

Abstract
Identifying genomic regions that have been targets of natural selection remains one of the most important and challenging areas of research in genetics. To this end, we report an analysis of 26,530 single nucleotide polymorphisms (SNPs) with allele frequencies that were determined in three populations. Specifically, we calculated a measure of genetic differentiation, FST, for each locus and examined its distribution at the level of the genome, the chromosome, and individual genes. Through a variety of analyses, we have found statistically significant evidence supporting the hypothesis that selection has influenced extant patterns of human genetic variation. Importantly, by contrasting the FST of individual SNPs to the empirical genome-wide distribution of FST, our results are not confounded by tenuous assumptions of population demographic history. Furthermore, we have identified 174 candidate genes with distribution of genetic variation that indicates that they have been targets of selection. Our work provides a first generation natural selection map of the human genome and provides compelling evidence that selection has shaped extant patterns of human genomic variation.
[Supplemental material is available online at www.genome.org.]
Natural selection, which can be defined as the differential contribution of genetic variants to future generations (Aquadro et al. 2001), is the driving force of Darwinian evolution. Despite intense research, only a relatively small number of regions and genes have been directly implicated as targets of selection in the human genome (Kitano and Saitou 1999; Rana et al. 1999; Huttley et al. 2000; Hollox et al. 2001; Hull et al. 2001; Hurst and Pal 2001; Koda et al. 2001; Sullivan et al. 2001; Tishkoff et al. 2001; Baum et al. 2002; Fullerton et al. 2002; Gilad et al. 2002; Hamblin et al. 2002). A more comprehensive and genomic understanding of how and where natural selection has shaped patterns of genetic variation may provide important insights into the mechanisms of evolutionary change (Otto 2000), guide selection of loci for inclusion in population genetic studies (Vitalis et al. 2001), facilitate the annotation of functionally significant genomic regions (Nielsen 2001), and help elucidate genotype-phenotype correlations in complex diseases (Przeworski et al. 2000; Nielsen 2001).
Detecting unambiguous evidence for natural selection remains challenging because the effect of selection on the distribution of genetic variation can be mimicked by population demographic history (i.e., the size, structure, and mating pattern of a population). For instance, both adaptive hitchhiking and population expansion can cause an excess of rare variants observed in DNA sequence data compared with what is expected under a standard neutral model (Tajima 1989; Przeworski et al. 2000). Despite these difficulties, the recent deluge of publicly available single nucleotide polymorphisms (SNPs) provides an exciting opportunity to identify genome-wide signatures of selection (Sunyaev et al. 2000; Fay et al. 2001; Sachidanandam et al. 2001).
To this end, examining the variation in SNP allele frequencies between populations, which can be quantified by the statistic FST, is a promising strategy for detecting signatures of natural selection (Lewontin and Krakauer 1973; Rana et al. 1999; Hollox et al. 2001; Fullerton et al. 2002; Gilad et al. 2002; Hamblin et al. 2002). Under selective neutrality, FST is determined by genetic drift, which will affect all loci across the genome in a similar and predictable fashion. On the other hand, natural selection is a locus-specific force that can cause systematic deviations in FST values for a selected gene and nearby genetic markers. For example, geographically restricted directional selection may lead to an increase in FST of a selected locus, whereas balancing or species-wide directional selection may lead to a decrease in FST compared with neutrally evolving loci (Cavalli-Sforza 1966; Bowcock et al. 1991; Andolfatto 2001). Previous studies that have attempted to identify natural selection based on patterns of population differentiation relied on simulations to obtain the expected distribution of FST under selective neutrality (Lewontin and Krakauer 1973; Bowcock et al. 1991; Beaumont and Nichols 1996). However, the simulated distribution of FST strongly depends on the assumed population demographic history, which is rarely known with any degree of certainty.
As an expanding number of SNPs are genotyped across multiple populations, a complimentary approach that does not require tenuous assumptions about population demographic history is now becoming feasible. Specifically, by sampling a large number of SNPs throughout the genome, loci that have been affected by natural selection can simply be identified as outliers in the extreme tails of the empirical distribution of FST (Cavalli-Sforza 1966; Black et al. 2001; Goldstein and Chikhi 2002). Recently, this strategy has been used to infer natural selection in the CAPN10 gene; however, the empirical distribution of FST contained <100 loci (Fullerton et al. 2002).
In this work, we describe an analysis of 26,530 SNPs with allele frequencies that were determined in three populations: African-American, East Asian, and European-American. The density of this SNP allele frequency map provides a unique and powerful opportunity to interrogate the genome for signatures of natural selection. Through a variety of analyses, we have found statistically significant evidence supporting the hypothesis that selection has influenced extant patterns of human genetic variation. Furthermore, we have identified 174 candidate genes that demonstrate signatures of selection when contrasted to the empirical genome-wide distribution of FST. This analysis provides the conceptual foundation for constructing a high-resolution natural selection map, which will be an important resource in understanding the recent evolutionary history of our species, and will facilitate detailed studies on the identified candidate genes.
RESULTS
SNP Characteristics and Data Quality
In total, 26,530 SNPs were identified from The SNP Consortium (TSC) allele frequency project in which allele frequency data was available for three populations. The average intermarker distance, excluding the Y chromosome, was 132 kb. Because the SNP allele frequencies were determined by six genotyping labs that used different sample sizes and genotyping methods (see Methods), we performed several tests to assess data quality and identify sources of experimental variation.
First, we compared the distribution of common and uncommon SNPs with a previously reported estimate in the same three population samples (Table 1; Marth et al. 2001). The proportion of SNPs common (minor allele frequency <0.20) in 0, 1, 2, or 3 populations was not significantly different compared with the previous estimate based on 502 SNPs (χ32 = 2.02, P = 0.57), indicating no gross deviations of allele frequency in this expanded data set.
Table 1.
Allele Frequency Distribution of SNPs
| SNP | Current data | Previously reported data |
|---|---|---|
| Total number | 26,530 | 502 |
| Uncommon | 1,342 (5.1%) | 30 (6.0%) |
| Common ≥1 population | 21,101 (79.5%) | 385 (76.7%) |
| Common ≥2 populations | 15,029 (56.6%) | 263 (52.4%) |
| Common in all 3 populations | 7,908 (29.8%) | 135 (27.0%) |
A common SNP is defined in which the minor allele frequency is ≥20%. This frequency threshold was used so that the current data could be compared with previously reported estimates of allele frequency (Marth et al. 2001).
Second, we identified two sources of redundant allele frequency data: (1) 828 SNPs were genotyped by at least two of the six genotyping labs, and (2) the Sanger Centre genotyped 3145 SNPs in two independent European-American population samples of size 12 and 96 individuals. Although some markers demonstrated considerable variation, we observed a strong correlation in allele frequencies between the duplicated SNPs (ρ = 0.89 and 0.97 for the data sets containing 828 and 3145 duplicated SNPs, respectively).
Third, we investigated the effect of genotyping errors on estimates of FST in this data set. Specifically, we used a simple deterministic formula derived by Ohta and Kimura (1969) to describe the frequency of a SNP allele in the presence of genotyping errors, which we denote as P‘A (described in Methods; see also Akey et al. 2001). Next, we calculated P‘A for all 26,530 SNPs, assuming different genotyping error rates, and then reestimated FST using the new allele frequencies (i.e., P‘A). Obviously, the assumption that the original SNP allele frequencies are error free is incorrect, but our goal is to simply assess how an increasing error rate affects estimates of FST in this data set. On average, genotyping errors tend to decrease FST, and even modest error rates (2%–5%) can begin to have appreciable effects (Fig. 1). These observations are consistent with the effect of genotyping errors on estimates of LD (Akey et al. 2001). As a guide to interpret the impact of genotyping errors on this data set, the Whitehead Institute estimates a 0.5% genotyping error rate (see http://snp.cshl.org/). If this error rate is representative of the other laboratories, then genotyping errors have likely had a limited impact on our estimates of FST.
Figure 1.

The effect of genotyping errors on estimates of FST. The genotyping error rates μ and ν were assumed to be equal (see Methods for details).
Finally, we performed multiple regression analysis to estimate how much of the total variation in FST was attributable to variation in sample size and genotyping laboratory (which will reflect the use of slightly different samples and genotyping error rates across laboratories). Although highly significant, the variation in sample size and genotyping laboratory accounted for only 3.8% of the total variation in FST (F6,26523 = 45.7, P < 0.0001; adjusted R2 = 3.8%). Overall, the allele frequency data and estimates of FST across the six genotyping laboratories appears to be quite robust.
Empirical Genome-Wide Distribution of FST
To examine interlocus variation in allele frequencies, we constructed the empirical genome-wide distribution of FST for all autosomal markers (Fig. 2). The average FST for the 25,549 autosomal SNPs was 0.123, which lies within the range of previously reported estimates (Bowcock et al. 1991; Tishkoff et al. 2000). There is considerable variation around the mean, and a high proportion of markers are located in the tails of the distribution; ∼11% of SNPs have FST = 0.0, and 6% of SNPs have FST ≥ 0.40. To determine if the observed distribution of FST was consistent with selective neutrality, we performed coalescent simulations that assumed the only forces affecting variation in allele frequencies were genetic drift, mutation, and migration. Specifically, 25,549 SNPs were simulated in three constant-sized populations under an island model of migration, conditioning on the observed sample size, and average FST.
Figure 2.

Genome-wide distribution of FST. Solid bars show the observed distribution of FST for 25,549 autosomal SNPs. The X chromosome was not included in this analysis because it has a different effective population size compared with that of autosomal markers. Lightly shaded bars represent the simulated distribution of FST. The inset figure shows the observed and simulated distributions of FST for values ≥0.5.
The simulated distribution of FST was significantly different compared with the empirical distribution (Kolmogorov-Smirnov test, D = 0.058, P < 0.0001). In concordance with previous studies (Bowcock et al. 1991), we observed an excess of both high- and low-FST values (Fig. 2), which is consistent with the action of natural selection. For instance, adaptation to a local environmental pressure will cause a change in allele frequencies for the selected locus in a particular subpopulation and, hence, lead to a higher than expected level of population differentiation (FST). Anomalously high levels of population differentiation have been observed at several genes mediating local adaptation to traits such as disease resistance (Tishkoff et al. 2001; Hamblin et al. 2002), lactose intolerance (Hollox et al. 2001), skin pigmentation (Rana et al. 1999), and perhaps behavioral phenotypes (Gilad et al. 2002). Conversely, balancing selection maintains allelic variation between subpopulations and therefore leads to lower levels of population differentiation. Examples of balancing selection may include genes in the major histocompatibility complex (MHC) (Meyer and Thomson 2001) and β-globin region (Currat et al. 2002), FUT2 (Koda et al. 2001), and GYPA (Baum et al. 2002).
Alternatively, the deviation between the observed and simulated distribution of FST may not be owing to selection, but may merely reflect the highly simplified model of human demographic history that we used in the coalescent simulations (i.e., island model of migration, constant population size, etc.) to obtain the theoretical distribution of FST under neutrality. Therefore, the results based on this single analysis should be interpreted with caution. In the sections below, we present additional analyses to test this data set for signatures of selection that are not confounded by assumptions regarding human demographic history, because the only comparisons made are within the observed data itself and not in reference to simulations or analytical formulations.
Chromosomal Distribution of FST
The empirical genome-wide distribution of FST indicates that natural selection has operated on the human genome. To further test this hypothesis and identify specific genomic regions containing signatures of selection, we examined the distribution of FST across chromosomes (Fig. 3). The average FST for autosomal and X-linked SNPs was significantly different (0.123 and 0.195, respectively; t test, t = 14.1, P < 10−20). A higher average FST for X-chromosome SNPs is expected because of its smaller effective population size compared with that of the autosomes, which makes it more sensitive to demographic events and/or natural selection.
Figure 3.

Chromosomal distribution of FST. For each chromosome, chromosomal position in Mb is shown on the X-axis, and FST is plotted on the Y-axis. FST values for individual SNPs are shown in blue, and the average FST for nonoverlapping 1 Mb bins is plotted in yellow. The red horizontal lines in each panel provide a guide for identifying exceptionally high-FST values (corresponding to the upper 2.5% of the empirical distribution of FST; notice the higher threshold for the X chromosome). SNP density is proportional to the line spacing, and although the overall density is high, several large gaps were observed (e.g., see chromosomes 1, 9, 16, and 20). These gaps correspond to heterochromatic staining regions found near the centromeres of these chromosomes. The Y chromosome contained only seven SNP markers with allele frequency data and therefore was not included in subsequent analyses.
A striking feature that emerges when examining the distribution of FST across a chromosome is that FST values tend to cluster together (Fig. 3). In other words, estimates of FST for adjacent SNPs appear to be correlated. We formally tested this observation by calculating the correlation coefficient, ρ, between FST values as a function of physical distance between SNPs. A modest, yet statistically significant positive correlation between FST values of linked SNPs exists, which extends to ∼200 kb (Fig. 4). To assess whether this result is consistent with neutral expectations, we performed coalescent simulations. The simulated data shows a much weaker correlation compared with the observed data, which is nearly three times higher for closely linked markers (Fig. 4). Specifically, the relationship between FST values and physical distance in the observed data is significantly greater than that of the simulated data until 30 kb, at which point the two curves overlap and become statistically indistinguishable (except for the two points at 50 and 100 kb).
Figure 4.

Correlation between FST values as a function of physical distance. Intermarker distance was calculated between adjacent SNPs across the genome. Marker pairs were then separated into various bins (shown on the X-axis) according to their intermarker distance, and ρ was calculated for each bin. In the observed data, ρ was calculated for unlinked markers by comparing FST values on different chromosomes. Vertical bars represent 95% confidence intervals.
How can these results be explained? In the coalescent simulations, the relationship between FST values for linked markers is dictated solely by population demography and recombination. In the observed data, we propose that some additional evolutionary force is responsible for driving the correlation upward for closely linked loci. It may be that a more complex demographic model could lead to a higher predicted correlation. However, simulations incorporating population expansion and a range of migration rates indicate that alternative demographic histories do not account for the observed correlation between FST and physical distance (data not shown).
Moreover, in our simulations we assumed that recombination was uniformly distributed at a rate of 1 cM/Mb. However, several recent studies indicate that the distribution of recombination is highly punctuated and can vary substantially across genomic regions (Daly et al. 2001; Jeffreys et al. 2001). Thus, one may argue that the higher observed correlation simply reflects regions of recombination “deserts” (Yu et al. 2001). A close examination of Figure 4, however, argues against this hypothesis. Specifically, consider the observed and simulated correlations for an intermarker distance of 1 kb in Figure 4, which in practice corresponds to a recombination desert. Even under this condition of essentially zero recombination, the simulated correlation coefficient is ∼0.12, whereas the observed empirical correlation is ∼0.33. Therefore, in the absence of recombination, the correlation in FST values for the observed data is statistically different (higher) than what is expected based on neutrality. Thus, adaptive hitchhiking and/or background selection (Andolfatto 2001) provides the most parsimonious explanation for the increased correlation in FST between closely linked SNPs relative to a neutral model. Furthermore, the observed data indicate that the average unit of background selection and/or adaptive hitchhiking is ∼20 kb.
Distribution of FST in Genes
To further interrogate the genome for signatures of selection, we classified autosomal SNPs according to functional category (coding, intronic, and noncoding; see Methods) and then compared the average FST between groups (Table 2). As expected, the largest difference in average FST was observed between coding and noncoding SNPs, which is consistent with purifying selection. Furthermore, although small, the difference in average FST between intronic and noncoding SNPs is also significant, perhaps indicating some degree of functional constraint on intronic SNPs.
Table 2.
Average FST as a Function of SNP Category
| Category | No. | Average FST | SE | Significance of difference in average FSTa | |
|---|---|---|---|---|---|
| Coding | Intronic | ||||
| Coding | 238 | 0.107 | 0.008 | – | – |
| Intronic | 5,455 | 0.118 | 0.002 | 0.094 | – |
| Noncoding | 13,615 | 0.123 | 0.001 | 0.024 | 0.008 |
Empirical P values were determined by randomly permuting FST values between SNP categories 10,000 times and then counting the number of permutations with difference in average FST equal to or greater than the original difference.
Identification of Candidate Genes Subject to Selection
To identify candidate genes that have been subject to natural selection, we mapped 8862 SNPs to gene-associated regions. Using the empirical distribution of FST and the criteria described in the Methods, we identified 174 candidate selection genes: 156 that demonstrate unusually high levels of FST, and 18 that exhibit unusually low levels of FST (Supplemental Tables A, B, respectively). The 174 candidate selection genes encompass 253 SNPs, and include 17 genes underlying known mendelian (such as the CFTR gene, which is associated with cystic fibrosis, OMIM 219700) or complex diseases (such as the PPARG gene, which is associated with type 2 diabetes, OMIM 125853; see Supplemental Tables A, B).
To better understand the molecular functions that these genes perform, we examined their gene ontogeny (GO) classifications (Ashburner et al. 2000). The candidate selection genes participate in a broad range of molecular functions and biological processes (Tables 3, 4, respectively). Although sample sizes across the GO categories are too small to make meaningful statistical comparisons, several interesting trends emerge. For example, the proportion of proteins that perform a defense/immunity function (GO:0003793) is nearly four times higher in the low-FST candidate genes compared with the high-FST candidates (Table 3). This observation is consistent with balancing selection, a well-known force affecting genes involved in immunity (Richman 2000). Conversely, molecular functions that appear to be more predominant in high-FST candidate genes include enzymes (GO:0003824) and transporters (GO:0005215). These trends are also seen in the GO biological process terms (Table 4). Obviously, although interesting, these observations need to be explored and verified in replicate samples preferably of larger size.
Table 3.
Molecular Function of Candidate Selection Genes
| Gene ontogeny term | High FST | Low FST |
|---|---|---|
| Total number terms | 183 | 31 |
| Apoptosis regulator | 1 (0.5%) | 0 (0.0%) |
| Cell adhesion molecule | 4 (2.2%) | 0 (0.0%) |
| Cell growth and/or maintenance | 2 (1.1%) | 0 (0.0%) |
| Chaperone | 2 (1.1%) | 0 (0.0%) |
| Defense/immunity protein | 3 (1.6%) | 2 (6.5%) |
| Enzyme | 50 (27.3%) | 5 (16.1%) |
| Hydrolase | 11 (6.0%) | 3 (9.7%) |
| Kinase | 11 (6.0%) | 0 (0.0%) |
| Transferase | 12 (6.6%) | 1 (3.2%) |
| Enzyme regulator | 4 (2.2%) | 0 (0.0%) |
| Ligand binding or carrier | 57 (31.1%) | 9 (29.0%) |
| Calcium binding | 7 (3.8%) | 0 (0.0%) |
| Nucleic acid binding | 23 (12.6%) | 1 (3.2%) |
| Protein binding | 3 (1.6%) | 6 (19.4%) |
| Motor | 0 (0.0%) | 1 (3.2%) |
| Signal transducer | 27 (14.8%) | 10 (32.3%) |
| Ligand | 4 (2.2%) | 1 (3.2%) |
| Receptor | 14 (7.7%) | 7 (22.6%) |
| Structural molecule | 6 (3.3%) | 2 (6.5%) |
| Transcriptional regulator | 9 (4.9%) | 1 (3.2%) |
| Transporter | 18 (9.8%) | 1 (3.2%) |
In the Gene Oncology (GO) classification system, a parent term can have multiple subcategories, or children terms (indented text). For instance, hydrolase, kinase, and transferase are the children of the parent term enzyme. A single gene can have multiple parent and children terms (see Ashburner et al. 2000 for more specific information). Note that percentages sum to 100% for parent terms only.
Table 4.
Biological Processes of Candidate Selection Genes
| Gene ontogeny term | High FST | Low FST |
|---|---|---|
| Total number of terms | 123 | 39 |
| Behavior | 2 (1.6%) | 1 (2.6%) |
| Cell communication | 38 (30.9%) | 15 (38.5%) |
| Cell adhesion | 6 (4.9%) | 2 (5.1%) |
| Cell-cell signaling | 2 (1.6%) | 1 (2.6%) |
| Response to external stimulus | 7 (5.7%) | 3 (7.7%) |
| Immune response | 1 (0.8%) | 2 (5.1%) |
| Perception of external stimulus | 6 (4.9%) | 0 (0.0%) |
| Signal transduction | 21 (17.1%) | 7 (18.0%) |
| Cell growth and/or maintenance | 69 (56.1%) | 11 (28.2%) |
| Metabolism | 43 (35.0%) | 6 (15.4%) |
| Protein metabolism and modification | 15 (12.2%) | 0 (0.0%) |
| Transcription | 9 (7.3%) | 2 (5.1%) |
| Transport | 12 (9.8%) | 0 (0.0%) |
| Death | 1 (0.8%) | 2 (5.1%) |
| Developmental processes | 10 (8.1%) | 5 (12.8%) |
| Embryogenesis and morphogenesis | 3 (2.4%) | 5 (12.8%) |
| Epigenetic control of gene expression | 2 (1.6%) | 0 (0.0%) |
| Reproduction | 1 (0.8%) | 0 (0.0%) |
| Physiological processes | 3 (2.4%) | 5 (12.8%) |
| Pregnancy | 1 (0.8%) | 0 (0.0%) |
See notes to Table 3.
To better recognize the signatures that natural selection imparts on a locus and closely linked markers, we examined the distribution of FST in the candidate genes (which we refer to as FST profiles; examples are shown in Fig. 5). Strikingly different FST profiles are observed across the candidate selection genes. Specifically, several candidate genes contain contiguous SNPs with FST values that are consistently low (Fig. 5 A,B) or consistently high (Fig. 5 C,D), or a complex pattern of both (Fig. 5 E,F). For example, CMAH (Fig. 5 E) demonstrates statistically significant signatures of both high- and low-FST candidate genes, perhaps indicating that this locus has been subject to multiple types of selective pressures.
Figure 5.

FST profiles for six genes showing signatures of natural selection. For each gene, FST is plotted on the Y-axis, and chromosomal position in Kb is plotted on the X-axis. The genes shown here include guanine nucleotide exchange factor for Rap1 (GFR; (A)), tropomodulin 3 (TMOD3; (B)), apolipoprotein B (APOB; (C)), phosphoinositide-3-kinase, catalytic, β-polypeptide (PIK3CB; (D)), cytidine monophosphate-N-acetylneuraminic acid hydroxylase (CMAH; (E)), and oligophrenin 1 (OPHN1; (F)). The location of SNPs within each gene is denoted as boxes: introns (black), exons (open), 5′ UTR (grey), 5′ upstream (vertically striped), and 3′ downstream (hatched). Intron and exon numbers are noted within each box where appropriate.
Finally, it is notable that for many of the high-FST candidate selection genes, the population pair-wise FST values reveal that a high FST often results from one population showing a large difference in allele frequency relative to the other two (Supplemental Table A). For example, the SNP rs1806931 results in a Ser171Phe substitution in the gene OR10H2, which has an overall FST of 0.524 (Supplemental Table A). The pair-wise FST values are 0.523, 0.576, and 0.008, corresponding to East Asian/European American, East Asian/African American, and African American/European American comparisons, respectively. This pattern would be expected under adaptive evolution, in which in a unique environment, one particular allele is favored over the other (Bowcock et al. 1991), in this case during or after the settling of East Asia.
DISCUSSION
We have identified signatures of natural selection by compiling and analyzing a high-density SNP allele frequency map. The various analyses that we used to detect selection included both direct and indirect approaches (Fay and Wu 2001). Although direct approaches are often viewed as powerful evidence for selection, indirect approaches have been criticized because of their strong dependence on population demographic history (Nielsen 2001). Our indirect tests of selection include (1) comparing the observed and simulated distributions of FST and (2) comparing the observed and simulated correlation of FST values. In addition, because of the large number of SNP markers, we were also able to pursue direct tests of selection by (1) comparing the average FST between coding, intronic, and noncoding SNPs, and (2) identifying candidate selection genes based on the empirical distribution of FST. More specifically, by comparing the FST of individual loci to the empirical distribution, it was possible to distinguish between the consequences of genetic drift and natural selection without invoking any assumptions regarding population demography (Black et al. 2001; Hamblin et al. 2002). Therefore, when all of our analyses are collectively interpreted, a consistent signature of natural selection emerges.
Candidate Selection Genes
We have identified 174 genes with a pattern of FST that indicates that they have been subject to natural selection. Of the 174 candidate selection genes, 156 demonstrate unusually high levels of FST, and 18 exhibit unusually low levels of FST. Because of the large proportion of SNPs with a FST = 0.0, a more stringent threshold was applied to the selection of low-FST candidate genes. Therefore, it is important to note that in the present study, the discrepancy between the number of high- and low-FST candidate selection genes is a consequence of the different approaches used to identify them rather than some underlying evolutionary force. Additional studies will be required to establish the prevalence of different types of selection that have operated on the human genome. For example, when genotype frequencies are available, analytical methods based on FIS may be more sensitive to detect loci subject to balancing selection (Black et al. 2001).
Furthermore, to our knowledge, only two of the candidate selection genes have been implicated/confirmed in previous studies (CFTR [Slatkin and Bertorelle 2001] and F5 [Lindqvist et al. 1998]). Moreover, 18 candidate selection genes are themselves candidate genes that have been identified by computational predictions. Thus, more detailed and direct studies need to be performed in order to confirm the preliminary signatures of selection that we have identified in these 174 genes.
Limitations
In critically evaluating our results, it is important to note that our analyses, and hence interpretations, are subject to several limitations. First, many of our analyses rely on data derived from publicly available databases with contents that are, and will continue to be for some time, in a state of change. For example, in our comparison of the difference in average FST between SNPs located in coding, intronic, and noncoding regions, some coding and intronic SNPs lie within predicted genes and thus may not actually be coding or intronic SNPs. Therefore, our results represent a snapshot based on currently available data, and ultimately, when the human genome annotation becomes more stable, it will be important to verify these results.
Second, the SNP allele frequencies were determined in a relatively small sample size (see Methods), and stochastic variation could affect the robustness of our conclusions. Although we observed a strong correlation in allele frequencies between duplicated SNP markers (Supplemental Fig. A), confirming these allele frequency estimates in a larger sample size will be important.
Third, the power of our analyses is limited by several factors. For instance, we have searched for signatures of natural selection by analyzing the distribution of allele frequency differences between populations, which is most powerful with a geographically diverse set of samples. Because allele frequencies were available for only three populations (and the African Americans are an admixed population; Parra et al. 1998), we have likely only captured a fraction of the available evidence for natural selection. Furthermore, our study design is most powerful for detecting geographically restricted directional selection; although when migration between subpopulations is limited, it can identify species-wide selective pressures (Slatkin and Wiehe 1998; Majewski and Cohan 1999). Moreover, although we have compiled and analyzed the highest-density SNP allele frequency map constructed to date, even more markers, particularly in gene-associated regions, will be necessary to systematically identify targets of natural selection. For example, our list of candidate selection genes does not include Fy (Hamblin et al. 2002), which demonstrates one of the clearest known signatures of selection. The closest SNP (rs856042) in our data set to Fy is ∼80 kb upstream, which precluded our ability to detect a signal.
Finally, we have implicitly assumed no ascertainment bias (AB) of SNP markers, which has recently been demonstrated to affect estimates of several population genetic parameters such as the population mutation rate (Kuhner et al. 2000; Nielsen 2000), the population migration rate (Wakeley et al. 2001), and the population recombination rate (Nielsen 2000). One may hypothesize that because TSC SNPs were identified in a small number of chromosomes (Altschuler et al. 2000), FST will be underestimated. Specifically, the probability of discovering SNPs with a higher minor allele frequency is larger compared with SNPs with a lower minor allele frequency (Eberle and Kruglyak 2000). Thus, TSC SNPs may contain an over representation of common SNPs, which are expected to be shared across populations and therefore have smaller allele frequency differences. Preliminary simulations confirm this expectation (data not shown), and this issue merits further theoretical study. However, our empirical data shows an excess of both high- and low-FST values, which cannot be accounted for solely by AB.
Conclusions
In conclusion, our results provide a comprehensive assessment of how and where natural selection has shaped extant patterns of human genetic variation, and demonstrates the feasibility of constructing a high-density natural selection map of the human genome. Developing and ultimately integrating a selection map with other “genomic maps”—such as haplotype (Robertson 2001) and recombination maps (Yu et al. 2001)—will provide important insights into human evolution, genome function, and the mechanisms of evolutionary change.
METHODS
Data mining and Processing
We downloaded the SNP allele frequency data (genotypes were not available) from the TSC Web site (http://snp.cshl.org/allele_frequency_project/), which was generated by six genotyping labs: Sanger Centre, Orchid, Washington University, Celera, Whitehead Institute, and Motorola. The allele frequencies across these laboratories were based on a common set of DNA samples. Specifically, the allele frequency panels consist of 42 East Asian, 42 African-American, and 42 European-American individuals. The average sample size (number of individuals) across populations used to estimate allele frequencies varied between these six laboratories: Sanger Centre (n = 12), Washington University (n = 42), Celera (n = 30), Orchid (n = 41), Whitehead Institute (n = 53), and Motorola (n = 29). Note that in the Sanger data set, sample sizes for each locus were not available, so we assumed a fixed sample size of 12. Therefore, the different genotyping laboratories are using either identical or overlapping sets of identical individuals (see below). The only exception to this is that the Whitehead Institute is not using the same set of European-American samples. However, we expect that the additional variation introduced by a different set of European-American samples used by the Whitehead Institute is mitigated because (1) they are still using the same East Asian and African-American samples, and (2) they contributed only 1077 SNPs, or 4% of the total number of SNPs.
The full data set contained 63,658 SNPs. We removed markers that were not genotyped in all three populations, leaving a total of 26,530 SNPs. PERL scripts were written to retrieve dbSNP (rs# and ss#), and TSC identifiers when necessary, to map SNPs to approximate chromosomal coordinates (both National Center for Biotechnology Information [NCBI] and TSC coordinates) and to assign functional categories to SNPs (coding, noncoding, or intronic based on the Ensembl Human Genome annotation release 5.28; http://www.ensembl.org/). In addition, we also downloaded a second SNP allele frequency data set (http://snp.cshl.org/allele_frequency_project/) from the Sanger Center in which the allele frequencies were estimated in an additional sample of European individuals of size 96. SNPs that overlapped with original set of 26,530 were identified and used to assess data quality.
Assessing Data Quality
SNP markers that had been genotyped by more than one group were identified, and we retained the data that had the larger sample size for subsequent analyses. In addition, we also downloaded a second SNP allele frequency data set (http://snp.cshl.org/) from the Sanger Center in which allele frequencies were estimated in an additional sample of European individuals of size 96. SNPs that overlapped with original set of 26,530 were identified and used to assess data quality. Specifically, the correlation coefficient, ρ, was calculated for allele frequencies between duplicated SNP markers.
The potential impact of genotyping errors on FST was studied by simplified methods similar to Akey et al. 2001. If we denote the alleles at a SNP locus as A and a (and their frequencies in the absence of genotyping errors as PA and Pa, respectively), and assume that genotyping errors follow a model in which the genotyping error rate of A→a is μ and of a→A is ν, then the estimated frequency of A in the presence of genotyping errors, denoted as P‘A, is as follows: P‘A = PA + ν − [(μ + ν) PA]. This formula is identical to Equation 1 in Ohta and Kimura (1969), who derived it to describe the change of allele frequency owing to mutation. To gain a better appreciation of how genotyping errors affect estimates of FST, we calculated P‘A for all 26,530 SNPs, assuming different error rates (ν = μ = 0, 0.005, 0.01, 0.02,…, 0.05) and then reestimated FST using the new estimates of allele frequencies in the presence of genotyping errors (P‘A).
To explore potential sources of variation based on the differences in experimental design, standard multiple linear regression (conducted with SPSS, version 9.0) was performed in which the dependent variable was square root transformed FST values, and the independent variables were the genotyping laboratory and average sample size/SNP.
Estimates of FST and Other Genetic Distances
We calculated unbiased estimates of FST as described by Weir and Cockerham 1984 (see also Weir 1996). Specifically, consider i subpopulations (where i = 1,…, s), and denote the frequency of the SNP allele A in the ith subpopulation as pAi. Then FST can be estimated as follows:
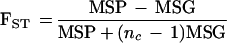 |
where, MSG denotes the observed mean square errors for loci within populations,
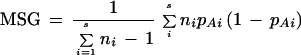 |
and MSP denotes the observed mean square errors for between populations,
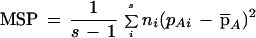 |
In the above formulae, ni denotes the sample size in subpopulation i, 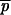 = nipAi/Σini (a weighted average of PA across subpopulations), and nc is the average sample size across samples that also incorporates and corrects for the variance in sample size over subpopulations (Weir 1996):
= nipAi/Σini (a weighted average of PA across subpopulations), and nc is the average sample size across samples that also incorporates and corrects for the variance in sample size over subpopulations (Weir 1996):
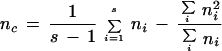 |
As originally defined (Wright 1951), the range of FST is between 0 and 1. However, it is possible for the above unbiased estimate of FST to assume negative values, which does not have a biological interpretation. Therefore, as indicated with other estimates of genetic distance, we set negative values of FST = 0.0 (Nei 1990). Other genetic distance measures were also calculated, including Nei’s minimum distance (Nei 1990), the allele frequency difference (Nei 1990), and genetic identity (Nei 1990). Because all of the distance measures were highly correlated (data not shown), we have only presented results based on FST.
Coalescent Simulations
We used coalescent theory (Fu and Li 1999) to obtain the genome-wide distribution of FST under a selectively neutral model. We simulated 25,549 SNPs from three subpopulations connected by migration using the program SIMCOAL (http://cmpg.unibe.ch/software/simcoal/; Excoffier et al. 2000). The simulated SNPs matched the characteristics of the observed data in terms of sample size, average FST, and center-specific average FST (i.e., the average FST for SNPs from each genotyping lab). In addition, we also performed coalescent simulations to study the relationship between the correlation coefficient of FST values and physical distance, assuming selective neutrality and 1 cM = 1 Mb. For this analysis, we used coalescent software available from Richard Hudson (http://home.uchicago.edu/∼rhudson1/source.html; see program mksamples) that simulates genealogies with recombination. For each intermarker distance in Figure 4, we simulated 50,000 SNP pairs (except for distances >200 Kb, in which 10,000 SNP pairs were simulated owing to computational constraints) and calculated the correlation coefficient for the resulting square root transformed FST values. Nonparametric Spearman rank correlations were also calculated and were nearly identical (average difference = 0.01) to the parametric correlation coefficient.
Identification of Candidate Selection Genes
We mapped all 26,530 SNPs to gene-associated regions by searching the Locus Link (http://www.ncbi.nlm.nih.gov/LocusLink/) and Ensembl databases (http://www.ensembl.org/). A SNP was considered located in a gene region if it mapped to either a 5′ upstream, 5′ UTR, coding, intronic, 3′ UTR, or 3′ downstream region. There were some discrepancies between Locus Link and Ensembl, as the latter included a larger 5′ upstream and 3′ downstream region. To minimize false positives, we took a conservative approach and only considered SNPs extending 5 kb into the upstream and downstream regions (see Fig. 4). After mapping SNPs to gene regions, we identified high- and low-FST candidate selection genes. For autosomal loci, a gene was considered a high-FST candidate selection gene if it contained at least one SNP with an FST ≥ 0.45. Based on the genome-wide distribution of FST, this corresponds to an empirical significance level of α = 0.026. To identify high-FST candidate selection genes on the X chromosome, we used a higher threshold (to compensate for the higher average FST compared with autosomal SNPs) of FST ≥ 0.59 (which corresponds to α = 0.0078 using the autosomal genome-wide distribution of FST, and α = 0.05 based on the empirical distribution of FST on the X chromosome). To identify low-FST candidate selection genes, an alternative approach was taken owing to the high proportion of FST values = 0 (11%). A gene was selected as a low-FST candidate selection gene if it contained two SNPs with an FST = 0 and one SNP with an FST ≤ 0.005. This threshold corresponds to a significance level of α = 0.03, as determined by coalescent simulations. Thus, the overall significance level for the identification of autosomal candidate selection genes was α = 0.056, which, although slightly anticonservative, is justified given the exploratory nature of this study.
Functional Characterization of Candidate Selection Genes
To characterize the molecular functions that the candidate selection genes perform, we retrieved the Swiss Protein accession number for each gene (http://www.expasy.ch/sprot/sprot-top.html; 39 genes did not have corresponding Swiss Protein identifications). The GO database was then queried by using QUICKGO (http://www2.ebi.ac.uk/ego/QuickGO), which accepts as input Swiss Protein accession numbers. For the 39 candidate genes that did not have Swiss Protein accession numbers, we scanned the protein with InterProScan (Zdobnov and Apweiler 2001; http://www.ebi.ac.uk/interpro/scan.html). The identified InterPro motifs were then used to query QICKGO. Genes that could not be assigned either Swiss Protein accession numbers or InterPro motifs were classified as “unknown” and are not included in Tables 3 or 4.
Data Availability
The entire data set, results, and supplementary information is available at http://cgi.uc.edu/∼jakey.
WEB SITE REFERENCES
http://snp.cshl.org/allele_frequency_project/; The SNP Consortium Home AFP home page.
http://www.ensembl.org/; Ensembl home page.
http://cmpg.unibe.ch/software/simcoal/; SIMCOAL software.
http://home.uchicago.edu/∼rhudson1/source.html; coalescent with recombination, see program mksamples.
http://www.ncbi.nlm.nih.gov/LocusLink/; Locus Link home page.
http://www2.ebi.ac.uk/ego/QuickGO; QuickGO home page.
http://www.ebi.ac.uk/interpro/scan.html; InterProScan home page.
http://cgi.uc.edu/∼jakey; summarized and raw SNP data presented in this article.
Acknowledgments
We thank Bing Su and Dayna Akey for critical reading of the manuscript and Ken Weiss, Ranajit Chakraborty, and Esteban Parra for productive discussions in the early phases of this project. This work was supported in part by grants from the NIH/NHGRI (HG002154) to M.D.S.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL mds17@psu.edu; FAX (814) 863-1474.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.631202.
REFERENCES
- Akey JM, Zhang K, Xiong M, Doris P, Jin L. The effect that genotyping errors have on the robustness of common linkage-disequilibrium measures. Am J Hum Genet. 2001;68:1447–1456. doi: 10.1086/320607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altshuler D, Pollar VJ, Cowles CR, Van Etten WJ, Baldwin J, Linton L, Lander ES. A SNP map of the human genome generated by reduced representation shotgun sequencing. Nature. 2000;407:513–516. doi: 10.1038/35035083. [DOI] [PubMed] [Google Scholar]
- Andolfatto P. Adaptive hitchhiking effects on genome variability. Curr Opin Genet Dev. 2001;11:635–641. doi: 10.1016/s0959-437x(00)00246-x. [DOI] [PubMed] [Google Scholar]
- Aquadro CF, Bauer DuMont V, Reed FA. Genome-wide variation in the human and fruitfly: A comparison. Curr Opin Genet Dev. 2001;1:627–634. doi: 10.1016/s0959-437x(00)00245-8. [DOI] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene ontology: Tool for the unification of biology: The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baum J, Ward RH, Conway DJ. Natural selection on the erythrocyte surface. Mol Biol Evol. 2002;19:223–229. doi: 10.1093/oxfordjournals.molbev.a004075. [DOI] [PubMed] [Google Scholar]
- Beaumont M, Nichols RA. Evaluating loci for use in the genetic analysis of population structure. Proc R Soc Lond B Biol Sci. 1996;263:1619–1626. [Google Scholar]
- Black WC, IV, Baer CF, Antolin MF, DuTeau NM. Population genomics: Genome-wide sampling of insect populations. Annu Rev Entomol. 2001;46:441–469. doi: 10.1146/annurev.ento.46.1.441. [DOI] [PubMed] [Google Scholar]
- Bowcock AM, Kidd JR, Mountain JL, Hebert JM, Carotenuto L, Kidd KK, Cavalli-Sforza LL. Drift, admixture, and selection in human evolution: A study with DNA polymorphisms. Proc Natl Acad Sci. 1991;88:839–843. doi: 10.1073/pnas.88.3.839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalli-Sforza LL. Population structure and human evolution. Proc R Soc Lond B Biol Sci. 1966;164:362–379. doi: 10.1098/rspb.1966.0038. [DOI] [PubMed] [Google Scholar]
- Currat M, Trabuchet G, Rees D, Perrin P, Harding RM, Clegg JB, Langaney A, Excoffier L. Molecular analysis of the β-globin gene cluster in the Niokholo Mandenka population reveals a recent origin of the β(S) Senegal mutation. Am J Hum Genet. 2002;70:207–223. doi: 10.1086/338304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daly MJ, Rioux JD, Schaffner SF, Hudson TJ, Lander ES. High-resolution haplotype structure in the human genome. Nat Genet. 2001;29:229–232. doi: 10.1038/ng1001-229. [DOI] [PubMed] [Google Scholar]
- Eberle MA, Kruglyak L. An analysis of strategies for discovery of single-nucleotide polymorphisms. Genet Epidemiol. 2000;19:S29–S35. doi: 10.1002/1098-2272(2000)19:1+<::AID-GEPI5>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Excoffier L, Novembre J, Schneider S. SIMCOAL: A general coalescent program for the simulation of molecular data in interconnected populations with arbitrary demography. J Hered. 2000;91:506–509. doi: 10.1093/jhered/91.6.506. [DOI] [PubMed] [Google Scholar]
- Fay JC, Wu CI. The neutral theory in the genomic era. Curr Opin Genet Dev. 2001;11:642–646. doi: 10.1016/s0959-437x(00)00247-1. [DOI] [PubMed] [Google Scholar]
- Fay JC, Wyckoff GJ, Wu CI. Positive and negative selection on the human genome. Genetics. 2001;158:1227–1234. doi: 10.1093/genetics/158.3.1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu YX, Li WH. Coalescing into the 21st century: An overview and prospects of coalescent theory. Theor Popul Biol. 1999;56:1–10. doi: 10.1006/tpbi.1999.1421. [DOI] [PubMed] [Google Scholar]
- Fullerton SM, Bartoszewicz A, Ybazeta G, Horikawa Y, Bell GI, Kidd KK, Cox NJ, Hudson RR, Di Rienzo A. Geographic and haplotype structure of candidate type 2 diabetes susceptibility variants at the calpain-10 locus. Am J Hum Genet. 2002;70:1096–1106. doi: 10.1086/339930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilad Y, Rosenberg S, Przeworski M, Lancet D, Skorecki K. Evidence for positive selection and population structure at the human MAO-A gene. Proc Natl Acad Sci. 2002;99:862–867. doi: 10.1073/pnas.022614799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein DB, Chikhi L. Human migrations and population structure: What we know and why it matters. Annu Rev Genomics Hum Genet. 2002;3:129–152. doi: 10.1146/annurev.genom.3.022502.103200. [DOI] [PubMed] [Google Scholar]
- Hamblin MT, Thompson EE, Di Rienzo A. Complex signatures of natural selection at the Duffy blood group locus. Am J Hum Genet. 2002;70:369–383. doi: 10.1086/338628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollox EJ, Poulter M, Zvarik M, Ferak V, Krause A, Jenkins T, Saha N, Kozlov AI, Swallow DM. Lactase haplotype diversity in the Old World. Am J Hum Genet. 2001;68:160–172. doi: 10.1086/316924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hull J, Ackerman H, Isles K, Usen S, Pinder M, Thomson A, Kwiatkowski D. Unusual haplotypic structure of IL8, a susceptibility locus for a common respiratory virus. Am J Hum Genet. 2001;69:413–419. doi: 10.1086/321291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurst LD, Pal C. Evidence for purifying selection acting on silent sites in BRCA1. Trends Genet. 2001;17:62–65. doi: 10.1016/s0168-9525(00)02173-9. [DOI] [PubMed] [Google Scholar]
- Huttley GA, Easteal S, Southey MC, Tesoriero A, Giles GG, McCredie MR, Hopper JL, Venter DJ. Adaptive evolution of the tumour suppressor BRCA1 in humans and chimpanzees: Australian Breast Cancer Family Study. Nat Genet. 2000;25:410–413. doi: 10.1038/78092. [DOI] [PubMed] [Google Scholar]
- Jeffreys AJ, Kauppi L, Neumann R. Intensely punctate meiotic recombination in the class II region of the major histocompatibility complex. NatGenet. 2001;29:217–222. doi: 10.1038/ng1001-217. [DOI] [PubMed] [Google Scholar]
- Kitano T, Saitou N. Evolution of Rh blood group genes have experienced gene conversions and positive selection. J Mol Evol. 1999;49:615–626. doi: 10.1007/pl00006583. [DOI] [PubMed] [Google Scholar]
- Koda Y, Tachida H, Pang H, Liu Y, Soejima M, Ghaderi AA, Takenaka O, Kimura H. Contrasting patterns of polymorphisms at the ABO-secretor gene (FUT2) and plasma α(1,3)fucosyltransferase gene (FUT6) in human populations. Genetics. 2001;158:747–756. doi: 10.1093/genetics/158.2.747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhner MK, Beerli P, Yamato J, Felsenstein J. Usefulness of single nucleotide polymorphism data for estimating population parameters. Genetics. 2000;156:439–447. doi: 10.1093/genetics/156.1.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewontin RC, Krakauer J. Distribution of gene frequency as a test of the theory of the selective neutrality of polymorphisms. Genetics. 1973;74:175–195. doi: 10.1093/genetics/74.1.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindqvist PG, Svensson PJ, Dahlback B, Marsal K. Factor V Q506 mutation (activated protein C resistance) associated with reduced intrapartum blood loss: A possible evolutionary selection mechanism. Thromb Haemost. 1998;79:69–73. [PubMed] [Google Scholar]
- Majewski J, Cohan FM. Adapt globally, act locally: The effect of selective sweeps on bacterial sequence diversity. Genetics. 1999;152:1459–1474. doi: 10.1093/genetics/152.4.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marth G, Yeh R, Minton M, Donaldson R, Li Q, Duan S, Davenport R, Miller RD, Kwok PY. Single-nucleotide polymorphisms in the public domain: How useful are they? Nat Genet. 2001;27:371–372. doi: 10.1038/86864. [DOI] [PubMed] [Google Scholar]
- Meyer D, Thomson G. How selection shapes variation of the human major histocompatibility complex: A review. Ann Hum Genet. 2001;65:1–26. doi: 10.1046/j.1469-1809.2001.6510001.x. [DOI] [PubMed] [Google Scholar]
- Nei M. Molecular evolutionary genetics. New York, NY: Columbia University Press; 1990. [Google Scholar]
- Nielsen R. Estimation of population parameters and recombination rates from single nucleotide polymorphisms. Genetics. 2000;154:931–942. doi: 10.1093/genetics/154.2.931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ————— Statistical tests of selective neutrality in the age of genomics. Heredity. 2001;86:641–647. doi: 10.1046/j.1365-2540.2001.00895.x. [DOI] [PubMed] [Google Scholar]
- Ohta T, Kimura M. Linkage disequilibrium at steady state determined by random genetic drift and recurrent mutation. Genetics. 1969;69:229–238. doi: 10.1093/genetics/63.1.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto SP. Detecting the form of selection from DNA sequence data. Trends Genet. 2000;16:526–529. doi: 10.1016/s0168-9525(00)02141-7. [DOI] [PubMed] [Google Scholar]
- Parra EJ, Marcini A, Akey J, Martinson J, Batzer MA, Cooper R, Forrester T, Allison DB, Deka R, Ferrell RE, et al. Estimating African-American admixture proportions by use of population-specific alleles. Am J Hum Genet. 1998;63:1839–1851. doi: 10.1086/302148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przeworski M, Hudson RR, Di Rienzo A. Adjusting the focus on human variation. Trends Genet. 2000;16:296–302. doi: 10.1016/s0168-9525(00)02030-8. [DOI] [PubMed] [Google Scholar]
- Rana BK, Hewett-Emmett D, Jin L, Chang BH, Sambuughin N, Lin M, Watkins S, Bamshad M, Jorde LB, Ramsay M, et al. High polymorphism at the human melanocortin 1 receptor locus. Genetics. 1999;151:1547–1457. doi: 10.1093/genetics/151.4.1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richman A. Evolution of balanced genetic polymorphism. Mol Ecol. 2000;9:1953–1963. doi: 10.1046/j.1365-294x.2000.01125.x. [DOI] [PubMed] [Google Scholar]
- Robertson D. Racially defined haplotype project debated. Nat Biotechnol. 2001;19:795–796. doi: 10.1038/nbt0901-795b. [DOI] [PubMed] [Google Scholar]
- Sachidanandam R, Weissman D, Schmidt SC, Kakol JM, Stein LD, Marth G, Sherry S, Mullikin JC, Mortimore BJ, Willey DL, et al. A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature. 2001;409:928–933. doi: 10.1038/35057149. [DOI] [PubMed] [Google Scholar]
- Slatkin M, Bertorelle G. The use of intraallelic variability for testing neutrality and estimating population growth rate. Genetics. 2001;158:865–874. doi: 10.1093/genetics/158.2.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slatkin M, Wiehe T. Genetic hitch-hiking in a subdivided population. Genet Res. 1998;71:155–160. doi: 10.1017/s001667239800319x. [DOI] [PubMed] [Google Scholar]
- Sullivan AD, Wigginton J, Kirschner D. The coreceptor mutation CCR5Δ32 influences the dynamics of HIV epidemics and is selected for by HIV. Proc Natl Acad Sci. 2001;98:10214–10219. doi: 10.1073/pnas.181325198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunyaev SR, Lathe WC, III, Ramensky VE, Bork P. SNP frequencies in human genes an excess of rare alleles and differing modes of selection. Trends Genet. 2000;16:335–337. doi: 10.1016/s0168-9525(00)02058-8. [DOI] [PubMed] [Google Scholar]
- Tajima F. The effect of change in population size on DNA polymorphism. Genetics. 1989;123:597–601. doi: 10.1093/genetics/123.3.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tishkoff SA, Pakstis AJ, Stoneking M, Kidd JR, Destro-Bisol G, Sanjantila A, Lu RB, Deinard AS, Sirugo G, Jenkins T, et al. Short tandem-repeat polymorphism/alu haplotype variation at the PLAT locus: Implications for modern human origins. Am J Hum Genet. 2000;67:901–925. doi: 10.1086/303068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tishkoff SA, Varkonyi R, Cahinhinan N, Abbes S, Argyropoulos G, Destro-Bisol G, Drousiotou A, Dangerfield B, Lefranc G, Loiselet J, et al. Haplotype diversity and linkage disequilibrium at human G6PD: Recent origin of alleles that confer malarial resistance. Science. 2001;293:455–462. doi: 10.1126/science.1061573. [DOI] [PubMed] [Google Scholar]
- Vitalis R, Dawson K, Boursot P. Interpretation of variation across marker loci as evidence of selection. Genetics. 2001;158:1811–1823. doi: 10.1093/genetics/158.4.1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakeley J, Nielsen R, Liu-Cordero SN, Ardlie K. The discovery of single-nucleotide polymorphisms and inferences about human demographic history. Am J Hum Genet. 2001;69:1332–1347. doi: 10.1086/324521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir BS. Genetic data analysis II. Sunderland, MA: Sinauer Associates; 1996. Population substructure; pp. 161–173. [Google Scholar]
- Weir BS, Cockerham CC. Estimating F-statistics for the analysis of population structure. Evolution. 1984;38:1358–1370. doi: 10.1111/j.1558-5646.1984.tb05657.x. [DOI] [PubMed] [Google Scholar]
- Wright S. The genetical structure of populations. Ann Eugen. 1951;15:323–354. doi: 10.1111/j.1469-1809.1949.tb02451.x. [DOI] [PubMed] [Google Scholar]
- Yu A, Zhao C, Fan Y, Jang W, Mungall AJ, Deloukas P, Olsen A, Doggett NA, Ghebranious N, Broman KW, et al. Comparison of human genetic and sequence-based physical maps. Nature. 2001;409:951–953. doi: 10.1038/35057185. [DOI] [PubMed] [Google Scholar]
- Zdobnov EM, Apweiler R. InterProScan: An integration platform for the signature-recognition methods in InterPro. Bioinformatics. 2001;17:847–848. doi: 10.1093/bioinformatics/17.9.847. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The entire data set, results, and supplementary information is available at http://cgi.uc.edu/∼jakey.