

Abstract
Ferryl (Fe(IV)=O) species are involved in key enzymatic processes with direct biomedical relevance; among others, the uncontrolled reactivities of ferryl Mb (myoglobin) and Hb (haemoglobin) have been reported to be central to the pathology of rhabdomyolysis and subarachnoid haemorrhage. Rapid-scan stopped-flow methods have been used to monitor the spectra of the ferryl species in Mb and Hb as a function of pH. The ferryl forms of both proteins display an optical transition with pK∼4.7, and this is assigned to protonation of the ferryl species itself. We also demonstrate for the first time a direct correlation between Hb/Mb ferryl reactivity and ferryl protonation status, simultaneously informing on chemical mechanism and toxicity and with broader biochemical implications.
Keywords: Compound II, ferryl, globin, haemoglobin, myoglobin, peroxidase
Abbreviations: 13-HPODE, (13S)-hydroperoxyoctadeca-(9Z,11E)-dienoic acid; CCP, cytochrome c peroxidase; CPO, chloroperoxidase; DFT, density functional theory; Hb, haemoglobin; HRP, horseradish peroxidase; Mb, myoglobin
INTRODUCTION
Ferryl species, formally [Fe4+=O2−]2+, are formed in haem and non-haem iron proteins on treatment with peroxides or related dioxygen species as part of their respective catalytic cycles/functions and/or as significant side reactions under physiological and pathophysiological conditions [1–9]. Such species have long been known to contain an oxo atom bound to Fe(IV), as supported by Mössbauer, resonance Raman, NMR, ENDOR (electron nuclear double resonance) and EXAFS spectroscopies and by X-ray crystallography (as recently reviewed in [1,2,10–13]). In haem proteins, the ferryl species is also referred to as ‘Compound II’; the related ‘Compound I’ species contain an additional oxidizing equivalent located on the porphyrin ring or an adjacent amino acid residue.
Recent experimental and theoretical results have suggested that at least some ferryl species may in fact be protonated, i.e. Fe(IV)–OH rather than Fe(IV)=O. A hydroxo ligand has been implied for the histidine-ligated ferryl group in Mb (myoglobin) Compound II [14] at acidic pH and in CCP (cytochrome c peroxidase) Compound I, based on ∼1.9 Å (1 Å =0.1 nm) iron–oxygen distances determined by atomic-resolution X-ray-diffraction crystallography [15] (CCP Compound I is an atypical Compound I in that the additional oxidizing equivalent is localized on a protein side chain rather than on the porphyrin moiety; in this respect, CCP Compound I is more akin to typical Compound II species). Hayashi and Yamazaki [16] have also shown that Compound II formation in peroxidases is accompanied by uptake of a proton (although they could not demonstrate the exact location of this proton). Green and co-workers have more recently challenged the crystallographic findings on Mb and CCP using EXAFS and resonance Raman spectroscopies [13,17–21]. Indeed, Green and co-workers have reported that the Fe–O bond in the quintessential peroxidase HRP (horseradish peroxidase) Compound II is ∼1.65 Å, consistent with a non-protonated ferryl group [17]. They have further shown that the Fe–O stretching frequency, as measured by resonance Raman spectroscopy, does not significantly change position in the range of pH from 4.5 to neutral and basic values for Mb Compound II [19,21]. On the other hand, the same spectroscopic tools, together with Mössbauer spectroscopy and DFT (density functional theory) calculations, have been used by Green and co-workers to establish that chloroperoxidase Compound II and cytochrome P450 Compound II are indeed protonated [13,19,22]. CPO (chloroperoxidase) and cytochrome P450 feature an endogenous anionic thiolate ligand to the haem group, instead of the neutral imidazole ligand found in HRP (though some researchers have interpreted NMR and resonance Raman data for the latter as evidence for imidazolate character [2,23]) or in Mb. Precisely this difference in axial ligation has been proposed to be the reason why HRP Compound II would, unlike CPO Compound II, be unprotonated [17]. A Mössbauer-spectroscopy report, complemented by DFT calculations, was also interpreted as evidence that catalase Compound II is at least partially protonated; catalase, like CPO and cytochrome P450, features an anionic ligand to the haem moiety, namely a tyrosinate group. [24] The accumulated body of evidence from spectroscopic techniques thus currently favours the view that ferryl protonation does not occur within physiologically relevant pH ranges with histidine-ligated haemoproteins, whereas the ferryl group is at least partially protonated in haemoproteins ligated by thiolate or tyrosinate. Nevertheless, as will be shown below, we believe that the entire body of spectroscopic evidence on histidine-ligated haemoprotein Compound II species may also bear an alternative interpretation.
Mb and Hb (haemoglobin) have long been known to form ferryl (Compound II) species upon treatment of the Fe(III) or Fe(II) state with peroxide [25–27]. Ferryl Mb and Hb are relatively stable at neutral and basic pH, but less so at acidic pH values [28]. This instability at acidic pH parallels an increased reactivity towards reducing substrates, and it has been proposed on several occasions that protonation of the ferryl group could be responsible for these trends in reactivity [7,8,28,29]. A recent crystal structure of Mb ferryl group at acidic pH has indeed shown a long Fe–O bond (1.9 Å), consistent with an OH ligand rather than the commonly accepted Fe=O description [14]. Although subsequent spectroscopic work has led to the conclusion that the crystal structure of Mb ferryl group must be in error and that Mb ferryl group is not protonated [17], the reason for the pH-dependence of ferryl Mb and ferryl Hb reactivity is still important, as this species has previously been detected in vivo and is known to be implicated in pathophysiological states involving acute oxidative stress, with this involvement being most manifest in compartments with lowered pH such as kidney and cerebrospinal fluid [7,8] (although, notably, even normal blood contains detectable amounts of by-products derived from ferryl Hb [9]). Below we present results that demonstrate that the optical absorption spectrum of ferryl haem is distinctly pH-dependent. This optical transition is rapid (submillisecond) and, in our view, is best interpreted as direct protonation of the ferryl haem species.
EXPERIMENTAL
Horse heart Mb (Sigma) and human Hb were used as previously described [7,8,28–30]. Ferryl Mb and ferryl Hb were prepared as previously described [28]; briefly, a 50 μM solution of metMb in 20 mM phosphate buffer, pH 8, was mixed with a 1.5-fold excess of H2O2 at room temperature; at this pH value, the only observable product is Compound II (as opposed to Compound I) [28]. Ferryl formation was monitored in the Soret region, as well as in the visible region, as previously described [28]; within ∼20 min, the UV–visible spectrum converted into ferryl (Compound II) and remained unchanged throughout the time required for subsequent stopped-flow measurements. Stopped-flow measurements were performed using an Applied Photophysics (Leatherhead, Surrey, U.K.) model SX-18MV instrument. The buffers used for pH-jump experiments were 200 mM phosphate (pH 2, 3, 7 and 8) and acetate (pH 4, 5 and 6); alternatively, for acidic pH values, citrate buffers were also used. Diode-array-collected data were analysed within the Pro-K II software (Applied Photophysics). Time courses recorded for up to 20 s following mixing of ferryl Mb/ferryl Hb with acidic-pH solutions could be fitted as an A→B→C sequence. The spectra of A and B were very similar to each other and to the spectrum of Mb ferryl/Hb ferryl at pH 8, and also resembled the first spectrum recorded at 1.28 ms after mixing. Spectra of species A are therefore shown in Figure 1 (below). On the other hand, species C, which accumulates on a slower timescale, featured broadened bands throughout the UV–visible spectrum, suggestive of protein unfolding, most likely accompanied by protonation and dissociation of the proximal histidine ligand. pH-dependence data were fitted to the Henderson–Hasselbach equation, assuming one-proton processes; for the kinetic data, initial guesses for maximal rates were chosen assuming a pKa of 4.5, after which both the maximal rates and the pKa values were optimized with a least-squares algorithm within Microsoft Excel 2003; calculated pKa values were ∼4.7 for all data sets shown in Figure 2 (below). The rates of decay of ferryl haem via reduction by ascorbate were measured by procedures identical with those used for the data in Figure 1 (below), with the exception that 1 mM ascorbic acid was added at the time of mixing pH 8 ferryl with the respective buffers. Turnover experiments with ascorbate were performed under conditions similar to those used in a standard assay for ascorbate peroxidase [31], using 600 μM ascorbate, 1 mM H2O2, and 5 μM Mb in 20 mM phosphate, pH 7, at room temperature (25 °C). Urate turnover experiments were similar to ascorbate turnover, except for the use of 500 μM urate instead of ascorbate, and 8 μM Hb instead of Mb. Turnover rates varied linearly with Mb/Hb concentrations over these ranges of concentrations (results not shown). DFT calculations (UBP86/6-31G**; this hybrid density functional employs the Becke three-parameter set [31a] with the non-local electron correlation provided by the Perdew86 functional [31b]) on unsubstituted iron porphyrin adducts listed in Table 1 were performed following protocols previously described; proton affinities were obtained in vacuo, but the trends were readily maintained in solvents [2,32]; direct computational prediction of pKa values, as opposed to simple proton affinities, is possible, but has inherent pitfalls, as recently reviewed in [33].
Figure 1. Spectra of ferryl Mb collected at known pH values (upper panel) and normalized pH-dependence of A529−A588 for Mb and Hb (lower panel).

Upper panel: spectra of ferryl Mb collected at known pH values; the corresponding haemoglobin ferryl spectra are shown as an inset. Lower panel: pH-dependence of A529−A588 for Mb and Hb normalized between maximum value (1) and minimum value (0); fits to the Henderson–Hasselbach equation yield pKa values of ∼4.7 for both proteins.
Figure 2. Comparison of the optically detected pKa values for Mb and Hb ferryl with pH-dependent kinetics of decay with or without exogenous substrates added.

The reactivity data points refer to reactions listed in text. Explanation of key captions: iso-P, isoprostane F2 formation by Mb [36]; HPODE, HPODE consumption by Mb [30]; ascorbate+ferryl, rate of ferryl reduction by ascorbate; haemin, HPODE consumption by free haemin [28]; Mb, ascorbate turnover and Hb, ascorbate turnover, catalytic ascorbate and urate oxidation by Mb and Hb respectively in the presence of H2O2.
Table 1. Room-temperature pKa values (at neutral pH and at room temperature for Mb where otherwise not specified, and for CPO) and calculated proton affinities for relevant deprotonation processes.
For models of the corresponding active sites, see the Experimental section. Abbreviations: Im, imidazole (imidazolate is not abbreviated); P450, cytochrome P450.
| Charge balance | Experimental protonation state and pKa | Calculated proton affinity (kcal/mol) |
|---|---|---|
| +1→0 | ||
| [Im–Fe(III)–OH2]1+→[Im–Fe(III)–OH]0 | Partially protonated (pKa ∼8 in Hb and 9 in Mb) [43] | 258 |
| [Im–Fe(IV)–OH]1+→[Im–Fe(IV)=O]0 | Partially protonated proposed interpretation of Figure 2 data | 256 |
| 0→−1 | ||
| [Im–Fe(III)–OH]0→[imidazolate–Fe(III)–OH]−1 | Protonated, pKa ∼11 [42] | |
| [Im–Fe(III)–OH]0→[Im–Fe(III)=O]−1 | Protonated | |
| [Im–Fe(II)–OO]0→[imidazolate–Fe(II)–OO]−1 | Protonated | 407 |
| [Im–Fe(III)–OOH]0→[Im–Fe(III)–OO]−1 | Protonated [46] | 374 |
| [Im–Fe(III)–OOH]0→[imidazolate–Fe(III)–OOH]−1 | Protonated [46] | 338 |
| [Im–Fe(III)–OO]0→[imidazolate–Fe(III)–OO]−1 | Protonated [46] | 373 |
| [Im–Fe(IV)=O]0→[imidazolate–Fe(IV)=O]−1 | Alternative interpretation of Figure 2 data based on [42] | 337 |
| [Thiolate–Fe(IV)–OH]0→[thiolate–Fe(IV)=O]−1 (CPO) | Protonated [17] | 345 |
| [Thiolate–Fe(III)–OH2]0→[thiolate-Fe(III)–OH]−1 (P450) | Protonated [3] |
RESULTS AND DISCUSSION
Figure 1 shows optical spectra collected on rapidly mixing ferryl Mb (produced by standard procedures at basic pH, where it is relatively stable) with solutions having various pH values in a stopped-flow spectrophotometer. Following mixing, the time courses of spectral changes were recorded, from which the initial spectra (t=0) could be constructed (see the Experimental section). The latter therefore report the spectra of the ferryl form at any given pH value before decay of the ferryl species or protein structural changes occurred. These spectra show a pH-dependence with a pKa of ≤5. Likewise, a pKa of ≤5 was obtained for Hb ferryl in similar pH-jump experiments.
The optically detected pKa values for the globin haem ferryl species are entirely consistent, as shown in Figure 2, with previously measured [7,8,28–30,34,35] pH profiles for Mb ferryl reactivity in autoreduction, or in oxidative reactions such as 13-HPODE [(13S)-hydroperoxyoctadeca-(9Z,11E)-dienoic acid] consumption, isoprostane formation or for ascorbate or urate reduction. These latter reactivity data hold direct physiological relevance; HPODE is generated in vivo from linoleic acid by lipoxygenases and has been used as a model lipid peroxide to explore the mechanism of metglobin-induced damage to lipid membranes [30]; isoprostane F2 formation from low-density lipoprotein has been implicated in Mb- and acidosis-driven renal failure [36]; finally, ascorbate and urate are known to detoxify met and ferryl states of Mbs and Hbs [9,37–39]. These pH profiles are also identical with those previously measured by Skibsted and co-workers for the reactivity of ferryl Mb towards ABTS [2,2′-azinobis-(3-ethylbenzothiazoline-6-sulfonic acid), a typical peroxidase substrate], as well as towards chlorogenate [40,41]. Free haemin in solution exhibits a pH-dependence identical with that of Mb in the HPODE assay [28], suggesting that this reactivity pattern is an intrinsic property of the haem group rather than being due to protonation of a protein residue. Taken together, these data establish a direct link between biologically relevant acid-induced reactivity of ferryl globins and a distinct spectral change in the optical spectrum of their haem groups.
The pH-dependent spectral changes shown in Figure 1 for ferryl Mb are consistent with spectra previously reported for the same species at basic pH versus pH 3.5 [42]. These differences were shown to be paralleled by changes in the magnetic CD spectra that were irreconcilable with simple changes in hydrogen-bonding patterns and could only be explained as arising from protonation of an iron ligand at acidic pH [42]. This ligand was proposed by Thomson and co-workers [42] to be the proximal histidine residue, that is:
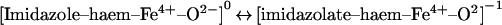 |
The pKa of this proximal histidine residue is ∼11 in ferric Mb, that is:
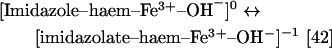 |
Thomson and co-workers have pointed out that a pKa shift of several pH units for the imidazole ligand may be expected on going from the ferric hydroxide state to the ferryl state [42]. Nevertheless, especially in view of the recent crystallographic data on Mb Compound II [14], an attractive alternative interpretation is that the Mb ferryl pKa may be due to protonation of the oxygen atom rather than to protonation of an imidazolate axial ligand. Notably, in the Mb Compound II crystal structure, besides the iron–oxygen bond length being consistent with a protonated ferryl, the iron–imidazole bond length is consistent with a protonated imidazole (i.e., it is ∼2.1 Å as opposed to the ∼1.9 Å predicted for an imidazolate group) [2,14].
In order better to estimate the relative likelihood of the two sites (imidazolate versus oxygen atom) being protonated in Mb ferryl, Table 1 shows a list of similar
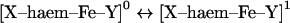 |
processes (i.e. similar charge balances), where either one of the haem axial ligands, X or Y, undergoes deprotonation in Mb as well as in selected related proteins. Table 1 lists known pKa values or known protonation states where experimental data is available, and it also lists calculated proton affinities for models of the Mb active site as well as of the chloroperoxidase active site (see the Experimental section for an account of how these models are treated computationally). For the cases where both experimental data and calculated proton affinities are available, theory and experiment appear to correlate well, without exception. Thus calculated proton affinities in excess of 330 kcal/mol (1 kcal=4.184 kJ) always correlate with a protonated state being observed at physiological pH, whereas calculated proton affinities of 250–260 kcal/mol appear consistent with the deprotonated state becoming populated in the physiological pH range. Table 1 thus notably shows that the proton affinity of [imidazolate–Fe(IV)=O]−1 is very similar to those of other species, with the same overall charge and present in the same protein pocket known to be protonated at neutral pH. Computations thus offer no underlying electronic structural reason to assign the ferryl Mb/ferryl Hb pKa as due to protonation of the histidine ligand, rather than the ferryl itself. Table 1 also shows that the proton affinity of the oxygen atom in an imidazole-ligated ferryl group, that is:
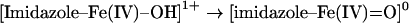 |
would be slightly lower than the proton affinity of the ferric-hydroxo adduct, that is:
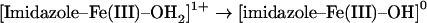 |
(comparing two protonation processes with the same overall charge balance). Ferric Mb and Hb feature an aqua ligand with a pKa of ∼8–9 [43], the pKa of the ferryl Mb/Hb should therefore be expected to be lower than that of ferric Mb/Hb. This prediction is in good agreement with the experimental data in Figures 1 and 2 (note that our interpretation of the computational results focuses on trends and relative values, whereas the exact value of the difference in pKa values is not quantitatively addressed by our gas-phase calculations). It thus follows that the iron ligand protonating at pH ∼5 in ferryl Mb is most likely the ‘oxo’ oxygen atom. This now provides an atomic-level insight into the origin of the known [7,8,28,29] physiologically relevant pH-dependence of the Mb ferryl reactivity. We have argued elsewhere that the alternative electronic description of Fe(IV)–OH as Fe(III)+hydroxyl radical would recommend protonated ferryl as a particularly reactive species [28,29,32].
As stated in the Introduction, a large body of spectroscopic evidence has recently been interpreted as evidence for a non-protonated ferryl in Mb and for a protonated ferryl in anion-ligated haemoproteins (CPO and cytochrome P450). We feel that this spectroscopic evidence bears an alternative interpretation that is also in line with our own data reported herein, as follows.
(1) Green and co-workers found that, using resonance Raman spectroscopy, they could detect the Fe=O (non-protonated) stretching frequency for ferryl Mb even at pH values as low as 4.5. This is entirely consistent with our measured pKa value of 4.7; there should indeed be significant amounts of unprotonated ferryl present, even at pH 5 [13,21,22].
(2) A history of decades of unsuccesful attempts with CPO has shown that detection of the Fe–OH stretching frequency for a protonated ferryl can be extremely problematic [2,21]. Remarkably, Green and co-workers have recently succeeded in detecting this Fe–OH frequency in CPO Compound II [21]. However, in parallel Mössbauer and DFT studies they also showed that ∼30% of CPO Compound II is in fact non-protonated [19]; neither they nor others have so far been able to detect this non-protonated CPO ferryl using resonance Raman spectroscopy. One lesson that we can therefore learn from CPO Raman spectroscopy is that detection of the ferryl iron–oxygen stretching frequency by this method can be problematic, even after sustained effort. The failure so far to detect a protonated ferryl group in Mb by resonance Raman spectroscopy is therefore not a strong argument against such protonation.
(3) Green and co-workers advocate that an anionic ligand (thiolate) in ferryl Compound II results in protonation of the ferryl form at physiological pH values [17]. If one were to interpret our measured pKa value in terms of the basic form of Mb Compound II containing an anionic imidazolate ligand, then we would also have to conclude, by analogy with thiolate-ligated CPO and cytochrome P450 (and perhaps also by analogy with tyrosinate-ligated catalases [24]), that the basic imidazolate-ligated form of Mb ferryl contains a hydroxide ligand. This, however, would be at odds with the resonance- Raman-spectroscopy data showing that, even at pH 4.5, at least part of the sites contain a non-protonated ferryl.
(4) EXAFS data on HRP Compound II has recently been interpreted to show an iron–oxygen bond of 1.65 Å [17]. This can be safely interpreted as proof for a non-protonated ferryl group. However, as previously discussed by one of us elsewhere [2] an older, independent EXAFS study cites a 1.9-Å distance for the same bond length, consistent now with a protonated ferryl [44]. Furthermore, a ∼0.1 Å difference in the other iron–axial ligand bond length (iron–histidine) also exists between the two EXAFS measurements [17,44]. In fact, the earlier-obtained EXAFS measurement, citing an Fe–O bond length at 1.9 Å and an iron–histidine bond length at 2.1 Å, is also entirely consistent with the atomic-resolution crystal structure of Mb ferryl [14], whereas the results of the more recent EXAFS study [17] is at odds with the crystallography results both in terms of the Fe–O distance and in terms of the Fe–histidine distance; the latter is important to note, as the larger number of atoms in the histidine ligand makes it less likely to be affected by crystallographic disorder or partial occupation compared with the Fe–O distance. There are now three reports of high-resolution crystal structures for three Compound II species (Mb, 1.35 Å resolution [14]; CCP, 1.3 Å [15]; HRP, 1.6 Å [45]), all of which concur in indicating a protonated ferryl group and an ∼2.1 Å iron–histidine bond length.
On the basis of arguments (1)–(4) presented above, our proposed interpretation of the Mb pKa as being due to ferryl protonation appears to be in reasonable agreement with other spectroscopic evidence, as well as with X-ray-crystallographic findings.
Thomson and co-workers have compared the magnetic CD spectra of acidic and basic ferryl Mb, and found that the acid form closely resembled typical peroxidase Compound II species, whereas the basic form did not [42]. If we accept that acid ferryl Mb contains an Fe–OH bond, the implication is that most histidine-ligated Compound II species are also protonated. It has long been known that Compound II formation in peroxidases is accompanied by uptake of a proton [16]; our findings now seem to indicate that this proton is located on the ferryl moiety. In Mb/Hb, protonation brings about increased reactivity (cf. Figure 2); a non-protonated ferryl Mb/Hb at neutral pH presents an evolutionary advantage, limiting potentially deleterious reactivity in this dioxygen-carrying protein. It is also an evolutionary advantage that the ferryl is protonated at neutral pH in peroxidases, enzymes the precise function of which is to form ferryl and efficiently use its oxidizing power. In peroxidases, the ferryl pKa is likely to be modulated by a carboxylate side chain hydrogen-bonded to the proximal imidazole group [5]; this added negative charge should increase proton affinity and shift the pKa towards basic values. By contrast, the proximal imidazole/histidine ligand in Mb is hydrogen-bonded to a neutral backbone carbonyl group, implying a smaller proton affinity. The general conclusion is that Nature gates the reactivity of ferryl species by controlling their protonation state.
Acknowledgments
Funding from the European Union-funded project Eurobloodsusbtitutes and from the BBSRC (Biotechnology and Biological Sciences Research Council) is gratefully acknowledged. We thank Dr Ioan Silaghi-Dumitrescu (Facultatea De Chimie Si Inginerie Chimicã, Universitatea Babeş-Bolyai, Cluj-Napoca, Romania) for helpful discussions. Miss Martine Rukengwa (formerly at the Department of Biological Sciences, University of Essex, and now at the School of Medicine, Health Policy and Practice, University of East Anglia, Norwich, U.K.) is thanked for assistance with preliminary experiments on ascorbate turnover.
References
- 1.Groves J. T. High-valent iron in chemical and biological oxidations. J. Inorg. Biochem. 2006;100:434–447. doi: 10.1016/j.jinorgbio.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 2.Silaghi-Dumitrescu R. The nature of the high-valent complexes in the catalytic cycles of hemoproteins. J. Biol. Inorg. Chem. 2004;9:471–476. doi: 10.1007/s00775-004-0543-2. [DOI] [PubMed] [Google Scholar]
- 3.Sono M., Roach M. P., Coulter E. D., Dawson J. H. Heme-containing oxygenases. Chem. Rev. 1996;96:2841–2888. doi: 10.1021/cr9500500. [DOI] [PubMed] [Google Scholar]
- 4.Dunford H. B. New York and Chichester: John Wiley; 1999. Heme peroxidases. [Google Scholar]
- 5.Hiner A. N. P., Raven E. L., Thorneley R. N. F., Garcia-Canovas G., Rodriguez-Lopez J. N. Mechanisms of Compound I formation in heme peroxidases. J. Inorg. Biochem. 2002;91:27–34. doi: 10.1016/s0162-0134(02)00390-2. [DOI] [PubMed] [Google Scholar]
- 6.Schlichting I., Berendzen J., Chu K., Sweet R. M., Ringe D., Petsko G. A., Sligar S. G. The catalytic pathway of cytochrome P450cam at atomic resolution. Science. 2000;287:1615–1622. doi: 10.1126/science.287.5458.1615. [DOI] [PubMed] [Google Scholar]
- 7.Reeder B. J., Sharpe M. A., Kay A. D., Kerr M., Moore K., Wilson M. T. Toxicity of myoglobin and haemoglobin: oxidative stress in patients with rhabdomyolysis and subarachnoid haemorrhage. Biochem. Soc. Trans. 2002;30:745–748. doi: 10.1042/bst0300745. [DOI] [PubMed] [Google Scholar]
- 8.Reeder B. J., Svistunenko D. A., Cooper C. E., Wilson M. T. The radical and redox chemistry of myoglobin and hemoglobin: from in vitro studies to human pathology. Antioxid. Redox Signalling. 2004;6:954–966. doi: 10.1089/ars.2004.6.954. [DOI] [PubMed] [Google Scholar]
- 9.Vollaard N. B., Reeder B. J., Shearman J. P., Menu P., Wilson M. T., Cooper C. E. A new sensitive assay reveals that hemoglobin is oxidatively modified in vivo. Free Radical Biol. Med. 2005;39:1216–1228. doi: 10.1016/j.freeradbiomed.2005.06.012. [DOI] [PubMed] [Google Scholar]
- 10.Terner J., Palaniappan V., Gold A., Weiss R., Fitzgerald M. M., Sullivan A. M., Hosten C. M. Resonance Raman spectroscopy of oxoiron(IV) porphyrin p-cation radical and oxoiron(IV) hemes in peroxidase intermediates. J. Inorg. Biochem. 2006;100:480–501. doi: 10.1016/j.jinorgbio.2006.01.008. [DOI] [PubMed] [Google Scholar]
- 11.Fujii H., Kurahashi T., Tosha T., Yoshimura T., Kitagawa T. 17O NMR study of oxo metalloporphyrin complexes: correlation with electronic structure of MO moiety. J. Inorg. Biochem. 2006;100:533–541. doi: 10.1016/j.jinorgbio.2006.01.009. [DOI] [PubMed] [Google Scholar]
- 12.Bollinger J. M., Jr, Krebs C. Stalking intermediates in oxygen activation by iron enzymes: motivation and method. J. Inorg. Biochem. 2006;100:586–605. doi: 10.1016/j.jinorgbio.2006.01.022. [DOI] [PubMed] [Google Scholar]
- 13.Behan R. K., Green M. T. On the status of ferryl protonation. J. Inorg. Biochem. 2006;100:448–459. doi: 10.1016/j.jinorgbio.2005.12.019. [DOI] [PubMed] [Google Scholar]
- 14.Hersleth H. P., Dalhus B., Gorbitz C. H., Andersson K. K. An iron hydroxide moiety in the 1.35 Å resolution structure of hydrogen peroxide derived myoglobin compound II at pH 5.2. J. Biol. Inorg. Chem. 2002;7:299–304. doi: 10.1007/s007750100296. [DOI] [PubMed] [Google Scholar]
- 15.Bonagura C. A., Bhaskar B., Shimizu H., Sundaramoorthy M., McRee D. E., Goodin D. B., Poulos T. L. High-resolution crystal structures and spectroscopy of native and compound I cytochrome c peroxidase. Biochemistry. 2003;42:5600–5608. doi: 10.1021/bi034058c. [DOI] [PubMed] [Google Scholar]
- 16.Hayashi Y., Yamazaki I. Heme-linked ionization in compounds I and II of horseradish peroxidases A2 and C. Arch. Biochem. Biophys. 1978;190:446–453. doi: 10.1016/0003-9861(78)90297-7. [DOI] [PubMed] [Google Scholar]
- 17.Green M. T., Dawson J. H., Gray H. B. Oxoiron(IV) in chloroperoxidase compound II is basic: implications for P450 chemistry. Science. 2004;304:1653–1656. doi: 10.1126/science.1096897. [DOI] [PubMed] [Google Scholar]
- 18.Stone K. L., Behan R. K., Green M. T. X-ray absorption spectroscopy of chloroperoxidase compound I: insight into the reactive intermediate of P450 chemistry. Proc. Natl. Acad. Sci. U.S.A. 2005;102:16563–16565. doi: 10.1073/pnas.0507069102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stone K. L., Hoffart L. M., Behan R. K., Krebs C., Green M. T. Evidence for two ferryl species in chloroperoxidase compound II. J. Am. Chem. Soc. 2006;128:6147–6153. doi: 10.1021/ja057876w. [DOI] [PubMed] [Google Scholar]
- 20.Green M. T. Application of Badger's rule to heme and non-heme iron-oxygen bonds: an examination of ferryl protonation states. J. Am. Chem. Soc. 2006;128:1902–1906. doi: 10.1021/ja054074s. [DOI] [PubMed] [Google Scholar]
- 21.Stone K. L., Behan R. K., Green M. T. Resonance Raman spectroscopy of chloroperoxidase compound II provides direct evidence for the existence of an iron(IV)-hydroxide. Proc. Natl. Acad. Sci. U.S.A. 2006;103:12307–12310. doi: 10.1073/pnas.0603159103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Behan R. K., Hoffart L. M., Stone K. L., Krebs C., Green M. T. Evidence for basic ferryls in cytochromes P450. J. Am. Chem. Soc. 2006;128:11471–11474. doi: 10.1021/ja062428p. [DOI] [PubMed] [Google Scholar]
- 23.Smulevich G., Feis A., Howes B. D. Fifteen years of Raman spectroscopy of engineered heme containing peroxidases: what have we learned? Acc. Chem. Res. 2005;38:433–440. doi: 10.1021/ar020112q. [DOI] [PubMed] [Google Scholar]
- 24.Horner O., Oddou J. L., Mouesca J. M., Jouve H. M. Mössbauer identification of a protonated ferryl species in catalase from Proteus mirabilis: density functional calculations on related models. J. Inorg. Biochem. 2006;100:477–479. doi: 10.1016/j.jinorgbio.2005.12.010. [DOI] [PubMed] [Google Scholar]
- 25.Svistunenko D. A. Reaction of haem containing proteins and enzymes with hydroperoxides: the radical view. Biochim. Biophys. Acta. 2005;1707:127–155. doi: 10.1016/j.bbabio.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 26.Kelso King N., Winfield M. E. The mechanism of metmyoglobin oxidation. J. Biol. Chem. 1963;238:1520–1528. [PubMed] [Google Scholar]
- 27.Kobert R. Beiträge zur Kenntniss der Methämoglobine. Pflügers Arch. Gesamte Physiol. Menschen Tiere. 1900;82:603–630. [Google Scholar]
- 28.Reeder B. J., Wilson M. T. The effects of pH on the mechanism of hydrogen peroxide and lipid hydroperoxide consumption by myoglobin: a role for the protonated ferryl species. Free Radical Biol. Med. 2001;30:1311–1318. doi: 10.1016/s0891-5849(01)00534-2. [DOI] [PubMed] [Google Scholar]
- 29.Reeder B. J., Svistunenko D. A., Sharpe M. A., Wilson M. T. Characteristics and mechanism of formation of peroxide-induced heme to protein cross-linking in myoglobin. Biochemistry. 2002;41:367–375. doi: 10.1021/bi011335b. [DOI] [PubMed] [Google Scholar]
- 30.Reeder B. J., Wilson M. T. Mechanism of reaction of myoglobin with the lipid hydroperoxide hydroperoxyoctadecadienoic acid. Biochem. J. 1998;330:1317–1323. doi: 10.1042/bj3301317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lad L., Mewies M., Basran J., Scrutton N. S., Raven E. L. Role of histidine 42 in ascorbate peroxidase: kinetic analysis of the H42A and H42E variants. Eur. J. Biochem. 2002;269:3182–3192. doi: 10.1046/j.1432-1033.2002.02998.x. [DOI] [PubMed] [Google Scholar]
- 31a.Becke A. D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993;98:5648. [Google Scholar]
- 31b.Perdew J. P. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Condens. Matter Mater. Phys. Rev. B. 1986;33:8822–8824. doi: 10.1103/physrevb.33.8822. [DOI] [PubMed] [Google Scholar]
- 32.Silaghi-Dumitrescu R., Cooper C. E. Transient species involved in catalytic dioxygen/peroxide activation by hemoproteins: possible involvement of protonated Compound I species. Dalton Trans. 2005:3477–3482. doi: 10.1039/b505440k. [DOI] [PubMed] [Google Scholar]
- 33.Silaghi-Dumitrescu R., Silaghi-Dumitrescu I. Computational Inorganic Chemistry – a useful tool, and more. Chemtracts – Inorg. Chem. 2005:684–708. [Google Scholar]
- 34.Velianou J. L., Wilson S. H., Reeder G. S., Caplice N. M., Grill D. E., Holmes D. R., Jr, Bell M. R. Decreasing mortality with primary percutaneous coronary intervention in patients with acute myocardial infarction: the Mayo Clinic experience from 1991 through 1997. Mayo Clin. Proc. 2000;75:994–1001. doi: 10.4065/75.10.994. [DOI] [PubMed] [Google Scholar]
- 35.Holt S., Reeder B., Wilson M., Harvey S., Morrow J. D., Roberts L. J., 2nd, Moore K. Increased lipid peroxidation in patients with rhabdomyolysis. Lancet. 1999;353:1241. doi: 10.1016/S0140-6736(98)05768-7. [DOI] [PubMed] [Google Scholar]
- 36.Moore K. P., Holt S. G., Patel R. P., Svistunenko D. A., Zackert W., Goodier D., Reeder B. J., Clozel M., Anand R., et al. A causative role for redox cycling of myoglobin and its inhibition by alkalinization in the pathogenesis and treatment of rhabdomyolysis-induced renal failure. J. Biol. Chem. 1998;273:31731–31737. doi: 10.1074/jbc.273.48.31731. [DOI] [PubMed] [Google Scholar]
- 37.Cooper C. E., Jurd M., Nicholls P., Wankasi M. M., Svistunenko D. A., Reeder B. J., Wilson M. T. On the formation, nature, stability and biological relevance of the primary reaction intermediates of myoglobins with hydrogen peroxide. Dalton Trans. 2005:3483–3488. doi: 10.1039/b505786h. [DOI] [PubMed] [Google Scholar]
- 38.Ames B. N., Cathcart R., Schwiers E., Hochstein P. Uric acid provides an antioxidant defense in humans against oxidant- and radical-caused aging and cancer: a hypothesis. Proc. Natl. Acad. Sci. U.S.A. 1981;78:6858–6862. doi: 10.1073/pnas.78.11.6858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Frei B., England L., Ames B. N. Ascorbate is an outstanding antioxidant in human blood plasma. Proc. Natl. Acad. Sci. U.S.A. 1989;86:6377–6381. doi: 10.1073/pnas.86.16.6377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Carlsen C. U., Skovgaard I. M., Skibsted L. H. Pseudoperoxidase activity of myoglobin: kinetics and mechanism of the peroxidase cycle of myoglobin with H2O2 and 2,2-azino-bis(3-ethylbenzthiazoline-6-sulfonate) as substrates. J. Agric. Food Chem. 2003;51:5815–5823. doi: 10.1021/jf030067g. [DOI] [PubMed] [Google Scholar]
- 41.Carlsen C. U., Kroger-Ohlsen M. V., Bellio R., Skibsted L. H. Protein binding in deactivation of ferrylmyoglobin by chlorogenate and ascorbate. J. Agric. Food Chem. 2000;48:204–212. doi: 10.1021/jf9908906. [DOI] [PubMed] [Google Scholar]
- 42.Foote N., Gadsby P. M. A., Greenwood C., Thomson A. J. pH-dependent forms of the ferryl haem in myoglobin peroxide analysed by variable-temperature magnetic circular dichroism. Biochem. J. 1989;261:515–522. doi: 10.1042/bj2610515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Antonini E., Brunori M. Hemoglobin and myoglobin in their reactions with ligands. In: Neuberger A., Tatum E. L., editors. Frontiers in Biology. Amsterdam and London: North-Holland Publishing Company; 1971. pp. 1–52. [Google Scholar]
- 44.Chance B., Powers L., Ching Y., Poulos T., Schonbaum G. R., Yamazaki I., Paul K. G. X-ray absorption studies of intermediates in peroxidase activity. Arch. Biochem. Biophys. 1984;235:596–611. doi: 10.1016/0003-9861(84)90234-0. [DOI] [PubMed] [Google Scholar]
- 45.Berglund G. I., Carlsson G. H., Smith A. T., Szoke H., Henriksen A., Hajdu J. The catalytic pathway of HRP at atomic resolution. Nature. 2002;417:463–468. doi: 10.1038/417463a. [DOI] [PubMed] [Google Scholar]
- 46.Davydov R., Satterlee J. D., Fujii H., Sauer-Masarwa A., Busch D. H., Hoffman B. M. A superoxo-ferrous state in a reduced oxy-ferrous hemoprotein and model compounds. J. Am. Chem. Soc. 2003;125:16340–16346. doi: 10.1021/ja037037e. [DOI] [PubMed] [Google Scholar]