

Abstract
TRIF is an adaptor protein associated with the signaling by Toll-like receptor (TLR)3 and TLR4 for the induction of type I IFNs. Here, we demonstrate a mechanism by which TLR signaling controls cell proliferation and survival. We show that TLR3 and TLR4 can induce cell cycle entry via TRIF, which targets the cell cycle inhibitor p27kip1 for relocalization, phosphorylation by cyclin/cdk complexes, and proteasome degradation. These events are antagonized by type I IFN induced by the TRIF pathway. Furthermore, in human dendritic cells treated with TLR3, TLR4, or TLR5 ligands, we demonstrate that IFN signaling modulates p27kip1 degradation and apoptosis, identifying an immunoregulatory “switching” function of type I IFNs. These findings reveal a previously uncharacterized function of TLR signaling in cell proliferation and survival.
Keywords: cell cycle, p27
Toll-like receptors (TLRs) are expressed in many hematopoietic cell types, and their role in immune responses has been well documented (1–4). However, TLRs are also expressed in nonhematopoietic cells and are likely to play an important role in tissue homeostasis. Activation of the TLR adapter MyD88-dependent signaling pathway results in induction of NF-κB leading to production of inflammatory cytokines and chemokines. In the case of TLRs 3 and 4, the IFN regulatory factor (IRF)-3 pathway is induced through the adapter TRIF and leads to IFN-β secretion (5). In certain cell types, TRIF-dependent TLR signaling results in apoptosis with a mechanism that, in part, depends on the production of type I IFN (6–9). However, in addition to apoptosis, TLR signaling may also result in cellular proliferation (10–13).
The mechanisms responsible for the control of these outcomes are poorly defined. Type I IFNs can exert a direct antiproliferative response by controlling the expression of proteins that regulate cell cycle entry and exit. IFNs target key regulators of cell proliferation and survival by reducing levels of the oncoprotein c-myc, as well as increasing the expression of cyclin-dependent kinase inhibitors, namely p27Kip1 (p27) (14). The tumor suppressor p27 plays a critical role in regulating progression through the G1–S phases of the cell cycle. p27 protein levels are modulated posttranslationally principally by degradation, sequestration, and distribution between the nucleus and cytoplasm (15, 16). The TLR4-agonist lipopolysaccharide (LPS) induces in nonproliferating dendritic cells (DCs) up-regulation of p27 that is followed by apoptosis (17, 18). Thus, TLR-signaling also influences immune responses by specific interference with the life span of these cells.
Here, we report that TLR3 and 4 induction of IFN-β regulated p27 localization and degradation in fibroblasts. Unlike TLR5 that induces p27 degradation and phosphorylation at the C-terminal Thr-187 site through MyD88/AKT activity (11), we show here that TRIF-dependent phosphorylation of p27 depends on cyclin/cdk complexes formed on the same Thr-187 site. Furthermore, we show that TLR activation and endogenous type I IFN are involved in the regulation of p27 levels in human DCs. Not only does TLR signaling control DC survival, but this function is also counterregulated by the secretion or not of type I IFN, which switches the effect on cell viability and survival. This differential effect of TLR3 and -4 depends on the TRIF–TBK-1 pathway, revealing p27 as a target in TRIF signaling. Thus, our results indicate that TLR signaling affects proliferation and survival through complex mechanisms in both hematopoietic and nonhematopoietic cells.
Results
TLR3 and -4 Triggering Overcomes p27-Induced Growth Arrest When Type I IFN Signaling Is Blocked.
To elucidate the mechanisms of TLR3- and -4-induced cellular proliferation, we examined by immunoblotting the levels of several cell cycle regulators in serum-starved Rat-1 cells treated with TLR agonists in the presence or absence of neutralizing antibodies against the type I IFN receptor (IFNARab). Unlike flagellin, the TLR3 and -4 ligands poly (I:C) and LPS, which are unable to induce cell cycle entry (11), did not decrease p27 levels, but rather increased them. In addition, we observed neither cyclin A or D1 synthesis nor AKT phosphorylation. As a control, we used IFNARab in the absence of TLR agonists, and we observed no cell cycle entry of serum-starved Rat-1 cells. However, when using TLR3 and -4 ligands in the presence of IFNARab, cell cycle regulators were modulated with kinetics similar to that seen in Rat-1 cells stimulated with flagellin only (Fig. 1a).
Fig. 1.

Cell cycle-related protein expression in serum-starved Rat-1 cells stimulated with poly (I:C) (10 μg/ml), LPS (50 ng/ml), and flagellin (100 ng/ml) in the presence or absence of IFNARab. The IFNARab was added at the same time as the respective TLR ligand. (a) Expression of cell cycle proteins examined by Western blots in total cellular extracts. (b) (Upper) Immunoprecipitation of CDK2 and p27 complexes: anti-CDK2 immunoprecipitates from total cellular extracts of stimulated cells were immunoblotted with anti-CDK2 or p27 antibodies. (Lower) CDK2 kinase activity was measured from total cell extracts from stimulated cells by using histone H1 as a substrate.
Because p27 inhibits cell cycle by forming complexes with CDK2/CE at G0, we examined by coimmunoprecipitation the presence of the CDK2/p27 complex in Rat-1 cells after stimulation with flagellin, LPS or poly (I:C) in the presence or absence of IFNARab (Fig. 1b). We found that in starved cells (0 h) p27 was immunoprecipitated as a complex with CDK2. Stimulation with flagellin induced the loss of this complex in both the presence or absence of IFNARab, but poly (I:C) and LPS provoked its dissociation only in the presence of IFNARab. In agreement with these data, the kinase activity of CDK2 in cells exposed to poly (I:C) and LPS was only observed when type I IFN signaling was blocked (Fig. 1b). These data indicate that the ability of poly (I:C) and LPS to disrupt CDK2/p27 complexes and to induce the kinase activity of CDK2 in fibroblasts is suppressed by endogenous type I IFN induced by TLR3 and TLR4.
TLR Agonists Induce Accumulation of Endogenous p27 in the Cytoplasm.
The cytoplasmic distribution of p27 is important for the rapid clearance of this protein that accompanies the entry of cells into cell cycle (19–21). In serum-starved Rat-1 cells, p27 accumulated in the nucleus, but after stimulation with flagellin, the majority of p27 was redistributed to the cytoplasm (Fig. 2A). IFNARab treatment alone did not affect the nuclear localization of p27 (data not shown). Furthermore, p27 localization remained restricted to the nucleus in serum-starved Rat-1 cells stimulated with poly (I:C) or LPS (Fig. 2 B and C, respectively). However, blocking IFNAR signaling resulted in cytoplasmic localization of p27 after poly (I:C) and LPS stimulation (Fig. 2 B and C). These results demonstrated that the endogenous type I IFN induced by TLR3 and -4 ligands prevented the nuclear export of p27 during the cell cycle.
Fig. 2.
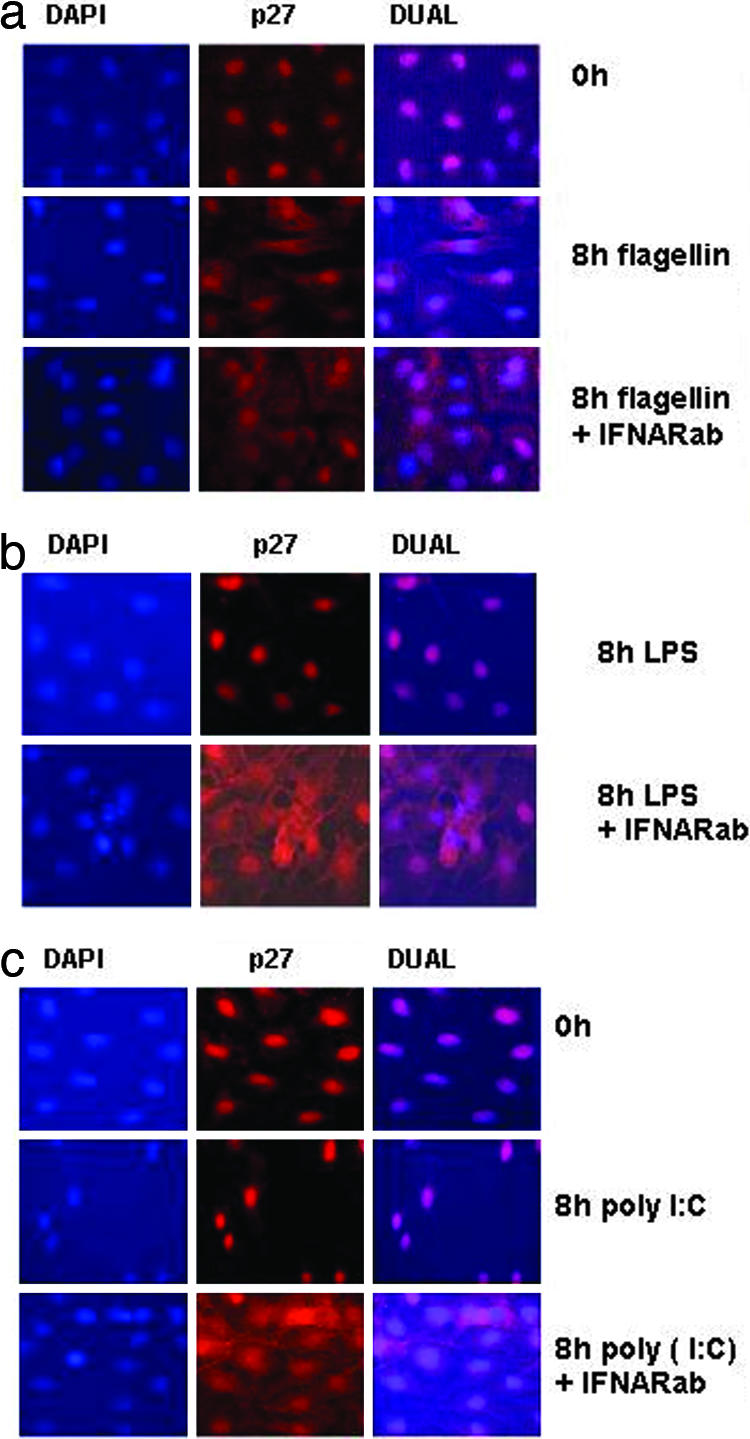
TLR3 and 4 induces cytoplasmic localization of endogenous p27 in the presence of IFNARab. Cells stimulated in the indicated conditions were stained with anti-p27 Ab together with Alexa Fluor 594-conjugated secondary Ab. Nuclei were counterstained with DAPI. (a) Localization of p27 in Rat-1 cells stimulated with flagellin (100 ng/ml) at 0 h and 8 h with or without 30 μg/ml of IFNARab. (b) Localization of p27 in Rat-1 cells stimulated with poly (I:C) (10 μg/ml) and 8 h with/without IFNARab. (c) Localization of p27 in Rat-1 cells stimulated with LPS (50 ng/ml) at 0 h and 8 h with/without IFNARab.
TRIF Mediates Activation of both NF-κB and IFN-β Promoters in Rat-1 Cells.
To confirm that poly (I:C) signaling in Rat-1 cells proceeds through the TRIF-dependent TLR3 pathway, rather than the TRIF-independent MDA-5 pathway (22), we performed analyses of TLR induced reporter assays, using siRNA to eliminate the functions of TRIF and MyD88. The results of these experiments confirmed that only poly (I:C) and LPS induced IFN-β promoter, and this induction was TRIF-dependent. Flagellin, on the other hand, activated NF-κB, but not IFN-β reporter, and this response depended on MyD88, but not on TRIF [supporting information (SI) Fig. 8].
TRIF, MyD88, and Their Downstream Signaling Molecules Induce Degradation of P27 That Is Prevented by Endogenous or Exogenous Type I IFN.
Among the four TIR adaptors known to positively regulate TLR signaling, only overexpression of MyD88 into HEK293T cells was capable of reducing the levels of p27; in particular, TRIF was unable to do so (11). It is possible that the inability of TRIF to modulate p27 levels may be due to IFN secretion. To test this hypothesis, we transfected HEK293T cells with a p27 expression vector in combination with either MyD88 or TRIF in the presence or absence of IFNARab (Fig. 3a). TRIF reduced the levels of p27 but, unlike MyD88, only in the presence of IFNARab. This reduction of p27 through TRIF was even more pronounced than that mediated by MyD88. In addition, cotransfection of the two kinases IKKε and TBK-1, which have an essential role in TRIF-mediated IFN-β induction (23) as well as RIG-I and MDA5 (24), also resulted in the reduced expression of cotransfected p27 but only in the presence of IFNARab (Fig. 3b). Transfection of dominant-negative mutants of these kinases had no effect. Interestingly, in contrast to IKKε and TBK-1, transfection of IKKα affected p27 levels independently of the presence of IFNARab (Fig. 3b). Furthermore, addition of exogenous recombinant IFN-β blocked p27 degradation induced by transfection of either MyD88 (Fig. 3c) or dominant-active AKT (DaAKT). Therefore, these data indicate that IFN-β inhibits MyD88 and/or AKT-induced cell cycle entry (Fig. 3c).
Fig. 3.

TRIF, MyD88 or their downstream signaling induces degradation of p27 that is prevented by endogenous or exogenous type I IFN. Data are mean values ± SEM. from one experiment (as described in Methods) representative of at least three independent experiments. (a) HEK293T cells were transfected with p27, TRIFHA, or MyD88FLAG expression plasmids as indicated. Twenty-four hours after transfection, cells cultured in the presence or absence of IFNARab were harvested and analyzed by Western blotting for p27, TRIFHA, and MyD88FLAG. (b) HEK293T cells were transfected with combinations of p27, IKK-ε-FLAG (WT or mutant), TBK-1FLAG (WT or mutant), and IKK-α-FLAG. At 6 h after transfection, IFNARab was added to the indicated samples. At 24 h after transfection, cells were harvested and analyzed by Western blotting for p27, MyD88FLAG, and DaAktHA. (c) HEK293T cells were transfected with combinations of p27, MyD88HA, and/or DaAKTHA. At 6 h after transfection, IFN-β was added to the indicated samples. At 24 h after transfection, cells were harvested and analyzed by Western blotting for p27, DaAKTHA, and MyD88FLAG. (d) HEK293T cells were transfected with p27, MyD88FLAG, and/or TRIFHA. Six hours later, IFNARab was added to the samples transfected with TRIFHA, and 8 h later, the proteasome inhibitor CBZ at 10 μM was added to the indicated samples. At 24 h after transfection, cells were harvested and analyzed by Western blotting for p27 and adapter proteins. In all experiments, GFP expression plasmid was cotransfected for normalization of transfection efficiency.
Proteosome-Mediated Degradation Is Involved in the p27 Down-Regulation in Response to both MyD88 and TRIF.
We next examined whether TRIF-induced p27 down-regulation was mediated by the proteosome-mediated degradation. HEK293T cells were cotransfected with either TRIF (in the presence of IFNARab) or MyD88 in the presence or absence of the proteasome inhibitor CBZ. The inhibitor restored the levels of p27 after MyD88 transfection. Similarly, TRIF transfection in the presence of IFNARab (Fig. 3d) also restored p27, thus demonstrating that TRIF does indeed induce proteosome-mediated p27 degradation.
TRIF-Mediated p27 Degradation Requires Kinase Activity of Cdk–Cyclin Complexes.
p27 phosphorylation and degradation can occur via two different mechanisms. The first involves CDK2/CE complexes that phosphorylate p27 at Thr-187 (25, 26). Phosphorylated p27 then forms a complex with ubiquitin ligase SCFSkp2, leading to 26S proteasome-mediated degradation of p27 in proliferating cells (27). Alternatively, p27 is also phosphorylated by AKT at Thr-157, 187, and 198 and Ser-10 (19, 20, 28). To establish whether the regulation of p27 by TRIF in the absence of type I IFN signaling depended on CDK2/CE or AKT, HEK293T cells were transfected with TRIF or MyD88 with or without IFNARab and with WT p27 or p27 mutants at the cyclin/cdk contact site (p27c-k) or at the cdk target site (p27V187). The expression levels of the different forms of p27 were determined by immunoblotting (Fig. 4). As described (11) and used here as a control, MyD88 expression promoted degradation of WT p27 and of the double cyclin/cdk contact mutant (Fig. 4). Transfection of TRIF alone did not have any effect on the levels of p27 but, in the presence of IFNARab, induced degradation of WT p27 (Fig. 4). However, the double cyclin/cdk contact mutant of p27, or the TPKK motif mutant (p27 Thr-187) were not affected. Therefore TRIF-dependent phosphorylation of p27 depends on cyclin/cdk complexes formed on the Thr-187 site and not AKT, as in the case of MyD88.
Fig. 4.

Degradation of p27 induced by TRIF/IFNARab is prevented by p27c-k and p27 Thr-187 mutations. HEK293T cells were cotransfected with p27 or mutants p27c-k and p27V187 together with MyD88 or TRIF in the presence or absence of IFNARab. Protein lysates were analyzed for expression of p27 and adapter proteins by Western blotting.
TLR Triggering Regulates p27 Expression in Dendritic Cells.
The differential effect on p27 as a novel target for the TLR-TRIF-TBK-1 pathway has been shown here to be essential in the cellular proliferation of nonhemopoietic cells. However, as we next sought to determine whether p27 regulation by TLR signaling also affects immune regulation, we extended our study to dendritic cells, a cell type that is crucial for initiation of acquired immunity. Human monocyte-derived DCs express several TLRs, including TLR3, -4, and -5 (29). Survival and longevity of DCs are critical factors influencing the outcome of immune responses (18, 30). It has been reported that, in the nonproliferating monocyte-derived DCs, up-regulation of p27 was not linked to cell cycle arrest in G0/G1 but, rather, associated with growth factor withdrawal-induced apoptosis and that p27 down-regulation increased survival (15). IFN secretion by DCs is also associated with increased levels of p27 that correlated with induction of apoptosis (17).
In agreement with Sangfelt et al. (31), we observed that the addition of IFN-β to growth factor-deprived DCs induced p27 accumulation, STAT-1 phosphorylation of isoforms α and β (upper and lower bands respectively), but not IκB or AKT phosphorylation (Fig. 5a). Treatment of DCs with TLR3 or -4 ligands, poly (I:C) or LPS, respectively, induced p27 accumulation (Fig. 5 b and c), phosphorylation of STAT-1, and phosphorylation and complete degradation of IκB. A delayed phosphorylation of AKT was observed only in DCs treated with LPS, consistent with studies in other cell types showing TLR4-induced AKT phosphorylation via GSK3 and/or PI3K (32–34). Unlike poly (I:C) and LPS, the TLR5 ligand flagellin rapidly induced phosphorylation of AKT Ser-473 in DCs and decreased the levels of p27 within 30 min. As expected, no STAT-1 phosphorylation was observed in flagellin-treated DCs, indicating that the IFN pathway was not activated (Fig. 5d). Flagellin-induced p27 degradation and AKT phosphorylation were drastically delayed but not abolished in the presence of added IFN-β, indicating that the TLR5 signal was able to override the IFN-driven p27 accumulation. Because MyD88-mediated degradation of p27 was prevented by IFN-β (Fig. 3c), flagellin may induce p27 degradation and AKT activation through a MyD88-dependent IFN-sensitive pathway as well as through an IFN-insensitive pathway.
Fig. 5.

p27 levels in human MonoDCs stimulated with TLR-ligands and/or human recombinant IFNβ (1,000 units/ml). Expression of p27, pSTAT1 tyr, STAT1, IKBα, pIKBα, and pAKT Ser-473 were examined by Western blots in total cellular extracts from MonoDCs at the indicated time points after stimulation.
Blocking Type I IFN Signaling in Monocyte-Derived DCs Prevents Apoptosis Induced by Poly (I:C) and LPS.
In mouse DCs treated with agonists for various TLRs, the genetic deficiency for type I IFN receptors prevented STAT-1 phosphorylation, an essential component of the signaling cascade of both type I and type II IFN receptors (35). Here, we show that IFNARab also blocked STAT-1 phosphorylation in human DCs stimulated with poly (I:C) and LPS (Fig. 6a). Unlike poly (I:C) and LPS, flagellin treatment of DCs down-regulated p27 in the presence or absence of IFNARab. These data suggest that, when the signaling through endogenous type I IFN is prevented, TLR signaling may prevent apoptosis and prolong DCs survival.
Fig. 6.

IFNARab blocks poly I:C and LPS induced p27 accumulation and prevents Caspase 7 and PARP cleavage in MonoDCs. MonoDCs were stimulated for the indicated times with poly (I:C), LPS, or Flagellin in the absence or presence of IFNRab. (a) p27, pSTAT tyr, and total STAT levels were analyzed by Western blotting in the total cellular extract of the stimulated cells. (b) Stimulated MonoDCs were stained for PARP cleavage or for NF-κB p65. Blue indicates DAPI-stained cells (nucleus); white indicates cellular morphology (negative for PARP); FITC (green) stained cells are positive for PARP or NF-κB. (c) PARP and Caspase 7, intact and cleaved, were detected by Western blotting in total cellular extract from the stimulated cells. Western blotting and immunofluorescence was performed three times with three different donors; data here present one of the three experiments.
Because induction of p27 is associated with the subsequent stimulation of caspase-7 (36) and then cleavage of PARP, we stained for PARP cleavage as a marker of apoptosis. Cleaved PARP fragments were detected by immunofluorescence, and Western blot analysis was also used to detect intact and cleaved caspase 7 and PARP. As depicted in Fig. 6b Bottom, stimulation of DCs with poly (I:C) resulted in the generation of cleaved PARP fragments. PARP was prevented when type I IFN signaling was blocked with IFNARab. These data were confirmed by Western blotting of caspase 7 and PARP cleavage. Caspase 7 cleavage was observed starting at 1 h and in cells treated with poly (I:C) (Fig. 6c). Similar results were obtained by using LPS (data not shown). Poly (I:C) stimulation in the presence or absence of the IFNARab did not affect NF-κB nuclear translocation (Fig. 6b). In DCs treated with flagellin, PARP was not cleaved while NF-κB nuclear translocation occurred (Fig. 6 b and c).
Cell viability can be measured in its ability to reduce resazurin into resorufin, which is highly fluorescent (37). To determine cell viability on DCs treated with ligands for TLRs3, -4 and -5 with or without IFNARab, cells were harvested after 24 and 48 h, and their metabolic activity was measured. Addition of resazurin to treated DCs induced a modest increase in the cell viability percentage in cells treated with poly (I:C) and LPS in the presence of IFNARab compared with those treated with IgG1 mouse sera and flagellin treated cells (SI Fig. 9). At 48 h, the viability of cells stimulated with TLRs 3 or -4 ligands and treated with IFNARab was clearly increased over that of nontreated cells, although cell viability decreased overall at the later time point. These data together suggest that stimulation of DCs with flagellin alone or stimulation with poly (I:C) or LPS when IFN signaling is blocked increases their survival.
Discussion
Our results point to the association of TLR signaling with cell cycle control and provide information on the molecular mechanisms involved. In addition they indicate that the specificity of TLR ligands expressed by bacteria and viruses has a discriminating effect on fibroblast proliferation or DC life span and that endogenous type I IFN has a switching function in determining the functional outcome of TLR signaling.
In particular, p27 plays a central role in the TLR mechanisms controlling these events. We showed that, when signaling through endogenous type I IFN induced by TLR3 and -4 agonists was prevented, the cell cycle arrest and/or apoptosis induced by triggering of these TLRs was reversed. Blocking type I IFN signaling in either fibroblasts or DC treated with TLR3 or -4 ligands resulted in p27 cytoplasmic sequestration and degradation. This was mediated by the TRIF/IKKε/TBK-1 signaling cascade that lead to p27 phosphorylation mediated by both CDK-cyclin and AKT and subsequent p27 down-regulation. These findings unveil functions of IKKε/TBK-1 kinases that were previously known only for the induction of IRF3 (38). They are consistent with reports indicating that both type I and II IFN strongly repressed the activity of CDK2 and -4 in a number of cell types by decreasing their levels or by increasing CDK inhibitors such as p27 (14, 39). During cell cycle regulation, p27 is degraded after a SCFSkp2-dependent ubiquitination in the nucleus, whereas the newly synthesized p27 is retained in the cytoplasm through AKT or CDK/cyclin complexes (20, 40). Here, we show that, whereas TLR5 induces p27 degradation and cytoplasmic retention through AKT, TLR3 and -4 induce the same effect on p27 through both CDK/cyclin- and AKT-dependent mechanisms. Even in the presence of added IFN-β, flagellin maintained some ability to reduce the p27 level, suggesting that TLR5 agonists may always favor cell survival and not induce cell death as seen in the case of the TLR3 and -4 agonists poly (I:C) and LPS. The possible mechanisms by which TLR3 and -4 pathways diverge from that of TLR5 are diagrammatically depicted in Fig. 7.
Fig. 7.

Diagram of the cell cycle regulatory mechanisms mediated through TLRs and analyzed in this study. (Left) TLR3 and -4 inductions of type I IFN drives cellular apoptosis through p27. Blocking the responses promotes cell survival. In both cases, TRIF and TBK-1/IKKε are required to mediate Aktser473–CDK2 regulation of p27. (Right) Role of TLR5 in cellular proliferation and survival, mediated by MyD88/IRAK complex, which targets Aktser473–p27 degradation.
These mechanisms unmask additional functions of these receptors that control cell survival. TRIF-mediated signaling by TLR3 and -4 in various cell types induces an apoptotic response that depends on the production of endogenous type I IFN (6, 8, 41–44). In the case of antigen-presenting cells such as DCs, TLR3 and -4 signaling may induce apoptosis before or after pathogen-derived peptides have been presented at DC surface, and this process may be critical for controlling the delicate balance between antigen cross-presentation and DC maturation. These processes, in addition to effective immunity against pathogens, may be involved in maintenance of self-tolerance and prevention of autoimmunity (45). The ability of the induced type I IFN to prevent the proliferative effect of signaling through these TLRs, and to contribute to the induction of apoptosis may cripple the ability of viruses or other pathogens to replicate more efficiently in proliferating cells (46–48).
Furthermore, in another physiological setting, the proliferation of nonhematopoietic cells induced by TLR ligands may have a significant role in biologically relevant situations related to both tissue homeostasis and resistance to infections and tissue damage (12). The ability of TLR5-induced MyD88-dependent signaling to promote proliferation would be advantageous in response to tissue damage, for example, in the intestine where flagellin stimulation by commensal bacteria might promote tissue repair.
The similarity of molecular mechanisms that we characterized in two representative models of nonhematopoietic and hematopoietic cells strongly suggests that they represent homeostatic mechanisms common to most cell types. Finally, our demonstration of the ability of certain TLRs to induce IFNs that mediate growth-inhibitory responses and of others to induce cellular proliferation through p27 regulation increases our understanding of the functions and diversity of these receptors.
Methods
Cell Culture.
HEK293T were obtained from American Type Culture Collection (Manassas, VA), and Rat-1 cells were obtained from the Imperial Cancer Research Fund (London, U.K.). Cells were maintained as described (11). Human blood samples were obtained according to institutional guidelines. Peripheral blood mononuclear cells were purified by Ficoll-Hypaque centrifugation. Monocyte-derived DC (MonoDCs) were prepared as described (13). For growth factor and Western blot studies, MonoDCs were arrested by washing three times with PBS and were then replenished with serum, GM-CSF, and IL-4-free medium 24 h before activation.
TLR Ligands and Reagents.
Poly I:C and LPS were purchased from InvivoGen (San Diego, CA), Flagellin from Salmonella munchen (Calbiochem, San Diego, CA) and were used at indicated concentrations. The IFNARab (PBL, Paris, France) (see below) was added at the same time as the respective TLR ligand. Rat and human IFN-β was purchased from PBL and used at 1,000 units.
Plasmid Constructs.
MyD88 and TRIF (TICAM-1) were amplified from human cDNA library and cloned into pCMV vectors. pCMVp27 and pBabePuro-p27 as well as pBabePuro-p27ck and V187 mutants were cloned as described (11). The dominant-active Thr-308/Ser-473 mutant of AKT (AcAKT) was obtained from Brian Hemmings (Friedrich Miescher Institute, Basel, Switzerland). IKK-ε and TBK-1 flagged WT and mutant constructs were kind gifts from Kate Fitzgerald (University of Massachusetts, Amherst, MA). Bakary Sylla (IARC) donated the IKK-α flag construct. The luciferase promoter NF-κB was purchased from Clontech, and IFN-β was cloned as described (49).
Luciferase Assay.
Rat-1 cells were transiently transfected as described (11).
Cell Viability Assay.
Mono-DCs (at D4) were plated into a 96-well plate to give a final cell count of 2 × 104 per well and were then treated with TLRs ligands with or without IFNARab. As a control for cell viability, cells were treated with increasing concentrations of DMSO. Treated cells were harvested after 24- and 48-h stimulation, and 20 μl of cell-titer-blue reagent was added (Promega, Madison, WI). Cells were incubated for 4 h at 37 C and then transferred into fluorometry plates, and fluorescence was read at 560 ex/590 em by using the Ascent fluorimeter.
SiRNA Treatment of Cells.
SiRNA duplexes targeting the coding region of TRIF and MyD88 were synthesized by Dharmacon (Lafayette, CO) (RNF138 and M-099508–00, respectively). TRIF mRNA expression levels were evaluated by performing a PCR on reverse-transcribed (Fermentas, Strausbourg, France) RNA from cells treated with siRNA for TRIF and scramble (50). For reporter assays, 24 h after siRNA treatment, cells were transfected with NF-κB or IFN-β and stimulated with TLR ligands as described (11).
Transfection of HEK293T.
Cells were transiently transfected by using FuGene (Roche, Meylan, France), with 500 ng of the indicated plasmid together with 250 ng of pCMVGFP for normalization of expression levels. Twenty-four hours post transfection, HEK293T cells were lysed for Western blot analysis. For IFN blocking studies, cells were washed 6–8 h after transfection, and medium was replenished with the addition of 10 μg/ml of IFNARab; control cells were treated with IgG1 mouse sera.
Biochemical Analysis.
Between 10 and 40 μg of total cellular protein (determined by Bradford assay; Bio-Rad, Hercules, CA), and Western blot analysis was performed (49).
Immunofluorescence Analysis.
Rat-1 cells or MonoDCs were divided into a six-well plate containing polylysine-coated 22 mm × 22 mm coverslips, and staining was performed as described (11).
Antibodies.
The following antibodies were used against p27: C19 (sc528; Santa Cruz Biotechnology, Santa Cruz, CA), CDK2: M2 (sc-163; Santa Cruz Biotechnology), cyclin A: H-432 (sc751; Santa Cruz Biotechnology), cyclin D1 (2926; Cell Signaling Technology, Beverly, MA), pAKTser473 (9271s; Cell Signaling Technology) type I IFNARab receptor (21385-1; PBL), anti-HA (Roche), anti-FLAG M2 (Sigma, St. Louis, MO), anti-GFP (Roche), goat anti-rabbit Alexa Fluor 594 (Molecular Probes, Invivogen, Cergy Pontoise, France) STAT-1 (9172; Cell Signaling Technology), pSTAT-1 (9171; Cell Signaling Technology), IkBα (9241; Cell Signaling Technology), pIKBα (9242; Cell Signaling Technology), cleaved PARP (9546; Cell Signaling Technology), total PARP (9542; Cell Signaling Technology), Caspase 7 (9492; Cell Signaling Technology), and NF-κB p65 (sc-8008; Santa Cruz Biotechnology).
Acknowledgments
We thank Pierre Garrone for advice on the MonoDCs experiments, Kate Fitzgerald and Bakary Sylla for the plasmid constructs, and Ruslan Medzhitov for all scientific discussions regarding TLRs and their other roles. This work was supported by grants from La Ligue Contre le Cancer (Comité de Savoie) and the grant “Applied Tumour Virology” German–French cooperation, German Cancer Research Center (DKFZ), Heidelberg–Cancéropôle du Grand-Est, Besançon.
Abbreviations
- DC
dendritic cell
- IFNARab
antibodies against the type I IFN receptor
- LPS
lipopolysaccharide
- MonoDC
monocyte-derived DC
- TLR
Toll-like receptor.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0700664104/DC1.
References
- 1.Yamamoto M, Takeda K, Akira S. Mol Immunol. 2004;40:861–868. doi: 10.1016/j.molimm.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 2.Takeda K, Akira S. J Dermatol Sci. 2004;34:73–82. doi: 10.1016/j.jdermsci.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 3.Takeda K, Akira S. Semin Immunol. 2004;16:3–9. doi: 10.1016/j.smim.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 4.Takeda K, Kaisho T, Akira S. Annu Rev Immunol. 2003;21:335–376. doi: 10.1146/annurev.immunol.21.120601.141126. [DOI] [PubMed] [Google Scholar]
- 5.Doyle SE, O'Connell R, Vaidya SA, Chow EK, Yee K, Cheng G. J Immunol. 2003;170:3565–3571. doi: 10.4049/jimmunol.170.7.3565. [DOI] [PubMed] [Google Scholar]
- 6.Han KJ, Su X, Xu LG, Bin LH, Zhang J, Shu HB. J Biol Chem. 2004;279:15652–15661. doi: 10.1074/jbc.M311629200. [DOI] [PubMed] [Google Scholar]
- 7.Jung DY, Lee H, Jung BY, Ock J, Lee MS, Lee WH, Suk K. J Immunol. 2005;174:6467–6476. doi: 10.4049/jimmunol.174.10.6467. [DOI] [PubMed] [Google Scholar]
- 8.Kaiser WJ, Offermann MK. J Immunol. 2005;174:4942–4952. doi: 10.4049/jimmunol.174.8.4942. [DOI] [PubMed] [Google Scholar]
- 9.Rasschaert J, Ladriere L, Urbain M, Dogusan Z, Katabua B, Sato S, Akira S, Gysemans C, Mathieu C, Eizirik DL. J Biol Chem. 2005;280:33984–33991. doi: 10.1074/jbc.M502213200. [DOI] [PubMed] [Google Scholar]
- 10.Abreu MT, Fukata M, Arditi M. J Immunol. 2005;174:4453–4460. doi: 10.4049/jimmunol.174.8.4453. [DOI] [PubMed] [Google Scholar]
- 11.Hasan UA, Trinchieri G, Vlach J. J Biol Chem. 2005;280:20620–20627. doi: 10.1074/jbc.M500877200. [DOI] [PubMed] [Google Scholar]
- 12.Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Cell. 2004;118:229–241. doi: 10.1016/j.cell.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 13.de Bouteiller O, Merck E, Hasan UA, Hubac S, Benguigui B, Trinchieri G, Bates EE, Caux C. J Biol Chem. 2005;280:38133–38145. doi: 10.1074/jbc.M507163200. [DOI] [PubMed] [Google Scholar]
- 14.Brierley MM, Fish EN. J Interferon Cytokine Res. 2002;22:835–845. doi: 10.1089/107999002760274845. [DOI] [PubMed] [Google Scholar]
- 15.Olashaw N, Bagui TK, Pledger WJ. Cell Cycle. 2004;3:263–264. doi: 10.4161/cc.3.3.721. [DOI] [PubMed] [Google Scholar]
- 16.Rodier G, Montagnoli A, Di Marcotullio L, Coulombe P, Draetta GF, Pagano M, Meloche S. EMBO J. 2001;20:6672–6682. doi: 10.1093/emboj/20.23.6672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hashimoto SI, Suzuki T, Nagai S, Yamashita T, Toyoda N, Matsushima K. Blood. 2000;96:2206–2214. [PubMed] [Google Scholar]
- 18.Woltman AM, van der Kooij SW, Coffer PJ, Offringa R, Daha MR, van Kooten C. Blood. 2003;101:1439–1445. doi: 10.1182/blood-2002-06-1688. [DOI] [PubMed] [Google Scholar]
- 19.Fujita N, Sato S, Katayama K, Tsuruo T. J Biol Chem. 2002;277:28706–28713. doi: 10.1074/jbc.M203668200. [DOI] [PubMed] [Google Scholar]
- 20.Liang J, Zubovitz J, Petrocelli T, Kotchetkov R, Connor MK, Han K, Lee JH, Ciarallo S, Catzavelos C, Beniston R, et al. Nat Med. 2002;8:1153–1160. doi: 10.1038/nm761. [DOI] [PubMed] [Google Scholar]
- 21.Shin I, Yakes FM, Rojo F, Shin NY, Bakin AV, Baselga J, Arteaga CL. Nat Med. 2002;8:1145–1152. doi: 10.1038/nm759. [DOI] [PubMed] [Google Scholar]
- 22.Gitlin L, Barchet W, Gilfillan S, Cella M, Beutler B, Flavell RA, Diamond MS, Colonna M. Proc Natl Acad Sci USA. 2006;103:8459–8464. doi: 10.1073/pnas.0603082103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fitzgerald KA, McWhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT, Coyle AJ, Liao SM, Maniatis T. Nat Immunol. 2003;4:491–496. doi: 10.1038/ni921. [DOI] [PubMed] [Google Scholar]
- 24.Bowie AG, Fitzgerald KA. Trends Immunol. 2007;28:147–150. doi: 10.1016/j.it.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 25.Blanchard DA, Affredou MT, Vazquez A. J Immunol. 1997;158:3054–3061. [PubMed] [Google Scholar]
- 26.Frost V, Delikat S, Al-Mehairi S, Sinclair AJ. J Gen Virol. 2001;82:3057–3066. doi: 10.1099/0022-1317-82-12-3057. [DOI] [PubMed] [Google Scholar]
- 27.Yang X, Menon S, Lykke-Andersen K, Tsuge T, Di X, Wang X, Rodriguez-Suarez RJ, Zhang H, Wei N. Curr Biol. 2002;12:667–672. doi: 10.1016/s0960-9822(02)00791-1. [DOI] [PubMed] [Google Scholar]
- 28.Liang J, Slingerland JM. Cell Cycle. 2003;2:339–345. [PubMed] [Google Scholar]
- 29.Visintin A, Mazzoni A, Spitzer JH, Wyllie DH, Dower SK, Segal DM. J Immunol. 2001;166:249–255. doi: 10.4049/jimmunol.166.1.249. [DOI] [PubMed] [Google Scholar]
- 30.Marx J. Science. 2006;311:1086. doi: 10.1126/science.311.5764.1086a. [DOI] [PubMed] [Google Scholar]
- 31.Sangfelt O, Strander H. Med Oncol. 2001;18:3–14. doi: 10.1385/mo:18:1:3. [DOI] [PubMed] [Google Scholar]
- 32.Vivarelli MS, McDonald D, Miller M, Cusson N, Kelliher M, Geha RS. J Exp Med. 2004;200:399–404. doi: 10.1084/jem.20040446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li X, Tupper JC, Bannerman DD, Winn RK, Rhodes CJ, Harlan JM. Infect Immun. 2003;71:4414–4420. doi: 10.1128/IAI.71.8.4414-4420.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee JY, Ye J, Gao Z, Youn HS, Lee WH, Zhao L, Sizemore N, Hwang DH. J Biol Chem. 2003;278:37041–37051. doi: 10.1074/jbc.M305213200. [DOI] [PubMed] [Google Scholar]
- 35.Gautier G, Humbert M, Deauvieau F, Scuiller M, Hiscott J, Bates EE, Trinchieri G, Caux C, Garrone P. J Exp Med. 2005;201:1435–1446. doi: 10.1084/jem.20041964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Don MJ, Chang YH, Chen KK, Ho LK, Chau YP. Mol Pharmacol. 2001;59:784–794. doi: 10.1124/mol.59.4.784. [DOI] [PubMed] [Google Scholar]
- 37.O'Brien J, Wilson I, Orton T, Pognan F. Eur J Biochem. 2000;267:5421–5426. doi: 10.1046/j.1432-1327.2000.01606.x. [DOI] [PubMed] [Google Scholar]
- 38.Fitzgerald KA, Rowe DC, Barnes BJ, Caffrey DR, Visintin A, Latz E, Monks B, Pitha PM, Golenbock DT. J Exp Med. 2003;198:1043–1055. doi: 10.1084/jem.20031023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sangfelt O, Erickson S, Castro J, Heiden T, Gustafsson A, Einhorn S, Grander D. Oncogene. 1999;18:2798–2810. doi: 10.1038/sj.onc.1202609. [DOI] [PubMed] [Google Scholar]
- 40.Sekimoto T, Fukumoto M, Yoneda Y. EMBO J. 2004;23:1934–1942. doi: 10.1038/sj.emboj.7600198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rasschaert J, Ladriere L, Urbain M, Dogusan Z, Katabua B, Sato S, Akira S, Gysemans C, Mathieu C, Eizirik DL. J Biol Chem. 2005;280:33984–33991. doi: 10.1074/jbc.M502213200. [DOI] [PubMed] [Google Scholar]
- 42.De Trez C, Pajak B, Brait M, Glaichenhaus N, Urbain J, Moser M, Lauvau G, Muraille E. J Immunol. 2005;175:839–846. doi: 10.4049/jimmunol.175.2.839. [DOI] [PubMed] [Google Scholar]
- 43.Ruckdeschel K, Pfaffinger G, Haase R, Sing A, Weighardt H, Hacker G, Holzmann B, Heesemann J. J Immunol. 2004;173:3320–3328. doi: 10.4049/jimmunol.173.5.3320. [DOI] [PubMed] [Google Scholar]
- 44.Han KJ, Su X, Xu LG, Bin LH, Zhang J, Shu HB. J Biol Chem. 2004;279:15652–15661. doi: 10.1074/jbc.M311629200. [DOI] [PubMed] [Google Scholar]
- 45.Chen M, Wang YH, Wang Y, Huang L, Sandoval H, Liu YJ, Wang J. Science. 2006;311:1160–1164. doi: 10.1126/science.1122545. [DOI] [PubMed] [Google Scholar]
- 46.Nowak AK, Lake RA, Marzo AL, Scott B, Heath WR, Collins EJ, Frelinger JA, Robinson BW. J Immunol. 2003;170:4905–4913. doi: 10.4049/jimmunol.170.10.4905. [DOI] [PubMed] [Google Scholar]
- 47.Thorley-Lawson DA. Nat Rev Immunol. 2001;1:75–82. doi: 10.1038/35095584. [DOI] [PubMed] [Google Scholar]
- 48.Gao SJ, Deng JH, Zhou FC, Thorley-Lawson DA. J Virol. 2003;77:9738–9749. doi: 10.1128/JVI.77.18.9738-9749.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hasan UA, Dollet S, Vlach J. Biochem Biophys Res Commun. 2004;321:124–131. doi: 10.1016/j.bbrc.2004.06.134. [DOI] [PubMed] [Google Scholar]
- 50.Sato N, Takahashi N, Suda K, Nakamura M, Yamaki M, Ninomiya T, Kobayashi Y, Takada H, Shibata K, Yamamoto M, et al. J Exp Med. 2004;200:601–611. doi: 10.1084/jem.20040689. [DOI] [PMC free article] [PubMed] [Google Scholar]