA-binding domain) (E) | 25 | 62 | 52 | 1.19 | Together with nuclear receptor ligand-binding domains (see above). Frequently found in proteins of developmental genes. LIM domain proteins deregulated in cancer cell-lines. |
| Homeodomain-like | 8 | 131 | 142 | 0.92 | Different homeobox proteins (transcription factors), particularly important in early embryogenesis. Some homeobox genes are oncogenes. |
| Protein kinase-like (PK-like) | 4 | 246 | 291 | 0.85 | About 100 different associated disease entries (e.g., different cancers). Range of kinases such as MAP or PKC (signal transduction). |
| RNA-binding domain | 6 | 76 | 255 | 0.30 | RNA splice factors (alternative splicing), rapid degradation of mRNAs in particular from cytokines and protooncogenes. Involved in spermatogenesis related to male infertility, for example. |
| RING-finger domain, C3HC4 (E) | 13 | 43 | 163 | 0.26 | Zinc-finger–like domain associated with protein-protein interaction, often found in transcription regulatory proteins. Linked to apoptosis inhibitors, breast cancer gene BRACA1, acute leukemia, for example. |
| Classic zinc finger, C2H2 | 2 | 135 | 549 | 0.25 | Nucleic-acid binding, range of transcription factors, cell proliferation and differentiation, early development, some are protooncogenes. |
| Tetratricopeptide repeat (TRP) | 19 | 25 | 121 | 0.21 | Interaction partner of regulatory proteins, subunit of G-proteins. Involved in a range of biological functions such as cell-cycle, activation of apoptosis, chromatin assembly, actin binding, cancer. |
| Ankyrin repeat | 12 | 33 | 187 | 0.18 | Protein-protein interaction domain. Found at least 17 different OMIM entries describing, e.g., inhibitor of NFkB and cyclin-dep. kinase inhibitors, interaction with p53 in apoptosis. Cooccurrence with other interaction and regulatory domains such as DEATH and SH3. |
| eL30-like | 58 | 5 | 45 | 0.11 | Ribosomal protein L30, translation termination. |
| Pyk2-associated protein β ARF-GAP domain (E) | 91 | 1 | 31 | 0.03 | RIP protein that assists HIV in replication by facilitating the nuclear export of mRNA. Corresponds to the putative GTP-ase activating protein for Arf in PFAM. Nondisease proteins are often associated with PH-domains or ankyrin repeats and may have a range of biological function. |
For each SCOP superfamily, the rank order (R) of superfamily occurrences in sequences of the human proteome is given (see text for details), followed by the sequence frequency in disease genes (ND) and the frequency in nondisease genes (NnD). The ratio (f) of these occurrences is then given as ND/NnD. The double horizontal line separates over-represented from underrepresented superfamilies. Taking all SCOP domains together, the two populations (disease and nondisease) are significantly different (>99.9% confidence) as calculated by a χ2 test. For each SCOP superfamily, the frequency ratio compared to the others was significant at >95% confidence, after allowing for the number of SCOP domains tested (testing domains of each superfamily against all remaining domains). Bold letters in braces in the superfamily field indicate that this superfamily is specific for eukaryotes (E), metazoans (M), or vertebrates (V). The Description field gives an overview of the broad biological functions associated with the disease genes.
Superfamilies over-represented in proteins of disease genes are mainly associated with regulation having biological functions in development, differentiation, and proliferation, and not being directly involved in metabolism. Overall, the overrepresented superfamilies can be put into the categories immune response, immune regulation, growth factors, and transcription factors. The main biological relevance of the underrepresented superfamilies may be summarized as transcription factors, protein-protein interaction domains involved in signaling and transcription (other than transcription factors), and translation. However, many of the superfamilies are involved in a wide range of biological functions and may be placed in more than one category, e.g., the interleukin 8–like chemokines are not only involved in immune response but also play a regulatory role during development.
The most over-represented superfamilies (with a ratio >2) are biased toward small, mainly extracellular single or two-domain messenger proteins (interleukin, cystine-knot cytokines, and 4-helical cytokines), whereas three of the seven strongly underrepresented superfamilies (with a ratio ≤0.3) are involved in regulation via protein-protein interaction, and another three superfamilies are involved in transcription and translation. Further, the five most overrepresented superfamilies are specific for human, metazoa, or at least eukaryota, whereas in the set of underrepresented superfamilies only two eukaryotic-specific superfamilies are found. On the other hand, eight of the nine under-represented superfamilies are in the list of the top 20 superfamilies in human sequences, four within the top 10. None of the overrepresented superfamilies is found within the top 20 ranks. The overrepresented superfamily with lowest rank (highest frequency) in human is the “winged helix” DNA-binding domain (rank 21).
Taking the above observations together, the most overrepresented superfamilies in disease genes are those likely to have evolved within the metazoan branch of evolution and that are moderately expanded in human (average sequence rank of 65 of 463). The homeodomain-like and protein kinase-like superfamilies are just slightly but significantly underrepresented, and are found with high overall frequencies in both categories. These two superfamilies are associated with biological key functions in many regulatory pathways (see Table 3 for details). Our results suggest that it is, in general, unlikely to find abundant superfamilies with a massive bias toward disease proteins, possibly because the disruption of key functions may often be lethal. However, despite this general suggestion, we do not have any explanation why certain superfamilies are over- or underrepresented in disease genes. Our observations may encourage future work to formulate hypotheses that may lead to deeper insights into the relationship between disease and structural folds.
Transmembrane Proteins
Transmembrane regions in the proteomes were identified using the hidden Markov approach implemented in TMHMM-2 (Sonnhammer et al. 1998). Figure 6A shows the fraction of the proteomes that were predicted to occur as membrane-spanning regions. For the human proteome, only 3% of residues are predicted to be in transmembrane regions (the membrane-spanning part of the protein), which is a similar percentage as for yeast and fly but less than in worm and the average values for bacteria and archaea. The figure also shows that 13% of the proteome consists of globular regions (regions excluding coiled-coils, low-complexity regions, or signal peptides) that are part of a protein chain that spans a membrane (striped, light-grey bars). In human, only about 1% of the residues form short loops (<30 continuous residues) linking two membrane-spanning regions or occurring at a chain terminus of membrane proteins. The ratio between the globular part of transmembrane proteins and the membrane-spanning part is smaller in bacteria and archaea than in the four eukaryotes. This may be because of a larger fraction of proteins in bacteria and archaea that are completely membrane integral (i.e., proteins mainly built by membrane helices and connecting loops such as bacteriorhodopsin and probably those of membrane integral redox cascades). The proteome of C. elegans contains both the largest fraction and the largest absolute number of transmembrane proteins (28% of the proteome, 4559 membrane proteins). The high number of transmembrane proteins is mainly from an expansion of the family of seven helix transmembrane G-protein coupled receptors (Bargmann 1998).
Figure 6.


Distribution of transmembrane and globular regions in the proteomes. (A) Fractions of globular and nonglobular parts in membrane proteins. Globular denotes globular domains in nontransmembrane proteins, TM/Globular are globular regions within transmembrane helix containing proteins, TM/Loop are short loops in transmembrane proteins, and TM are the actual transmembrane helices. (B) Ratio of globular regions to transmembrane regions in membrane sequences classified according to the number of transmembrane regions. The diagram only shows ratios for which at least nine transmembrane proteins were found.
Figure 6B shows the ratio of residues in globular domains to residues in transmembrane regions for different membrane proteins as determined by the number of predicted membrane-spanning helices. The ratios are substantially different between species for proteins with one to three transmembrane regions and become more similar as the number of transmembrane regions increases. This shows that the full sequence of transmembrane proteins with only one to three membrane-spanning regions differ in length between the proteomes of the analyzed organisms reflecting a higher number of potential globular domains, with the fly having longer protein sequences for transmembrane proteins than the other organisms. In bacteria and archaea, the ratio drops below one (e.g., the majority of the protein is membrane integral) at about six to seven membrane segments. In contrast, eukaryotes have the majority of the residues of the protein in potential globular domains, suggesting additional functionality such as protein-protein interaction or receptor capabilities of these membrane proteins.
Table 4 reports the frequencies of SCOP superfamilies that occur in protein chains that span the membrane. We focus on the globular domains associated with transmembrane proteins and accordingly exclude completely membrane-integral proteins of the analyzed proteomes and do not consider the SCOP class of membrane proteins. The four superfamilies of highest rank are domains that can be found in cell-surface proteins involved in cell–cell interaction and receptor molecules. In human, the most common SCOP domain associated with membrane-spanning chains is the immunoglobulin superfamily, whereas in fly and worm this superfamily is at rank four and five, respectively. The cadherin is the most common SCOP superfamily in fly, and in worm the EGF/laminin is the most popular membrane-associated superfamily. The relative importance of superfamilies involved in cell–cell interaction and cell-surface proteins is also pointed out by the absence of these superfamilies in yeast (also see Table 1). All eight immunoglobulin domains found in yeast are located in soluble, probably intracellular proteins (no signal peptides could be found via prediction). In conclusion, the results of the transmembrane analysis reflects the multicellular environment of human, fly, and worm, where specialized systems for cell–cell communication and recognition are required in, for example, tissue formation.
Table 4.
SCOP Superfamilies Associated With Transmembrane Proteins
| SCOP Superfamily | Human | Fly | Worm | Yeast | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N | % | R | N | % | R | N | % | R | N | % | R | |
| Immunoglobulin | 463 | 38 | 1 | 126 | 26 | 4 | 74 | 16 | 5 | – | – | – |
| Cadherin | 440 | 72 | 2 | 206 | 93 | 1 | 114 | 84 | 2 | – | – | – |
| Fibronectin type III | 359 | 43 | 3 | 134 | 54 | 3 | 66 | 30 | 7 | – | – | – |
| EGF/laminin | 216 | 18 | 4 | 139 | 43 | 2 | 163 | 39 | 1 | – | – | – |
| Ligand-binding domain of low-density lipoprotein receptor | 126 | 51 | 5 | 106 | 54 | 5 | 79 | 55 | 4 | – | – | – |
| P-loop containing nucleotide triphosphate hydrolases | 87 | 10 | 6 | 89 | 15 | 6 | 91 | 18 | 3 | 41 | 10 | 1 |
| Protein kinase-like (PK-like) | 65 | 12 | 7 | 27 | 10 | 12 | 72 | 17 | 6 | – | – | – |
| Complement control module/SCR domain | 56 | 20 | 8 | 25 | 44 | 13 | 3 | 6 | 65 | – | – | – |
| C-type lectin-like | 53 | 36 | 9 | 3 | 8 | 54 | 34 | 11 | 8 | – | – | – |
| MHC antigen-recognition domain | 47 | 82 | 10 | – | – | – | – | – | – | – | – | – |
| TNF receptor-like | 38 | 73 | 11 | 2 | 100 | 67 | – | – | – | – | – | – |
| RNI-like | 34 | 35 | 12 | 31 | 35 | 8 | 14 | 38 | 23 | – | – | – |
| Serine protease inhibitors | 32 | 25 | 13 | 17 | 41 | 19 | 18 | 21 | 19 | – | – | – |
| Periplasmic binding protein-like I | 28 | 93 | 14 | 16 | 73 | 22 | 30 | 88 | 11 | – | – | – |
| ConA-like lectins/glucanases | 27 | 20 | 15 | 28 | 42 | 10 | 27 | 26 | 12 | 5 | 63 | 10 |
| RING-finger domain, C3HC4 | 25 | 12 | 16 | 17 | 16 | 19 | 20 | 16 | 18 | 5 | 15 | 10 |
| L domain-like | 25 | 21 | 16 | 25 | 26 | 13 | 23 | 16 | 15 | 1 | 8 | 38 |
| Spermadhesin, CUB domain | 24 | 19 | 18 | 42 | 50 | 7 | 23 | 13 | 15 | – | – | – |
| (Phosphotyrosine protein) phosphatases II | 23 | 21 | 19 | 7 | 17 | 30 | 14 | 14 | 23 | – | – | – |
| EF-hand | 23 | 8 | 19 | 15 | 9 | 24 | 10 | 8 | 29 | – | – | – |
| Metalloproteases (“zincins”), catalytic domain | 22 | 33 | 21 | 4 | 15 | 43 | 8 | 16 | 37 | – | – | – |
| POZ domain | 22 | 18 | 21 | 5 | 5 | 37 | 22 | 15 | 17 | – | – | – |
| C2 domain (Calcium/lipid-binding domain, CaLB) | 21 | 11 | 23 | 17 | 25 | 19 | 32 | 36 | 10 | 16 | 50 | 2 |
| Ankyrin repeat | 21 | 8 | 23 | 18 | 15 | 18 | 34 | 27 | 8 | 5 | 16 | 10 |
| Extracytoplasmic domain of cation-dependent mannose 6-phosphate receptor | 15 | 88 | 32 | – | – | – | – | – | – | 1 | 100 | 38 |
| SpoIIaa | 5 | 83 | 63 | 4 | 100 | 43 | 5 | 100 | 53 | 2 | 100 | 25 |
| Adenylyl and guanylyl cyclase catalytic domain | 16 | 67 | 29 | 28 | 76 | 10 | 26 | 70 | 13 | – | – | – |
| Blood coagulation inhibitor (disintegrin) | 18 | 67 | 26 | 3 | 43 | 54 | 4 | 67 | 58 | – | – | – |
| Periplasmic binding protein-like II | 16 | 62 | 29 | 30 | 77 | 9 | 12 | 92 | 25 | – | – | – |
| Syntaxin 1A N-terminal domain | 8 | 62 | 47 | 5 | 56 | 37 | 8 | 62 | 37 | 7 | 88 | 5 |
| L-2-Haloacid dehalogenase | 11 | 61 | 34 | 2 | 10 | 67 | 5 | 28 | 53 | 2 | 13 | 25 |
| Snake toxin-like | 5 | 56 | 63 | 2 | 100 | 67 | 1 | 50 | 98 | – | – | – |
| Metal-binding domain | 6 | 55 | 58 | 4 | 80 | 43 | 4 | 80 | 58 | 5 | 71 | 10 |
| Transferrin receptor ectodomain, apical domain | 7 | 54 | 53 | – | – | – | 2 | 50 | 79 | 3 | 75 | 20 |
The table gives the number (N) of domains in each superfamily that are found in sequences that have a transmembrane section. The “%” is percentage of the toal occurrence of each domain in the proteome (i.e., transmembrane and nontransmembrane chains, see Table 1). R denotes the rank of N. The lower part of the table details superfamilies with highest percentages in membrane proteins and with a frequency of at least five domains in human that are not reported in the upper part.
Table 4 also presents the fraction of the total domain frequency for each superfamily that is associated with membrane-spanning chains. Of the superfamilies with at least five domains in transmembrane proteins, only the MHC antigen-recognition domain and the periplasmic binding protein-like I have more than 80% of their representative domains in transmembrane proteins. Further down the list (bottom part of Table 4), several other superfamilies are found with more than 50% of their domains in transmembrane proteins. However, in worm we find all six representatives of the scavenger receptor cysteine-rich (SRCR) domain (found in membrane glycoproteins) and all spoIIa domains with five representatives (sulphate transports) in membrane proteins.
SCOP superfamilies that are frequently associated with transmembrane regions are also common in chains that do not span the membrane. This supports the view that domains are mobile elements that are not restricted to coevolve either always in association with a transmembrane section or always in a chain that does not span the membrane.
The top-ranking superfamilies in bacteria are different from those found in eukaryotes (data not shown in Table 4). These superfamilies are mainly associated with bacterial signaling (ATPase domain and homodimeric domain of signal transduction histidine kinase, PYP-like sensor domain, CheY-like) or with small molecule binding, probably as membrane-bound receptors or enzymes (P-loop containing nucleotide hydrolases, phosphatases/sulphatases, Rossmann-fold, nucleotide-diphospho-sugar transferases, FAD/NAD(P)-binding domain, metal-binding domain). In bacteria, we do not find any globular superfamily with more than two representatives (an average over the seven processed bacterial proteomes) that is exclusively found in membrane proteins. The list of most popular superfamilies found in transmembrane proteins for archaea is similar to those for the bacteria, but the frequencies of which domains are found are much lower, e. g., the top-ranking superfamily is the P-loop with only eight domains in the three archaea proteomes.
Figure 7 shows the frequencies of the overall top 10 human superfamilies (the same superfamilies as in Figure 4) with their number of domains in membrane proteins compared to the other processed proteomes (7A), and the same for the top-ranking bacterial superfamilies (7B, the P-loop is not shown). As expected the immunoglobulin, cadherin, fibronectin, and EGF/laminin are most expanded in human compared to fly and worm. Interestingly, the P-loop is found with very similar numbers in membrane proteins in all metazoan proteomes, compared to the overall expansion shown in Figure 4A. This suggests that, although there are more P-loops in human than in fly and worm, the additional duplications are associated with soluble proteins only.
Figure 7.


Expansion of SCOP superfamilies in membrane proteins. The number of domains in a superfamily that are found in proteins that have at least one transmembrane helix are shown for the different proteomes. The 10 overall most abundant superfamilies in human (A), as in Figure 4, and bacteria (B) are plotted. The P-loop is excluded from B, as it is already shown in A.
The top-ranking superfamilies in bacteria (7B) are rarely associated with membrane proteins in prokaryotes and yeast, and this trend also remains across the metazoans for seven of the 10 superfamilies (we did not find any CheY-like domains in human). Little expansion is observed in total numbers for three superfamilies compared to the figure in human (7A). We find only one periplasmic binding protein-like II domain on average in membrane proteins in bacteria, and although the total number of domains in this superfamily is higher than for the other proteomes (data not shown), membrane association has only been expanded in metazoa. However, the periplasmic binding protein-like II is a diverse superfamily that contains at least 10 different PFAM families, and in bacteria there seem to be many soluble extracellular members of this superfamily (suggested by signal peptide prediction). Most of the metazoan domains of this superfamily are ligand-gated, ion-channel domains and receptor family ligand-binding domains, both found in membrane proteins. In yeast, four of the five domains of this superfamily are part of presumably intracellular soluble proteins involved in pyrimidine biosynthesis. The divergence of the periplasmic binding protein-like II superfamily to produce different functional families in bacteria and metazoa seems to be coupled to some extent with different sub-cellular location (soluble and membrane bound).
Conclusion
We have performed an integrated analysis of the human proteome and compared the results to those of other proteomes. The key aspect of this study is the integration in the context of the different species of the following features: the extent and reliability of structural and functional annotations of the proteomes; the extent of domain duplication; change and expansion of the structural superfamily repertoire between different proteomes; the relationship between human disease genes and structural superfamilies; and the relationship between transmembrane proteins and their globular regions. The study included a structure-based analysis from which we were able to get evolutionary insights that could not be obtained from sequence-based methods alone. The structural analysis complements consideration of the extent of functional annotation. We assessed the role of structural knowledge in assisting functional annotation.
These general bioinformatics analyses require simplifications and are also subject to errors in the predictive methods. In particular, we have had to employ a simplified strategy to estimate the extent to which there is some functional information derivable by homology. However, this reflects the standard practice in obtaining an initial suggestion of protein function in the absence of characterized motifs such as PFAM. Automated proteome annotation, particularly in eukaryotes, is complex and the exact numbers reported in our analysis will need to be refined as the bioinformatics tools improve and more experimental data becomes available.
This study and related work by others (Koonin et al. 2000; Apic et al. 2001; Frishman et al. 2001; Iliopoulos et al. 2001) have highlighted the extent to which we still need structural information as a step toward understanding the function of the human and other proteomes. The experimental determination of the structures of these proteomes is the goal of structural genomics initiatives. Vitkup and coworkers have suggested that within 10 years we will have representatives of most protein superfamilies (Vitkup et al. 2001). However, today we have some structural information for about 40% of the human proteome that can be used to provide functional insights.
METHODS
Protein Sequences From Complete Genomes
Eukaryota: Saccharomyces cerevisiae, Caenorhabditis elegans, Drosophila melanogaster, Homo sapiens. Bacteria: Mycobacterium tuberculosis, Escherichia coli, Bacillus subtilis, Mycoplasma genitalium, Helicobacter pylori, Aquifex aeolicus, Vibrio cholerae. Archaea: Aeropyrum pernix, Pyrococcus horikoshii, Methanococcus jannaschii. The H. sapiens proteome is the ENSEMBL-0.8.0 confirmed peptide data set (http://www.ensembl.org). Other sequences were taken from the NCBI (ftp://ftp.ncbi.nlm.nih.gov/genbank/genomes/).
Sequence Analysis
Sequences, annotations, and results are stored in a relational database (MySQL, http://www.mysql.com), which serves as the back end for an automated processing pipeline running on a Linux computer farm. Our software and database system allows for updates of the data and results as well as comparisons across proteomes.
The sequences were first scanned for signal peptides (SignalP-1.1 [Nielson et al. 1997], http://www.cbs.dtu.dk/services/SignalP/); transmembrane helices (TMHMM-2.0 [Sonnhammer et al. 1998], http://www.cbs.dtu.dk/services/TMHMM-2.0/); coiled-coils (Coils2 [Lupas et al. 1991]); low-complexity regions (SEG [Wootton and Federhen 1996]); and repeats ( Prospero V1.3 [Mott 2000]).
Protein sequence database searches were performed using PSI-BLAST version 2.0.14 (Altschul et al. 1997). Sequences were masked for low-complexity regions, transmembrane regions, coiled-coils, and repeats. The e-value cutoff was 5 × 10−4 and the maximum number of iterations was 20. The sequence database used contained 634,179 different protein sequences from the NCBI NR-PROT (all nonredundant GenBank CDS translations, PDB, SwissProt, and PIR, the protein sequences of the genomes processed in this work, and the sequences from the SCOP-1.53 database). It has been shown (Park et al. 1998) that PSI-BLAST–detected relationships are not symmetric, that is, a query with sequence A might not have a significant match to B whilst searching with B could have a significant match to A. To address this problem, each SCOP sequence was run against our protein sequence database via PSI-BLAST to construct a position-specific scoring matrix (PSSM) that was used with the IMPALA program (Schaffer et al. 1999) to assign SCOP domains to each of the genome sequences. We found that this procedure increases the sensitivity without introducing many new false positives.
Examination of our initial results showed that there was a problem in PSI-BLAST detecting very short SCOP domains (<50 residues) because BLAST/PSI-BLAST e-values may not be significant for short alignments yet manual investigation of the region strongly suggested that it should be assigned to a particular SCOP domain. We developed a heuristic method to address this problem whereby an assignment to a SCOP domain was accepted with an e-value <10 for an IMPALA and BLAST hit and five for a PSI-BLAST hit if the domain is shorter than 50 residues and the sequence identity of the alignment satisfies the identity cut-off described by Rost (1999), which requires a much higher sequence identity for shorter than for longer alignments. If the identity condition was not satisfied, a SCOP domain was still accepted if the alignment shares a common Prosite pattern (Hofmann et al. 1999) between query and subject. All accepted SCOP domains must be present with at least 65% of their domain in the alignment, to avoid partial domain assignments that are in many cases false positives. This additional confidence measure was chosen by analyzing the distribution of true and false-positive alignments between SCOP domains (data not shown).
GAP-BLAST (Altschul et al. 1997) was run for those sequences that contain a transmembrane region, coiled-coil region, or a repeat, but without removing (masking) these regions, only low-complexity regions were masked. This was required to identify the close homologous for a query sequence for which PSI-BLAST would fail to identify homologs as a result of the sequence masking (e.g., membrane integral sequences). The e-value cutoff was 5 × 10−4. PFAM domains were assigned via HMMer (Eddy 1998) and the PFAM hidden Markov model library version 6.2. The e-value cutoff to accept a hit was 0.1 and a domain had to be present in the reported alignment with at least 75% of its entire length. It should be noted that residue-based calculations are based on the sequence comparison methods mentioned above. For the BLAST-based (and derivatives-based) assignments this means that ends of domains may not be correctly identified during the extension step of the algorithm. Also, we do not consider potential interdomain regions, so that even in theory, 100% residue-based assignment may not be reached. This affects the results shown in our bar plots. However, this is a systematic error on the algorithm level of the employed methods, and we assume that this affects the results of all the processed sequences equally, so that as first approximation, a comparison of residue-based fractions is still valid.
For the analysis of transmembrane proteins, sequences were truncated if the SignalP program could identify a potential signal peptide. This avoids false-positive predictions of transmembrane regions at the N terminus of a sequence.
Availability of Annotation
The results of our analysis are available as 3D-GENOMICS via our web page http://www.sbg.bio.ic.ac.uk. This includes query forms for database searches and the display of tables and alignments. We provide a special section with results from comparative analyses, including an application to generically list different domain properties such as repetitiveness, association with transmembrane proteins, or domain partners ranked by frequency in a selected “master” proteome.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
WEB SITE REFERENCES
http://supfam.mrc-lmb.cam.ac.uk/SUPERFAMILY/; results from the HMM superfamily analysis by Gough et al. (2001).
http://www.cbs.dtu.dk/services/SignalP/; SignalP-1.1 Web site.
http://www.cbs.dtu.dk/services/TMHMM-2.0/; TMHMM-2.0 Web site.
http://www.ensembl.org; ENSEMBL Web site.
http://www.mysql.com; MySQL relational database.
http://www.sbg.bio.ic.ac.uk; data and results for this article.
Footnotes
E MAIL m.sternberg@ic.ac.uk; FAX +44-(0)20-7594-5264.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.221202.
REFERENCES
- Aloy P, Querol E, Aviles FX, Sternberg MJE. Automated structure-based prediction of functional sites in proteins—Applications to assessing the validity of inheriting protein function from homology in genome annotation and to protein docking. J Mol Biol. 2001;311:395–408. doi: 10.1006/jmbi.2001.4870. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: A new generation of protein data base search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonarakis SE, McKusick VA. OMIM passes the 1,000-disease-gene mark. Nat Genet. 2000;25:11. doi: 10.1038/75497. [DOI] [PubMed] [Google Scholar]
- Apic G, Gough J, Teichmann SA. Domain combinations inarchaeal, eubacterial and eukaryotic proteomes. J Mol Biol. 2001;310:311–325. doi: 10.1006/jmbi.2001.4776. [DOI] [PubMed] [Google Scholar]
- Bargmann CI. Neurobiology of the Caenorhabditis elegans genome. Science. 1998;282:2028–2033. doi: 10.1126/science.282.5396.2028. [DOI] [PubMed] [Google Scholar]
- Bashton M, Chothia C. The geometry of domain combination in proteins. J Mol Biol. 2002;315:927–939. doi: 10.1006/jmbi.2001.5288. [DOI] [PubMed] [Google Scholar]
- Bateman A, Birney E, Durbin R, Eddy SR, Howe KL, Sonnhammer ELL. The Pfam protein families database. Nucleic Acid Res. 2000;28:263–266. doi: 10.1093/nar/28.1.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates PA, Sternberg MJE. Model building by comparison at CASP3: Using expert knowledge and computer automation. Proteins. 1999;Supplement 3:47–54. doi: 10.1002/(sici)1097-0134(1999)37:3+<47::aid-prot7>3.3.co;2-6. [DOI] [PubMed] [Google Scholar]
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The protein data bank. Nucleic Acid Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conte LL, Ailey B, Hubbard TJP, Brenner SE, Murzin AG, Chothia C. SCOP: A structural classification of proteins database. Nucleic Acid Res. 2000;28:257–259. doi: 10.1093/nar/28.1.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devos D, Valencia A. Practical limits of function prediction. Proteins. 2000;41:98–107. [PubMed] [Google Scholar]
- Eddy SR. Profile hidden Markov models. Bioinformatics. 1998;14:755–763. doi: 10.1093/bioinformatics/14.9.755. [DOI] [PubMed] [Google Scholar]
- Frishman D, Albermann K, Hani J, Heumann K, Metanomski A, Zollner A, Mewes HW. Functional and structural genomics using PEDANT. Bioinformatics. 2001;17:44–57. doi: 10.1093/bioinformatics/17.1.44. [DOI] [PubMed] [Google Scholar]
- Gerstein M. How representative are the known structures of the proteins in a complete genome? A comprehensive structural census. Fold Des. 1998;3:497–512. doi: 10.1016/S1359-0278(98)00066-2. [DOI] [PubMed] [Google Scholar]
- Gough J, Karplus K, Hughey R, Chothia C. Assignment of homology to genome sequences using a library of hidden Markov models that represent all proteins of known structure. J Mol Biol. 2001;313:903–919. doi: 10.1006/jmbi.2001.5080. [DOI] [PubMed] [Google Scholar]
- Hegyi H, Gerstein M. Annotation transfer for genomics: Measuring functional divergence in multi-domain proteins. Genome Res. 2001;11:1632–1640. doi: 10.1101/gr.183801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann K, Bucher P, Falquet L, Bairoch A. The PROSITE database, its status in 1999. Nucleic Acid Res. 1999;27:215–219. doi: 10.1093/nar/27.1.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliopoulos, I., Tsoka, S., Andrade, M.A., Janssen, P., Audit, B., Tramontano, A., Valencia, A., Leroy, C., Sander, C., and Ouzounis, C.A. 2001. Genome sequences and great expectations. Genome Biol. 2. [DOI] [PMC free article] [PubMed]
- Koonin EV, Wolf YI, Aravind L. Protein fold recognition using sequence profiles and its application in structural genomics. Adv Prot Chem. 2000;54:245–275. doi: 10.1016/s0065-3233(00)54008-x. [DOI] [PubMed] [Google Scholar]
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Lupas A, van Dyke M, Stock J. Predicting coiled coils from protein sequences. Science. 1991;252:1162–1164. doi: 10.1126/science.252.5009.1162. [DOI] [PubMed] [Google Scholar]
- Mott R. Accurate formula for P-values of gapped local sequence and profile alignments. J Mol Biol. 2000;300:649–659. doi: 10.1006/jmbi.2000.3875. [DOI] [PubMed] [Google Scholar]
- Nakamura S, Tsuji Y, Nakata Y, Komori S, Koyama K. Identification and characterization of a sperm peptide antigen recognized by a monoclonal antisperm autoantibody derived from a vasectomized mouse. Biochem Biophys Res Commun. 1994;205:1503–1509. doi: 10.1006/bbrc.1994.2837. [DOI] [PubMed] [Google Scholar]
- Nielson H, Engelbrecht J, Brunak S, von Heijne G. Identification of prokaryotic and eukaryotic signal peptides and prediction of their cleavage sites. Prot Eng. 1997;10:1–6. doi: 10.1093/protein/10.1.1. [DOI] [PubMed] [Google Scholar]
- Park J, Karplus K, Barrett C, Hughey R, Haussler D, Hubbard T, Chothia C. Sequence comparisons using multiple sequences detect three times as many remote homologues as pairwise methods. J Mol Biol. 1998;284:1201–1210. doi: 10.1006/jmbi.1998.2221. [DOI] [PubMed] [Google Scholar]
- Qian J, Luscombe NM, Gerstein M. Protein family and fold occurrence in genomes: Power-law behaviour and evolutionary model. J Mol Biol. 2001a;313:673–681. doi: 10.1006/jmbi.2001.5079. [DOI] [PubMed] [Google Scholar]
- Qian J, Stenger B, Wilson CA, Lin J, Jansen R, Teichmann SA, Park J, Krebs WG, Yu H, Alexandrov V, et al. PartsList: A web-based system for dynamically ranking protein folds based on disparate attributes, including whole-genome expression and interaction information. Nucleic Acids Res. 2001b;29:1750–1764. doi: 10.1093/nar/29.8.1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reboul J, Vaglio P, Tzellas N, Thierry-Mieg N, Moore T, Jackson C, Shin-i T, Kohara Y, Thierry-Mieg D, Thierry-Mieg J, et al. Nat Genet. 2001;27:332–336. doi: 10.1038/85913. [DOI] [PubMed] [Google Scholar]
- Rost B. Twilight zone of protein sequence alignments. Prot Eng. 1999;12:85–94. doi: 10.1093/protein/12.2.85. [DOI] [PubMed] [Google Scholar]
- Sanchez R, Sali A. Large-scale protein structure modeling of the Saccharomyces cerevisiae genome. Proc Natl Acad Sci. 1998;95:13597–13602. doi: 10.1073/pnas.95.23.13597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaffer AA, Wolf YI, Ponting CP, Koonin EV, Aravind L, Altschul SF. IMPALA: Matching a protein sequence against a collection of PSI-BLAST- constructed position-specific score matrices. Bioinformatics. 1999;15:1000–1011. doi: 10.1093/bioinformatics/15.12.1000. [DOI] [PubMed] [Google Scholar]
- Skovgaard M, Jensen LJ, Brunak S, Ussery D, Krogh A. On the total number of genes and their length distribution in complete microbial genomes. Trends Genet. 2001;17:425–428. doi: 10.1016/s0168-9525(01)02372-1. [DOI] [PubMed] [Google Scholar]
- Sonnhammer ELL, von Heijne G, Krogh A. A hidden Markov model for predicting transmembrane helices in protein sequences. Proc Int Syst Mol Biol. 1998;6:175–182. [PubMed] [Google Scholar]
- Sreekumar KR, Aravind L, Koonin EV. Computational analysis of human disease-associated genes and their protein products. Curr Opin Genet Dev. 2001;11:247–257. doi: 10.1016/s0959-437x(00)00186-6. [DOI] [PubMed] [Google Scholar]
- Teichmann SA, Park J, Chothia C. Structural assignments to the mycoplasma genitalium proteins show extensive gene duplications and domain rearrangements. Proc Natl Acad Sci. 1998;95:14658–14663. doi: 10.1073/pnas.95.25.14658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd AE, Orengo CA, Thornton JM. Evolution of function in protein superfamilies, from a structural perspective. J Mol Biol. 2001;307:1113–1143. doi: 10.1006/jmbi.2001.4513. [DOI] [PubMed] [Google Scholar]
- Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, Yandell M, Evans CA, Holt RA, et al. The sequence of the human genome. Science. 2001;291:1304–1351. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- Vitkup D, Melamud E, Moult J, Sander C. Completeness in structural genomics. Nat Struct Biol. 2001;8:559–566. doi: 10.1038/88640. [DOI] [PubMed] [Google Scholar]
- Wilson CA, Kreychman J, Gerstein M. Assessing annotation transfer for genomics: Quantifying the relations between protein sequence, structure and function through traditional and probabilistic scores. J Mol Biol. 2000;297:233–249. doi: 10.1006/jmbi.2000.3550. [DOI] [PubMed] [Google Scholar]
- Wolf YI, Brenner SE, Bash PA, Koonin EV. Distribution of protein folds in the three superkingdoms of life. Genome Res. 1999;9:17–26. [PubMed] [Google Scholar]
- Wood VR, Gwilliam MA, Rajandream M, Lyne R, Lyne A, Stewart J, Sgouros N, Peat J, Hayles S, Baker D, et al. The genome sequence of Schizosaccharomyces pombe. Nature. 2002;415:871–880. doi: 10.1038/nature724. [DOI] [PubMed] [Google Scholar]
- Wootton JC, Federhen S. Analysis of compositionally biased regions in sequence databases. Methods Enzymol. 1996;266:554–571. doi: 10.1016/s0076-6879(96)66035-2. [DOI] [PubMed] [Google Scholar]