

Abstract
The mechanisms underlying the effects of psychostimulants in attention deficit hyperactivity disorder (ADHD) are not well understood, but indirect evidence implicates D2 dopamine receptors. Here we dissect the components of dopaminergic neurotransmission in the hyperactive mouse mutant coloboma to identify pre- and postsynaptic elements essential for the effects of amphetamine in these mice. Amphetamine treatment reduced locomotor activity in coloboma mice, but induced a robust increase in dopamine overflow suggesting that abnormal regulation of dopamine efflux does not account for the behavioral effect. However, the D2-like dopamine receptor antagonists haloperidol and raclopride, but not the D1-like dopamine receptor antagonist SCH23390, blocked the amphetamine-induced reduction in locomotor activity in coloboma mice, providing direct evidence that D2-like dopamine receptors mediate the effect of amphetamine in these mice. With the precedent established that it is possible to directly antagonize this response, this strategy should prove useful for identifying novel therapeutics in ADHD.
Keywords: hyperactivity, coloboma, dopamine receptor, amphetamine, ADHD, microdialysis, SNAP-25, raclopride, adenylate cyclase, catalepsy, haloperidol, SCH23390
Introduction
Attention deficit hyperactivity disorder (ADHD) is characterized by inattention, impulsivity and hyperactivity. A familiar feature of ADHD is the response to psychostimulants such as methylphenidate (Ritalin) and D-amphetamine (Adderall). Both compounds are indirect agonists that increase extracellular monoamine concentrations (Ferris et al., 1972, Heikkila et al., 1975). In ADHD patients, low doses of stimulants produce beneficial behavioral effects by reducing excess motor activity and enhancing concentration. Although the efficacy of psychostimulants in ADHD was recognized nearly 70 years ago (Bradley, 1937), the biological mechanisms are not understood.
Many theories, including several that focus on dopaminergic transmission, have emerged to explain the effects of psychostimulants. The rate-dependency hypothesis suggests that the effect of psychostimulants in hyperactive children is a phenomenon driven by behavioral state (Glick and Milloy, 1973, Sahakian and Robbins, 1977). The locomotor response to psychostimulants is an inverted U-shaped function: low doses increase locomotor activity and high doses induce focused behaviors (stereotypy) that compete with and consequently reduce locomotor activity. Drug-naïve ADHD patients are at the highest point on this inverted U-shaped curve. Accordingly, the psychostimulant-induced reduction in motor activity is ascribed to an increase in stereotyped behavior. Others propose that low doses of stimulants reduce synaptic catecholamine concentrations or the amplitude of impulse-induced dopamine release (McCracken, 1991, Seeman and Madras, 1998, Solanto, 1998). Low doses of direct-acting dopamine agonists preferentially interact with presynaptic receptors to inhibit dopamine release and therefore reduce locomotor activity (Skirboll et al., 1979). By extension, it is proposed that low doses of the indirect agonists methylphenidate and amphetamine reduce locomotor activity in ADHD patients through a similar mechanism of action. Others suggest that psychostimulant therapy compensates for insufficient dopamine transmission in the frontal cortex while reducing nigrostriatal overactivity (Castellanos, 1997). Despite these and other well-developed hypotheses, there is little empirical evidence to explain the therapeutic mechanism of psychostimulants in ADHD.
Animal models of ADHD provide an opportunity to explore pathogenic and therapeutic mechanisms. The coloboma mouse mutant is one such model. These mice exhibit hyperactivity, inattention and delayed developmental milestones (Hess et al., 1992, Heyser et al., 1995, Bruno et al., 2007) plus abnormal monoaminergic regulation (Raber et al., 1997, Jones and Hess, 2003). These defects are attributable, at least in part, to a reduction in the expression of SNAP-25 (Hess et al., 1996), a protein essential for neurotransmitter exocytosis (Söllner et al., 1993). The phenotype results from a hemizygous deletion mutation that includes the Snap25 gene (Hess et al., 1992); several independent research groups have determined that there is also a genetic association between the SNAP25 gene and ADHD in humans (Barr et al., 2000, Brophy et al., 2002, Mill et al., 2002, Kustanovich et al., 2003, Brookes et al., 2006). Indeed, a recent meta-analysis also supports this association (Faraone et al., 2005). Similar to its effects on ADHD patients, amphetamine reduces locomotor activity in coloboma mice (Hess et al., 1996). Here we dissect the components of dopaminergic neurotransmission in coloboma mice to identify elements essential for the effect of amphetamine.
MATERIALS AND METHODS
Mice
Coloboma (Cm/+) mice were bred and housed in group cages at the Johns Hopkins University vivarium. In all experiments, coloboma mice and wild type control littermates (4–9 months of age) were age- and sex-matched, although there is no significant difference in the locomotor activity between males and females. Experiments were performed in accordance with the Guide for the Care and Use of Laboratory Animals as adopted by the National Institutes of Health.
Drugs and receptor nomenclature
Drugs were purchased from Sigma (St. Louis, MO) and injected intraperitoneally in a volume of 10 ml/kg; apomorphine was injected subcutaneously. Haloperidol and spiperone interact with D2, D3 and D4 dopamine receptors; both are butyrophenone compounds. Raclopride is a D2/D3 dopamine receptor antagonist but is a substituted benzamide. Quinpirole is a D2/D3 dopamine receptor agonist. SCH23390 is selective for D1-like dopamine receptors (D1 and D5 dopamine receptors). Apomorphine is a nonselective dopamine agonist. Although there are at least 5 different molecularly defined dopamine receptors, pharmacological agents generally discriminate only between the broad classes of D1-like and D2-like dopamine receptors.
Locomotor activity
Control and coloboma mice were tested in 16 automated photocell activity cages (29 × 50 cm) equipped with 12 infrared beams arranged in a 4 × 8 grid (San Diego Instruments; San Diego, CA). Computer-recorded beam breaks were accumulated every 10 min for the duration of the test period with changes in beam status assessed 18 times/sec. Control and coloboma littermates were tested in parallel on the same test days and were habituated to the test cages for at least 4 hr prior to drug injection. Mice had access to food and water ad lib during the entire test. Drug was injected 1 hr after the start of the dark cycle and locomotor activity was recorded for 2 hr after drug injection.
Within each experiment, mice were tested in a repeated measures design. For all behavioral tests, the order of drug doses and vehicle was pseudorandom with each mouse receiving every dose only once within an experiment. Mice were given a 4-day drug holiday between challenges to avoid supersensitivity. For amphetamine tests, mice were challenged with 4 mg/kg amphetamine after the entire experiment was complete to ensure that supersensitivity had not developed; in all groups, there was no evidence for supersensitivity as assessed by both locomotor activity and stereotypy scores. The dose-responses were analyzed using repeated measures ANOVAs followed by post-hoc paired t tests where appropriate.
Although there is no clear formula for converting human doses to mouse, 4 mg/kg amphetamine was selected based on the pharmacokinetic species differences and dose range in humans. The typical therapeutic dose range for amphetamine in humans is 0.2 – 0.5 mg/kg and drug half-life is ~6–8 hrs (Brown et al., 1979, Greenhill et al., 2003). In contrast, the half-life of amphetamine in mice is ~20 – 50 min (Miller et al., 1971, Fuller et al., 1972, Riffee et al., 1978); as much as 20-fold faster clearance than in humans.
Stereotypy
During the 2 hr locomotor activity tests, mice were rated for stereotypy under red light illumination every 10 min for 30 sec. A 0–5 behavioral scale was used to score stereotypy: 0 = sleeping; 1 = awake, inactive; 2 = active or exploring; 3 = hyperactive; 4 = hyperactive with bursts of stereotypic behavior; and 5 = continuous persistent stereotypy. The first stereotypy rating was made 10 min after drug injection.
Microdialysis
Microdialysis was performed in alert, freely moving mice. Concentric microdialysis probes were constructed as previously described (Page et al., 2003). Recovery rates averaged 10 ± 0.5%.
Mice were anesthetized with tribromoethanol (Avertin), positioned in a stereotaxic apparatus (Stoelting, Wood Dale, IL), and a microdialysis probe was implanted in the striatum (+0.6 AP, +1.7 ML, 4.5 DV). After surgery, the probe was perfused continuously with artificial cerebrospinal fluid (aCSF: 147 mM NaCl, 3.5 mM KCl, 1.2 mM CaCl2, 1.2 mM MgCl2, 1 mM NaH2PO4, 25 mM NaHCO3, pH 7.0–7.4) at a flow rate of 1.5 μl/min. For conventional microdialysis, samples were collected 14–16 hr after surgery at 20 min intervals. Four consecutive samples were collected as baseline. Then 10 samples were collected after injection of amphetamine. For no net flux microdialysis, the probe was perfused with aCSF containing 250 μM ascorbic acid at a flow rate of 0.3 μl/min overnight. Flow rate was then increased to 0.6 μl/min and the probe perfused with the aforementioned perfusate plus different concentrations of dopamine (0, 2, 10 or 20 nM; Cin) in pseudo-random order. Following a 25 min equilibration period, 3 samples were collected (Cout). The perfusate dopamine concentration was then switched and the process repeated until all dopamine concentrations were tested. After completion of the experiment, brains were removed and probe location was confirmed; only animals with probes in the striatum were included.
Samples were analyzed by HPLC (MD-150 column, 150 mm length; 3 mm I.D.; ESA, Chelmsford, MA) equipped with a 5014B coulometric microdialysis cell. The mobile phase consisted of 1.7 mM 1-octanesulfonic acid sodium salt, 25 μM EDTA, 75 mM NaH2PO4 and 8% acetonitrile (pH 2.9) at a flow rate of 0.6 ml/min. Neurotransmitters were identified by matching retention time to that of known standards. For conventional microdialysis, values were expressed as ng/ml after adjustment for recovery rate of the microdialysis probe. For no net flux microdialysis, data were subjected to a linear regression analysis plotting the gain or loss of dopamine from the perfusate (Cin − Cout) versus Cin. The x-intercept (C in − C out = 0) provides an unbiased estimate of extracellular dopamine concentration; the slope represents the extraction fraction, which is an indirect measure of dopamine uptake (Smith and Justice, 1994, Shippenberg et al., 1999). Data were analyzed using two-factor ANOVA or Student’s t test, where appropriate.
Catalepsy
Catalepsy was tested by gently placing both front paws of a mouse on a horizontal bar (0.5 cm diameter) 4 cm above the cage (28 ×17×13 cm) floor. Time was measured from the placement of the forepaws until both forepaws were removed from the bar or the mouse moved both forepaws to left or right on the bar. Cut-off time was 180 sec for each trial. Each mouse was tested in 3 consecutive trials per session unless one of the trials reached the cutoff time; in this case no further trials were run. The average of 3 trials or 180 sec, if cutoff was achieved, was used to calculate catalepsy. Catalepsy was tested in 20 min intervals for 2 hr starting 10 min after injection and analyzed using repeated measures ANOVAs.
Adenylate cyclase activity
Striata from individual mice were homogenized and centrifuged in buffer containing 10 mM imidazole (pH 7.4), 2 mM EDTA and 10% sucrose. The pellet was resuspended in buffer containing 10 mM imidazole (pH 7.4), 2 mM EDTA, 0.5 mM DTT and 10% sucrose. The reaction was performed (± quinpirole) in a 100 μl volume containing 80 mM Tris-HCl (pH 7.4), 150 mM NaCl, 4 mM MgCl2, 2 mM EDTA, 0.5 mM IBMX, 2 mM ATP, 20 mM phosphocreatine, 5 U of creatine phosphokinase, 1 mg BSA, 100 μM GTP, and 0.1 μM forskolin at 37°C for 7 min. About 15 μg membrane protein was added to each tube to start the reaction with triplicates for all conditions. The reaction was terminated by boiling the mixture for 3 min. After centrifuging the reaction mixture at 20,000 × g for 20 min, cAMP accumulation was determined by radioimmunoassay (Amersham Biosciences, Piscataway, NJ). Protein concentration was determined using a Bio-Rad protein assay (Bio-Rad, Hercules, CA) and adenylate cyclase activity was expressed as pmol cAMP/mg protein/min. Data were analyzed using two-factor ANOVA with repeated measures.
RESULTS
D2-like dopamine receptor antagonism blocks the amphetamine-induced reduction in locomotor activity
Consistent with previous results (Hess et al., 1996), 4 mg/kg amphetamine significantly reduced the locomotor hyperactivity exhibited by coloboma mice (p < 0.05; Fig. 1A). Stereotypic behavior was not observed at this dose of amphetamine (data not shown), suggesting that the reduction in locomotor activity in coloboma mice was not attributable to an increase in competing focused repetitive behaviors. To determine if the response to amphetamine is dependent on a specific dopamine receptor subtype, mice were challenged with amphetamine in the presence of subtype-selective antagonists. The D2-like dopamine receptor antagonist haloperidol blocked the amphetamine-induced reduction in locomotor activity in coloboma mice compared to amphetamine alone (Fig. 1A). In coloboma mice, 0.05 mg/kg haloperidol caused a significant 44% attenuation in amphetamine-mediated locomotor activity (p < 0.05). By itself, this low dose of haloperidol did not affect locomotor activity. Even 0.3 mg/kg haloperidol, which caused a marked reduction in baseline locomotor activity (p < 0.05), blocked amphetamine-attenuated locomotor activity in coloboma mice (p = 0.05).
Figure 1.
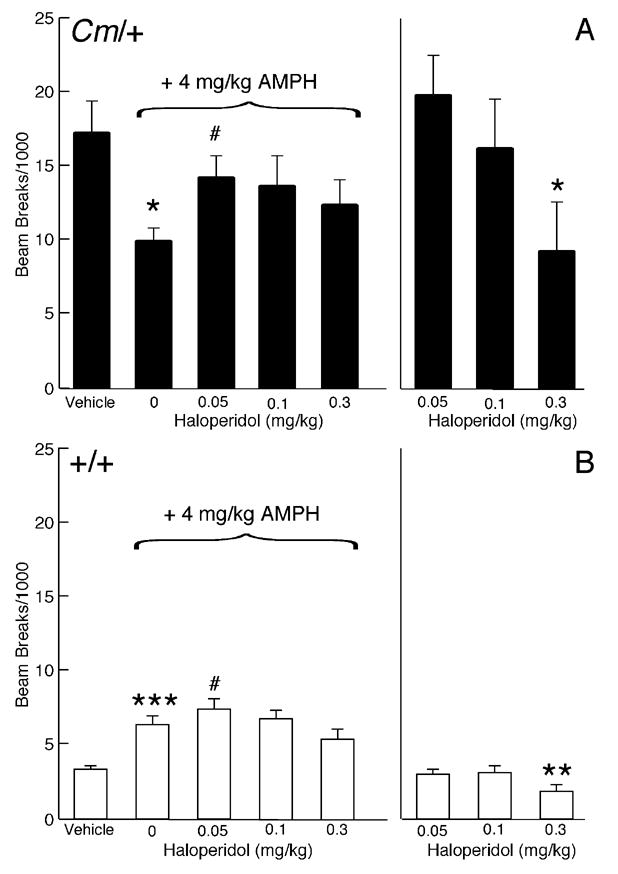
Effect of the D2-like dopamine receptor antagonist haloperidol on amphetamine-mediated locomotor activity. Coloboma (A) and control mice (B) were treated with saline or 4 mg/kg amphetamine and challenged with haloperidol. Compared to vehicle treatment, amphetamine significantly increased locomotor activity in control mice (***p < 0.001, paired Student’s t test) but significantly reduced locomotor activity in coloboma mice (*p<0.05, paired Student’s t test). Two-factor ANOVA with repeated measures revealed a significant effect of genotype (F1,14 = 17.77, p < 0.001) and dose of haloperidol (F3,42 = 4.00, p < 0.05) on amphetamine-mediated locomotor activity. Post hoc analyses using paired Student’s t tests demonstrated a significant increase in amphetamine-mediated locomotor activity after treatment with 0.05 mg/kg haloperidol in both control and coloboma mice (# p < 0.05). Treatment with haloperidol alone produced a significant genotype × dose interaction effect (two-factor ANOVA with repeated measures; F2,28 = 3.90, p < 0.05). Post hoc analyses (paired Student’s t tests) demonstrated a significant reduction in the locomotor activity of both control and coloboma mice after treatment with 0.3 mg/kg haloperidol alone compared to vehicle (*p < 0.05; **p < 0.01). Data are presented as beam breaks accumulated in 1 hr following drug treatment and are expressed as mean ± SEM (n = 8/genotype/dose).
In contrast to coloboma mice, amphetamine induced a two-fold increase in locomotor activity in control mice (p < 0.001). Surprisingly, 0.05 mg/kg haloperidol also produced a small, but significant increase (16%) in amphetamine-stimulated motor activity in wild type mice (p < 0.05; Fig. 1B). However, amphetamine-stimulated locomotor activity was significantly reduced with 0.3 mg/kg haloperidol (p < 0.001). Treatment with haloperidol alone dose-dependently reduced locomotor activity in both control and coloboma mice, although there was a significant genotype × dose interaction effect (p < 0.05) suggesting that there was a differential response to haloperidol between genotypes. Stereotypy was not observed with any drug treatment in either control or coloboma mice (data not shown).
To determine if the response to haloperidol, which is a butyrophenone compound, was selective for D2-like dopamine receptors or if the response was determined by the chemical and pharmacological profile of haloperidol per se, the specificity of the effect for D2-like dopamine receptors was tested using the substituted benzamide raclopride, a D2/D3 dopamine receptor antagonist. Similar to the effects of haloperidol, 0.3 mg/kg raclopride caused a significant 54% attenuation in amphetamine-mediated locomotor activity in coloboma mice (p < 0.05; Fig. 2A). By itself, raclopride at all doses significantly reduced coloboma mouse baseline activity (p < 0.005).
Figure 2.
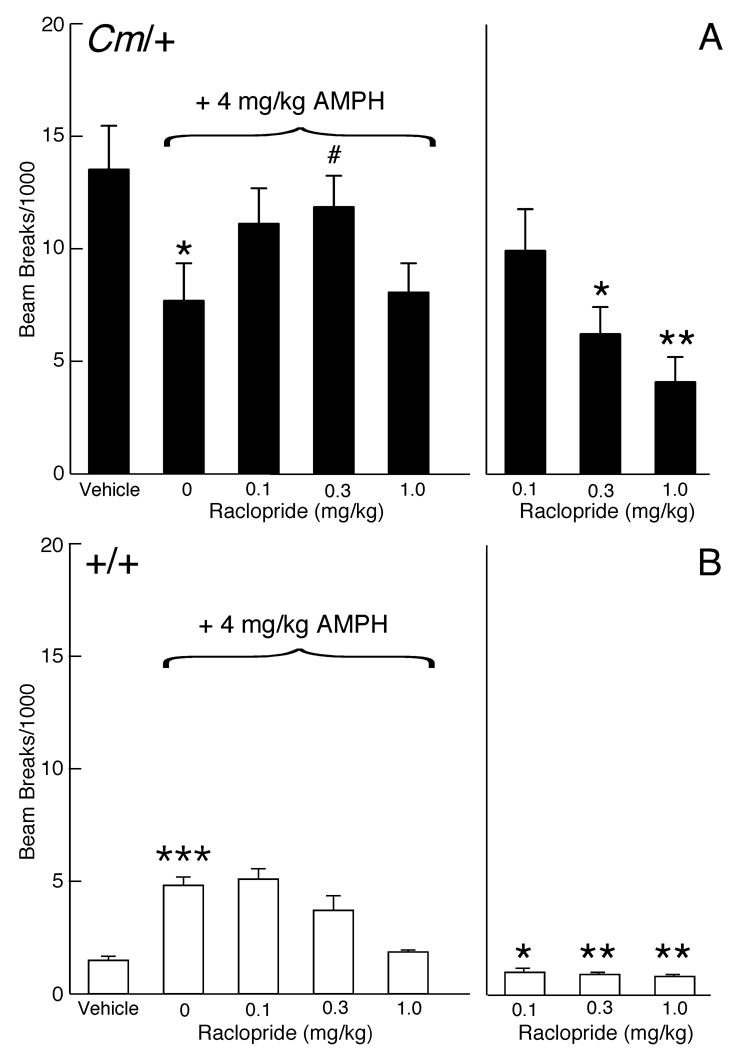
Effect of the D2/D3 dopamine receptor antagonist raclopride on amphetamine-mediated locomotor activity. Coloboma (A) and control mice (B) were treated with saline or 4 mg/kg amphetamine and challenged with raclopride. Compared to vehicle treatment, amphetamine significantly increased locomotor activity in control mice (***p < 0.001, paired Student’s t test) but significantly reduced locomotor activity in coloboma mice (*p<0.05, paired Student’s t test). Two-factor ANOVA with repeated measures revealed a significant genotype × dose interaction effect (F3,42 = 3.28, p < 0.05) on amphetamine-mediated locomotor activity. Post hoc analyses using paired Student’s t tests demonstrated a significant increase in amphetamine-mediated locomotor activity after treatment with 0.3 mg/kg raclopride in coloboma mice (# p < 0.05), but a trend (p = 0.06) for a reduction in the motor activity of normal mice at the same dose. Treatment with raclopride alone produced a significant genotype × dose interaction effect (two-factor ANOVA with repeated measures; F2,28 = 5.34, p < 0.05). Compared to vehicle treatment (paired Student’s t tests), all doses of raclopride significantly reduced locomotor activity in control mice (*p < 0.05; **p < 0.01); 0.3 and 1 mg/kg significantly reduced the locomotor activity of coloboma mice with a trend toward reduction with 0.1 mg/kg (p = 0.06). Data are presented as beam breaks accumulated in 1 hr following drug treatment and are expressed as mean ± SEM (n = 8/genotype/dose).
In contrast to coloboma mice, raclopride cause a dose-dependent reduction in the amphetamine-induced increase in locomotor activity in control mice (Fig. 2B). Treatment with raclopride alone dose-dependently reduced locomotor activity in both control and coloboma mice.
To determine if D1-like dopamine receptors also mediate the amphetamine-induced reduction in locomotor activity observed in coloboma mice, SCH23390, a D1/D5 dopamine receptor antagonist, was used in a similar series of amphetamine challenge experiments (Fig. 3). SCH23390 dose-dependently decreased amphetamine-mediated locomotor activity in both control and coloboma mice (p < 0.0001). The SCH23390-induced reduction in amphetamine-mediated locomotor activity occurred in parallel in control and coloboma mice whereby a significant reduction in amphetamine-mediated locomotor activity was observed with 0.2 mg/kg SCH23390 treatment in both genotypes (p < 0.005). Administration of 0.2 mg/kg SCH23390 alone also significantly reduced basal locomotor activity in both genotypes (p < 0.01), while 0.05 mg/kg SCH23390 alone significantly reduced locomotor activity in only coloboma mice (p < 0.05). At lower doses (0.0125 mg/kg or 0.00625 mg/kg, not shown), SCH23390 had no effect on basal or amphetamine-mediated locomotor activity.
Figure 3.
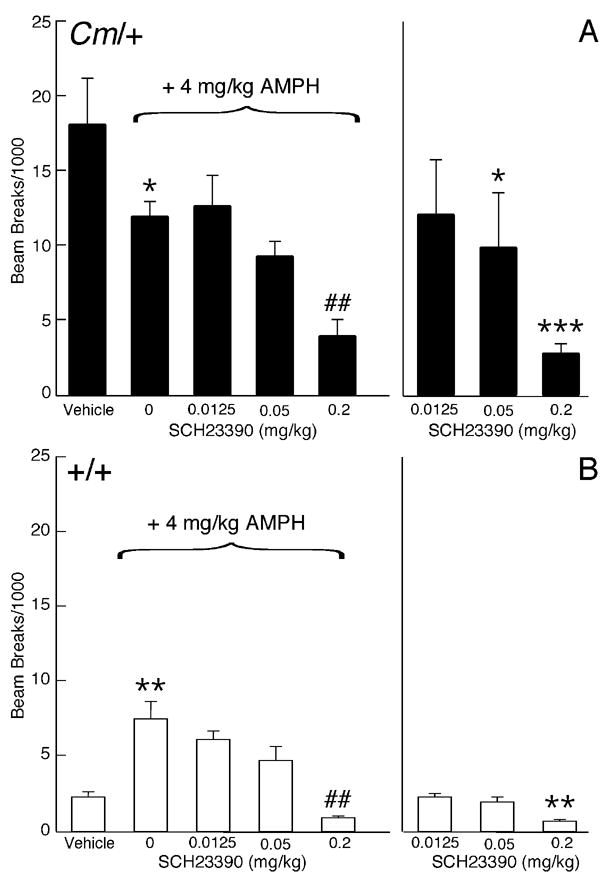
Effect of the D1/D5 dopamine receptor antagonist SCH23390 on amphetamine-mediated locomotor activity. Coloboma (A) and control mice (B) were treated with saline or 4 mg/kg amphetamine and challenged with SCH23390. Compared to vehicle treatment, amphetamine significantly increased locomotor activity in control mice (**p < 0.01, paired Student’s t test) but significantly reduced locomotor activity in coloboma mice (*p < 0.05, paired Student’s t test). Two-factor ANOVA with repeated measures revealed a significant effect of genotype (F1,12 = 25.22, p < 0.001) and dose of SCH23390 (F3,36 = 18.41, p < 0.0001) on amphetamine-mediated locomotor activity. 0.2 mg/kg SCH23390 resulted in a significant reduction in amphetamine-mediated behavior in both control and coloboma mice (##p < 0.01). Two-factor ANOVA with repeated measures for treatment with SCH23390 alone revealed significant main effects of genotype (F1,12 = 10.09, p < 0.01) and dose (F2,24 = 4.45, p < 0.05). Compared to vehicle treatment (paired Student’s t tests), 0.2 mg/kg SCH23390 significantly reduced locomotor activity in control and coloboma mice (**p < 0.01; ***p < 0.001); 0.05 mg/kg SCH23390 significantly reduced the locomotor activity of coloboma mice (*p < 0.05). Data represent total beam breaks in 1 hr after drug administration and are expressed as mean ± SEM (n=7/genotype/dose).
Amphetamine is a nonselective indirect agonist that increases the extracellular concentration of norepinephrine and serotonin, in addition to dopamine. Therefore, apomorphine, a direct-acting mixed D1/D2 dopamine receptor agonist was tested in a similar series of experiments to determine if selective activation of only dopamine receptors was sufficient to reproduce the effect. Responses to apomorphine are biphasic. Low doses of apomorphine act selectively at dopamine autoreceptors to reduce locomotor activity; we have previously demonstrated that the apomorphine-mediated autoreceptor response is normal in coloboma mice (Jones et al., 2001a). At higher doses, which were used in this experiment, apomorphine increases locomotor activity in normal rodents through its actions at postsynaptic dopamine receptors. As expected, 1 mg/kg apomorphine increased locomotor activity in control mice. At the same dose, apomorphine reduced locomotor activity in coloboma mice (p < 0.05), similar to the effects of amphetamine. Stereotypy was not observed in either control or coloboma mice (data not shown). Consistent with the amphetamine challenge experiments, 0.05 mg/kg haloperidol blocked the apomorphine-induced reduction in locomotor activity in coloboma mice (p < 0.05), while 0.0125 mg/kg SCH23390 had little effect on the apomorphine-induced reduction in locomotor activity (Fig. 4A). Neither haloperidol nor SCH23390 treatment significantly affected the apomorphine-induced increase in control mouse activity (Fig. 4B), although there was a trend toward a reduction in activity for both drugs.
Figure 4.
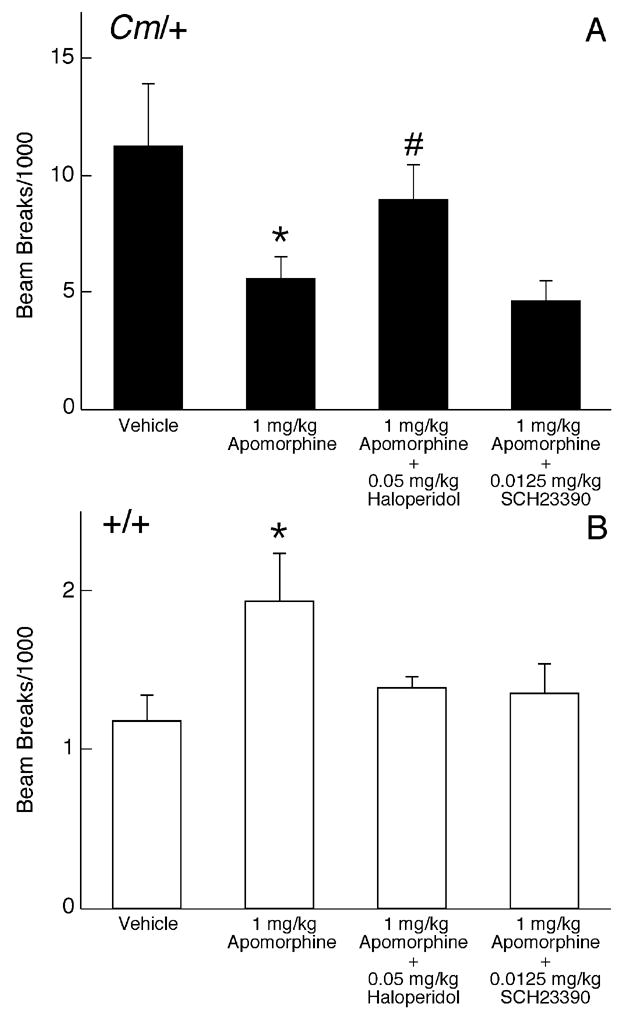
Effect of D1- and D2-like dopamine receptor antagonists on apomorphine-mediated locomotor activity. Coloboma (A) and control mice (B) were treated with saline or 1 mg/kg apomorphine and challenged with SCH23390 or haloperidol. Two-factor ANOVA with repeated measures revealed a significant genotype × treatment interaction effect (F3,42 = 4.95, p < 0.005). Post hoc analyses demonstrated that, compared to vehicle treatment, apomorphine significantly reduced locomotor activity in coloboma mice, but increased locomotor activity in control mice (*p < 0.05, paired Student’s t test). A significant increase in apomorphine-mediated locomotor activity was observed after treatment with 0.05 mg/kg haloperidol in coloboma mice (# p < 0.05). Data represent total beam breaks in 1 hr after drug administration and are expressed as mean ± SEM (n=8/genotype/dose).
Amphetamine-induced dopamine efflux is increased in the striatum of coloboma mice
Because the locomotor hyperactivity in coloboma mice results, at least in part, from a deficiency in SNAP-25, it is likely that presynaptic abnormalities contribute to the hyperactivity and abnormal response to amphetamine. Therefore, striatal dopamine efflux was assessed in alert freely moving mice. Basal extracellular dopamine concentrations were assessed using the no net flux method of microdialysis, which provides an unbiased estimate of transmitter concentration. Despite the reduction in SNAP-25 expression, the basal extracellular concentration of dopamine in coloboma mice exceeded that of control mice by more than 80% (Fig. 5A; p < 0.05). In contrast, the extraction fraction, which is an indirect measure of dopamine uptake (Smith and Justice, 1994), was comparable in control and coloboma mice (Fig. 5B) suggesting that the increase in the basal extracellular dopamine concentration in coloboma mice is attributable to abnormalities in transmitter release. Consistent with the no net flux method, conventional dialysis also demonstrated a significant increase (p < 0.05) in basal extracellular dopamine concentration in coloboma mice (data not shown). This increase in baseline extracellular dopamine in coloboma mice was not obvious with previous in vitro assays (Raber et al., 1997, Jones and Hess, 2003), where steady-state basal extracellular concentrations are not readily assessed.
Figure 5.
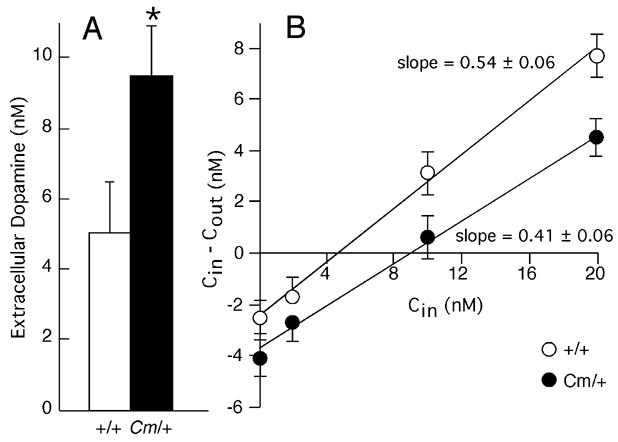
Basal extracellular dopamine concentrations in striatum. A, Striatal dopamine concentrations were measured by no net flux microdialysis in alert freely moving coloboma (n = 8) and control mice (n = 9). Values are expressed as mean ± SEM. * p < 0.05, Student’s t test. B. The extraction fraction, an indirect measure of dopamine reuptake, was determined by calculating the slope of the linear regression analysis of the perfused concentration of dopamine (Cin) versus the perfused dopamine concentration minus the dialysate dopamine concentration (Cin − Cout). Values are expressed as mean ± SEM. No significant difference was observed between the extraction fractions from control and coloboma mice.
Conventional microdialysis was used to assess the effects of amphetamine on dopamine efflux. Although amphetamine reduced locomotor activity in coloboma mice, a dose-dependent increase in striatal dopamine efflux was observed in response to amphetamine challenge in both control and coloboma mice (Fig. 6A–C). Amphetamine-induced dopamine efflux was significantly higher in coloboma mice (p < 0.001) even after normalizing for differences in basal extracellular dopamine concentrations (Fig. 6E).
Figure 6.
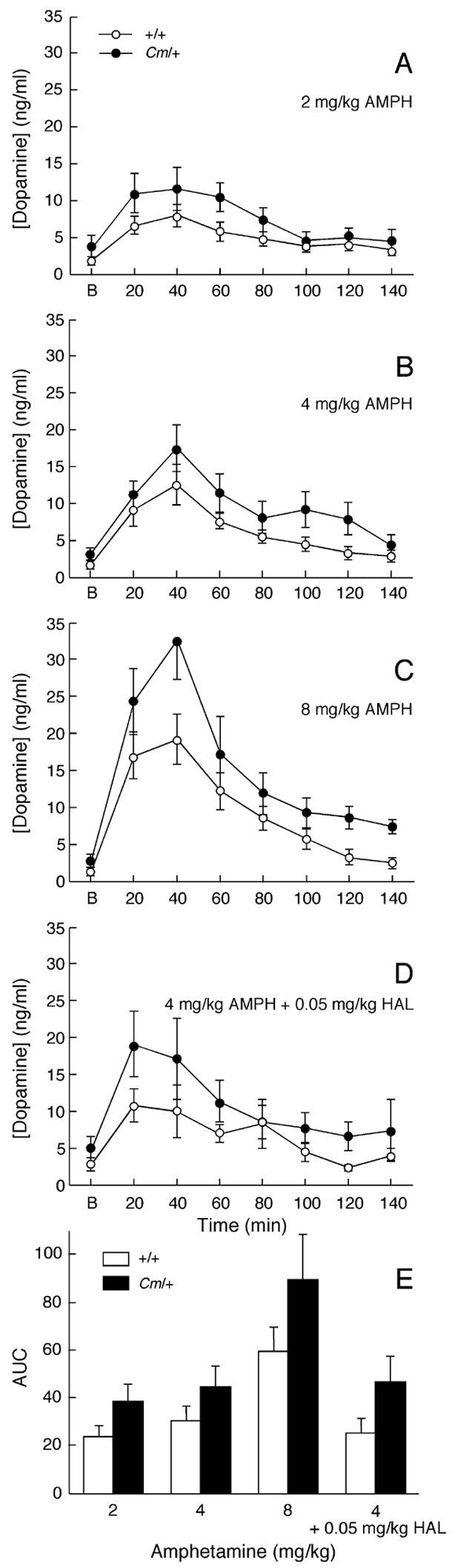
Amphetamine-induced dopamine efflux in striatum. A–D, Mice (n = 6–10/genotype/dose) were injected with 2 mg/kg (A), 4 mg/kg (B), 8 mg/kg (C) amphetamine or 4 mg/kg amphetamine plus 0.05 mg/kg haloperidol (D) and microdialysis samples collected in 20 min intervals. Basal values (‘B’) were calculated by averaging the dopamine concentrations from the 4 samples prior to amphetamine injection. Data are expressed as means ± SEM. E, The absolute amphetamine-induced increase in extracellular dopamine efflux in experiments A–D was determined by calculating the area under the curve (A.U.C.) after subtracting basal extracellular dopamine concentrations. Two-factor ANOVA revealed main effects of genotype (F1,53 = 7.58, p < 0.01) and treatment (F3,53 = 6.85, p < 0.001).
To determine if the effect of haloperidol on amphetamine-inhibited locomotor activity in coloboma mice was mediated presynaptically, amphetamine-induced dopamine efflux was assessed in the presence of the lowest dose of haloperidol that blocked the amphetamine effect in coloboma mice. Haloperidol (0.05 mg/kg) had no effect on amphetamine-induced dopamine efflux in either control or coloboma mice (Fig. 6D & E) nor did this very low dose of haloperidol alone significantly affect basal dopamine overflow (two-factor ANOVA with repeated measures, effect of drug, F1,14 = 1.626, NS).
D2-like dopamine receptor-mediated responses
A reduction in dopamine efflux, which might be expected in light of the reduction in SNAP-25 expression in coloboma mice, does not explain the reduction in locomotor activity observed in response to amphetamine. Further, the results presented in Figures 1 and 2 suggest that D2 dopamine receptors mediate the amphetamine-induced reduction in locomotor activity in coloboma mice. It is well established that persistent changes in extracellular dopamine alter dopamine receptor function. We have previously demonstrated that D2-like dopamine receptor densities and affinities are normal in coloboma mice (Jones et al., 2001b). This is consistent with studies of adult rodents subjected to monoaminergic insult early in development, including neonatal 6-hydroxydopamine-lesioned rats, mice lacking dopamine due to tyrosine hydroxylase deficiency in dopaminergic neurons (DA −/− mice) and norepinephrine transporter-deficient mice, demonstrating that receptor densities and affinities are normal (Breese et al., 1985, Kim et al., 2000, Xu et al., 2000). In contrast, behavioral responses to dopaminergic challenge are enhanced in these models, suggesting that there are functional changes that result in receptor supersensitivity (for review see Kostrzewa, 1995). Therefore, behavioral and biochemical measures were used to examine D2-like dopamine receptor function in coloboma mice.
Mice were challenged with the D2-like dopamine receptor antagonist spiperone, which induces akinesia or catalepsy. Because catalepsy tests time spent in an unnatural position, results from control and coloboma mice are quantitatively similar allowing the direct comparison of results from both genotypes. As shown in Figure 7A, coloboma mice were relatively insensitive to the cataleptogenic effects of the D2 dopamine receptor antagonist spiperone and required higher doses of spiperone to induce catalepsy than control mice (p < 0.05) suggesting that D2-like dopamine receptors are supersensitive in coloboma mice.
Figure 7.
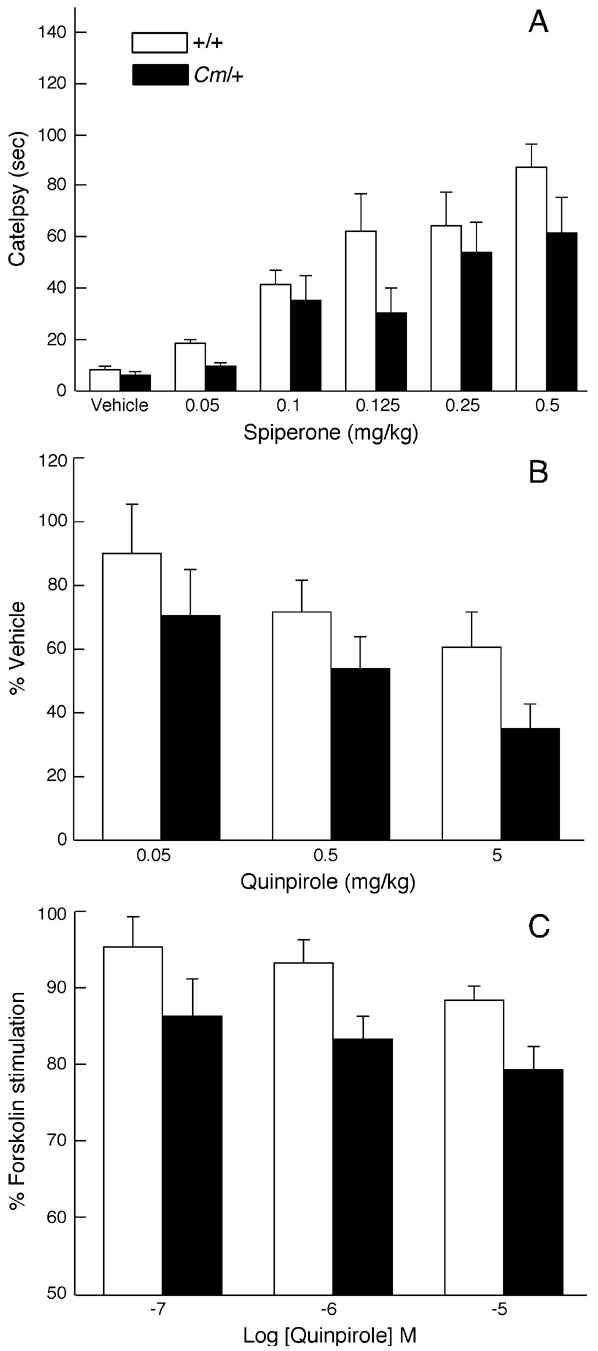
D2 dopamine receptor mediated responses. A, Control and coloboma mice (n = 11–12/genotype/dose) were injected with the D2-like dopamine receptor antagonist spiperone. Catalepsy was assessed every 20 min for 2 hr. Data represent average catalepsy time from the 6 test intervals and are expressed as mean ± SEM. Two-factor ANOVA with repeated measures revealed main effects of genotype (F1,18 = 5.28, p < 0.05) and dose (F5,90 = 17.37, p < 0.0001). B, Control and coloboma mice (n = 8/genotype/dose) were challenged with the D2/D3 dopamine receptor-selective agonist quinpirole and locomotor activity was assessed. Data are presented as percent of vehicle treatment to normalize for gross differences in baseline locomotor activity and are expressed as mean ± SEM. Two-factor ANOVA with repeated measures revealed main effects of genotype (F1,14 = 6.24, p < 0.05) and dose (F4,56 = 4.18, p < 0.01). C, Quinpirole-mediated inhibition of 0.1 μM forskolin-stimulated adenylate cyclase activity. Data are expressed as a percent of forskolin-induced adenylate cyclase activity without agonist. Data represent mean ± SEM (n = 8/genotype). Two-factor ANOVA with repeated measures revealed main effects of genotype (F1,14 = 5.65, p < 0.05) and dose (F2,28 = 4.73, p < 0.05).
Behavioral responses to agonist challenge were assessed using quinpirole, a D2/D3 dopamine receptor agonist. Quinpirole reduced locomotor activity in coloboma and control mice (Fig. 7B). However, coloboma mice were more sensitive to the effects of quinpirole: lower doses caused a greater reduction in locomotor activity in coloboma mice than control mice (p < 0.05), again reflecting supersensitivity. However, because baseline locomotor activity is much greater in coloboma mice than control mice, it is possible that the differences observed in quinpirole responses were simply a result of ceiling and floor effects, rather than a true reflection of receptor sensitivity.
To investigate functional changes in signal transduction, dopamine receptor-mediated adenylate cyclase activity was assessed. No difference in basal adenylate cyclase activity was observed between control (4.7 ± 0.7 pmol/min/mg) and coloboma mice (4.1 ± 0.9 pmol/min/mg). Forskolin-stimulated adenylate cyclase activity was also comparable (control mice, 260 ± 35 pmol/min/mg; coloboma mice, 299 ± 36 pmol/min/mg). The D2/D3 dopamine receptor agonist quinpirole dose-dependently inhibited forskolin-stimulated adenylate cyclase activity in both control and coloboma mice (Fig. 7C) but the inhibition was significantly greater in coloboma mice than control mice (p < 0.05). This suggests an increase in the functional sensitivity of D2-like dopamine receptors in accord with the behavioral drug challenges.
DISCUSSION
The indirect and direct dopamine agonists amphetamine and apomorphine reduced the hyperactivity exhibited by coloboma mice. This reduction in activity was not accompanied by an increase in stereotyped behavior, eliminating rate dependency as an explanation for the effect. An increase in basal extracellular dopamine was observed in coloboma mice, which may, in part, account for the hyperactivity inasmuch as increases in dopaminergic tone also occur in other models of hyperactivity (Giros et al., 1996, Zhuang et al., 2001). Despite causing a reduction in locomotor activity, amphetamine induced a dose-dependent increase in striatal dopamine efflux in coloboma mice that exceeded control mice. Thus, an abnormal reduction in neurotransmitter efflux, which might be expected in light of the reduction in SNAP-25 expression in coloboma mice, does not explain the reduction in locomotor activity induced by amphetamine. Instead, it is likely that abnormal receptor-mediated responses drive the response.
The D2-like dopamine receptor antagonists haloperidol and raclopride, but not the D1/D5 dopamine receptor antagonist SCH23390, blocked the amphetamine-induced reduction in locomotor activity in coloboma mice. In fact, haloperidol and raclopride restored amphetamine-attenuated locomotor activity to near baseline levels. Many theories and experiments in both humans and animals indirectly implicate D2 dopamine receptors in the effect of psychostimulants in ADHD (Seeman and Madras, 1998, Solanto, 1998, Ilgin et al., 2001, Zhuang et al., 2001, Viggiano et al., 2003, Lou et al., 2004). The results presented here provide the first direct evidence demonstrating that activation of D2-like, but not D1-like, dopamine receptors is necessary for the effect.
In control mice, amphetamine-induced locomotor activity was significantly increased after treatment with low doses of haloperidol, similar to previous reports (Salmi et al., 1998, O’Neill and Shaw, 1999), but this effect was not observed with raclopride. Haloperidol binds to all D2-like dopamine receptor subtypes (D2, D3 and D4) whereas raclopride interacts with only D2 and D3 dopamine receptor subtypes. Because blocking D2-like dopamine receptors with haloperidol increased the stimulatory effects of amphetamine in normal mice, it follows that there is a minor inhibitory element to the amphetamine response that is mediated by D2-like dopamine receptors. That raclopride did not have similar effects suggests D4 dopamine receptors mediate this inhibitory component of the amphetamine response. Indeed, mice lacking the D4 dopamine receptor subtype exhibit enhanced responses to amphetamine (Kruzich et al., 2004).
D2-like dopamine receptors were functionally supersensitive in coloboma mice as demonstrated across behavioral and second messenger responses. D2-like dopamine receptors are located pre- and postsynaptically. Autoreceptors on dopaminergic neurons inhibit firing activity and neurotransmitter release. One hypothesis explaining the effects of psychostimulants suggests that the dopamine efflux caused by low doses of psychostimulants acts preferentially at dopamine autoreceptors to attenuate dopaminergic transmission (Seeman and Madras, 1998, Solanto, 1998). However, results of single unit recording performed presynaptically in the substantia nigra pars compacta and postsynaptically in either the caudate or globus pallidus from normal animals demonstrate that neither methylphenidate nor amphetamine drives autoreceptor-selective stimulation, regardless of dose (Piercey et al., 1996, Ruskin et al., 2001). Further, although a large body of evidence supports a psychostimulant-induced increase in dopamine efflux in humans and animals (Imperato and Di Chiara, 1984, Kuczenski and Segal, 1989, Robinson and Camp, 1990, Volkow et al., 2001), there is little to suggest that low doses reduce overflow.
It is not likely that autoreceptors govern the amphetamine-induced reduction in locomotor activity in coloboma mice for several reasons. First, the locomotor response to an autoreceptor-selective dose of apomorphine is comparable in control and coloboma mice, suggesting that autoreceptor responses are normosensitive in these mutants (Jones et al., 2001a). Second, a robust amphetamine-induced increase in extracellular dopamine was observed in coloboma mice. Third, although the D2-like dopamine receptor antagonist haloperidol blocked the motor response to amphetamine in coloboma mice, the same dose had no obvious effect on amphetamine-induced dopamine efflux.
It is more likely that postsynaptic dopamine receptors regulate the effect of amphetamine. Stimulation of postsynaptic D2-like dopamine receptors generally reduces the excitability of medium spiny neurons in the striatum by blunting glutamatergic responses either directly at D2-like dopamine receptors expressed on medium spiny neurons or through heteroreceptors located on corticostriatal terminals that inhibit glutamate release (Brown and Arbuthnott, 1983, Cepeda et al., 1993, O’Donnell and Grace, 1994, West and Grace, 2002, Bamford et al., 2004). In contrast, most in vivo experiments and many in vitro studies suggest that D1-like dopamine receptors facilitate medium spiny neuron activity (Hu and Wang, 1988, Cepeda et al., 1993, Kiyatkin and Rebec, 1996, Gonon, 1997). The modest increase in extracellular dopamine in drug-naïve coloboma mice likely overstimulates both D1- and D2-like dopamine receptors, producing hyperactivity. Further increasing extracellular dopamine via amphetamine may shift the response to favor the supersensitive D2-like dopamine receptor response resulting in a net dampening of medium spiny neuron signaling and a concomitant reduction in activity. However, this explanation is probably too simplistic because other factors may contribute to the effect, including nigrostriatal firing patterns, and dysregulation of other transmitters.
Of course, these data do not exclude the possibility that other catecholamines contribute to the effect of amphetamine. Amphetamine elicits increases in synaptic norepinephrine concentrations, in addition to increasing synaptic DA. In normal animals, depletion of central norepinephrine or blockade of postsynaptic adrenergic receptors prevents the locomotor increase induced by amphetamine (Ogren et al., 1983, Archer et al., 1986, Darracq et al., 1998) suggesting a significant role for norepinephrine in the response. Further, we have found that norepinephrine contributes to the expression of locomotor hyperactivity and inattention in drug-naive coloboma mice via □2C-adrenergic receptors (Jones and Hess, 2003, Bruno and Hess, 2006, Bruno et al., 2007) suggesting that adrenergic receptors may also contribute to psychostimulant responses. Experiments similar in design to those presented here will directly test the involvement of adrenergic receptors in the response to amphetamine.
Amphetamine, but not methylphenidate, reduces locomotor activity in coloboma mice (Hess et al., 1996). Most ADHD patients respond to both amphetamine and methylphenidate (Efron et al 1997; Elia et al 1991). There are a couple possible explanations for the lack of response to methylphenidate in coloboma mice. First, there are important species differences in the neurochemical effects of psychostimulants. At clinically efficacious doses, amphetamine and methylphenidate induce comparable increases in extracellular dopamine in humans as assessed by positron emission tomography (PET) imaging of psychostimulant-induced displacement of [11C]raclopride binding (Breier et al., 1997, Volkow et al., 2001, Cardenas et al., 2004, Oswald et al., 2005, Udo de Haes et al., 2005). In contrast, even low doses of amphetamine elicit much larger increases in synaptic dopamine concentrations than extremely high doses of methylphenidate in rat (Kuczenski and Segal, 1997); we have observed similar effects in mice (unpublished observation). It is possible that the modest increase in extracellular dopamine elicited by even the highest doses of methylphenidate in rodents does not adequately stimulate the D2-like dopamine receptors given that activation of D2-like dopamine receptors requires relatively high concentrations of dopamine (Zheng et al., 1999). This hypothesis suggests that the differential response to methylphenidate and amphetamine may depend on level of D2-like dopamine receptor sensitivity or gain. Moderate D2-like dopamine receptor supersensitivity, as observed in coloboma mice, would require high concentrations of dopamine to inhibit motor activity whereas higher receptor supersensitivity would require less dopamine, such as that provided by methylphenidate, to produce the effect.
Alternatively, the coloboma mouse may model a subset of ADHD patients. Although many consider methylphenidate and amphetamine interchangeable for the treatment of ADHD, only approximately half of affected children respond to both methylphenidate and amphetamine with equal efficacy (Arnold et al., 1978, Elia et al., 1991, Efron et al., 1997). While the majority of children benefit from methylphenidate treatment, a small subset of children respond only to amphetamine, suggesting that underlying neurochemical differences define the response to psychostimulants. Since the etiology of hyperactivity is unlikely to be ascribed to a single molecular, neuroanatomical or neurochemical substrate, no single model is likely to account for the pathogenesis and therapeutic efficacy in all ADHD patients. Because D2-like dopamine receptors are generally viewed as negative regulators of activity, D2-like dopamine receptors were implicated in the efficacy of psychostimulants in ADHD for decades. Here, we provide direct evidence in an animal model and demonstrate that it is possible to directly antagonize the effect through D2-like dopamine receptors providing access to the mechanisms underlying the efficacy of psychostimulants in ADHD and targets for novel therapeutics.
Acknowledgments
We thank Drs. Michelle D. Jones and Irwin Lucki for consultation on the microdialysis experiments, Dr. Kristy J. Bruno for helpful discussion and Catherine Weisz for technical support. Supported by U.S. Public Health Service Grant R01 NS34845.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Archer T, Fredriksson A, Jonsson G, Lewander T, Mohammed AK, Ross SB, Soderberg U. Central noradrenaline depletion antagonizes aspects of d-amphetamine-induced hyperactivity in the rat. Psychopharmacology. 1986;88:141–146. doi: 10.1007/BF00652230. [DOI] [PubMed] [Google Scholar]
- Arnold LE, Christopher J, Huestis R, Smeltzer DJ. Methylphenidate vs dextroamphetamine vs caffeine in minimal brain dysfunction. Arch Gen Psychiatry. 1978;35:463–473. doi: 10.1001/archpsyc.1978.01770280073008. [DOI] [PubMed] [Google Scholar]
- Bamford NS, Zhang H, Schmitz Y, Wu NP, Cepeda C, Levine MS, Schmauss C, Zakharenko SS, Zablow L, Sulzer D. Heterosynaptic dopamine neurotransmission selects sets of corticostriatal terminals. Neuron. 2004;42:653–663. doi: 10.1016/s0896-6273(04)00265-x. [DOI] [PubMed] [Google Scholar]
- Barr CL, Feng Y, Wigg K, Bloom S, Roberts W, Malone M, Schachar R, Tannock R, Kennedy JL. Identification of DNA variants in the SNAP-25 gene and linkage study of these polymorphisms and attention-deficit hyperactivity disorder. Mol Psychiatry. 2000;5:405–409. doi: 10.1038/sj.mp.4000733. [DOI] [PubMed] [Google Scholar]
- Bradley C. The behavior of children receiving Benzedrine. Am J Psychiatry. 1937;94:577–585. [Google Scholar]
- Breese GR, Napier TC, Mueller RA. Dopamine agonist-induced locomotor activity in rats treated with 6-hydroxydopamine at differing ages: functional supersensitivity of D-1 dopamine receptors in neonatally lesioned rats. J Pharmacol Exp Ther. 1985;234:447–455. [PubMed] [Google Scholar]
- Breier A, Su TP, Saunders R, Carson RE, Kolachana BS, de Bartolomeis A, Weinberger DR, Weisenfeld N, Malhotra AK, Eckelman WC, Pickar D. Schizophrenia is associated with elevated amphetamine-induced synaptic dopamine concentrations: evidence from a novel positron emission tomography method. Proc Natl Acad Sci U S A. 1997;94:2569–2574. doi: 10.1073/pnas.94.6.2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brookes K, Xu X, Chen W, Zhou K, Neale B, Lowe N, Aneey R, Franke B, Gill M, Ebstein R, Buitelaar J, Sham P, Campbell D, Knight J, Andreou P, Altink M, Arnold R, Boer F, Buschgens C, Butler L, Christiansen H, Feldman L, Fleischman K, Fliers E, Howe-Forbes R, Goldfarb A, Heise A, Gabriels I, Korn-Lubetzki I, Marco R, Medad S, Minderaa R, Mulas F, Muller U, Mulligan A, Rabin K, Rommelse N, Sethna V, Sorohan J, Uebel H, Psychogiou L, Weeks A, Barrett R, Craig I, Banaschewski T, Sonuga-Barke E, Eisenberg J, Kuntsi J, Manor I, McGuffin P, Miranda A, Oades RD, Plomin R, Roeyers H, Rothenberger A, Sergeant J, Steinhausen HC, Taylor E, Thompson M, Faraone SV, Asherson P, Johansson L. The analysis of 51 genes in DSM-IV combined type attention deficit hyperactivity disorder: association signals in DRD4, DAT1 and 16 other genes. Mol Psychiatry. 2006;11:934–953. doi: 10.1038/sj.mp.4001869. [DOI] [PubMed] [Google Scholar]
- Brophy K, Hawi Z, Kirley A, Fitzgerald M, Gill M. Synaptosomal-associated protein 25 (SNAP-25) and attention deficit hyperactivity disorder (ADHD): evidence of linkage and association in the Irish population. Mol Psychiatry. 2002;7:913–917. doi: 10.1038/sj.mp.4001092. [DOI] [PubMed] [Google Scholar]
- Brown GL, Hunt RD, Ebert MH, Bunney WE, Jr, Kopin IJ. Plasma levels of d-amphetamine in hyperactive children. Serial behavior and motor responses. Psychopharmacology (Berl) 1979;62:133–140. doi: 10.1007/BF00427126. [DOI] [PubMed] [Google Scholar]
- Brown JR, Arbuthnott GW. The electrophysiology of dopamine (D2) receptors: a study of the actions of dopamine on corticostriatal transmission. Neuroscience. 1983;10:349–355. doi: 10.1016/0306-4522(83)90138-0. [DOI] [PubMed] [Google Scholar]
- Bruno KJ, Freet CS, Twining RC, Egami K, Grigson PS, Hess EJ. Abnormal latent inhibition and impulsivity in coloboma mice, a model of ADHD. Neurobiol Dis. 2007;25:206–216. doi: 10.1016/j.nbd.2006.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruno KJ, Hess EJ. The alpha(2C)-adrenergic receptor mediates hyperactivity of coloboma mice, a model of attention deficit hyperactivity disorder. Neurobiol Dis. 2006;23:679–688. doi: 10.1016/j.nbd.2006.05.007. [DOI] [PubMed] [Google Scholar]
- Cardenas L, Houle S, Kapur S, Busto UE. Oral D-amphetamine causes prolonged displacement of [11C]raclopride as measured by PET. Synapse. 2004;51:27–31. doi: 10.1002/syn.10282. [DOI] [PubMed] [Google Scholar]
- Castellanos FX. Toward a pathophysiology of attention-deficit/hyperactivity disorder. Clin Pediatr (Phila) 1997;36:381–393. doi: 10.1177/000992289703600702. [DOI] [PubMed] [Google Scholar]
- Cepeda C, Buchwald NA, Levine MS. Neuromodulatory actions of dopamine in the neostriatum are dependent upon the excitatory amino acid receptor subtypes activated. Proc Natl Acad Sci U S A. 1993;90:9576–9580. doi: 10.1073/pnas.90.20.9576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darracq L, Blanc G, Glowinski J, Tassin JP. Importance of the noradrenaline-dopamine coupling in the locomotor activating effects of D-amphetamine. J Neurosci. 1998;18:2729–2739. doi: 10.1523/JNEUROSCI.18-07-02729.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efron D, Jarman F, Barker M. Methylphenidate versus dexamphetamine in children with attention deficit hyperactivity disorder: A double-blind, crossover trial. Pediatrics. 1997;100:E6. doi: 10.1542/peds.100.6.e6. [DOI] [PubMed] [Google Scholar]
- Elia J, Borcherding BG, Rapoport JL, Keysor CS. Methylphenidate and dextroamphetamine treatments of hyperactivity: Are there true nonresponders? Psychiatry Res. 1991;36:141–155. doi: 10.1016/0165-1781(91)90126-a. [DOI] [PubMed] [Google Scholar]
- Faraone SV, Perlis RH, Doyle AE, Smoller JW, Goralnick JJ, Holmgren MA, Sklar P. Molecular genetics of attention-deficit/hyperactivity disorder. Biol Psychiatry. 2005;57:1313–1323. doi: 10.1016/j.biopsych.2004.11.024. [DOI] [PubMed] [Google Scholar]
- Ferris RM, Tang FL, Maxwell RA. A comparison of the capacities of isomers of amphetamine, deoxypipradrol and methylphenidate to inhibit the uptake of tritiated catecholamines into rat cerebral cortex slices, synaptosomal preparations of rat cerebral cortex, hypothalamus and striatum and into adrenergic nerves of rabbit aorta. J Pharmacol Exp Ther. 1972;181:407–416. [PubMed] [Google Scholar]
- Fuller RW, Molloy BB, Roush BW, Hauser KM. Disposition and behavioral effects of amphetamine and, -difluoroamphetamine in mice. Biochem Pharmacol. 1972;21:1299–1307. doi: 10.1016/0006-2952(72)90291-2. [DOI] [PubMed] [Google Scholar]
- Giros B, Jaber M, Jones SR, Wightman RM, Caron MG. Hyperlocomotion and indifference to cocaine and amphetamine in mice lacking the dopamine transporter. Nature. 1996;379:606–612. doi: 10.1038/379606a0. [DOI] [PubMed] [Google Scholar]
- Glick SD, Milloy S. Rate-dependent effects of d-amphetamine on locomotor activity in mice: Possible relationship to paradoxical amphetamine sedation in minimal brain dysfunction. Eur J Pharmacol. 1973;24:266–268. doi: 10.1016/0014-2999(73)90082-4. [DOI] [PubMed] [Google Scholar]
- Gonon F. Prolonged and extrasynaptic excitatory action of dopamine mediated by D1 receptors in the rat striatum in vivo. J Neurosci. 1997;17:5972–5978. doi: 10.1523/JNEUROSCI.17-15-05972.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenhill LL, Swanson JM, Steinhoff K, Fried J, Posner K, Lerner M, Wigal S, Clausen SB, Zhang Y, Tulloch S. A pharmacokinetic/pharmacodynamic study comparing a single morning dose of adderall to twice-daily dosing in children with ADHD. J Am Acad Child Adolesc Psychiatry. 2003;42:1234–1241. doi: 10.1097/00004583-200310000-00015. [DOI] [PubMed] [Google Scholar]
- Heikkila RE, Orlansky H, Mytilineou C, Cohen G. Amphetamine: evaluation of d- and l-isomers as releasing agents and uptake inhibitors for 3H-dopamine and 3H-norepinephrine in slices of rat neostriatum and cerebral cortex. J Pharmacol Exp Ther. 1975;194:47–56. [PubMed] [Google Scholar]
- Hess EJ, Collins KA, Wilson MC. Mouse model of hyperkinesis implicates SNAP-25 in behavioral regulation. J Neurosci. 1996;16:3104–3111. doi: 10.1523/JNEUROSCI.16-09-03104.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess EJ, Jinnah HA, Kozak CA, Wilson MC. Spontaneous locomotor hyperactivity in a mouse mutant with a deletion including the Snap gene on Chromosome 2. J Neurosci. 1992;12:2865–2874. doi: 10.1523/JNEUROSCI.12-07-02865.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyser CJ, Wilson MC, Gold LH. Coloboma hyperactive mutant exhibits delayed neurobehavioral developmental milestones. Brain Res Dev Brain Res. 1995;89:264–269. doi: 10.1016/0165-3806(95)00130-6. [DOI] [PubMed] [Google Scholar]
- Hu XT, Wang RY. Comparison of effects of D-1 and D-2 dopamine receptor agonists on neurons in the rat caudate putamen: an electrophysiological study. J Neurosci. 1988;8:4340–4348. doi: 10.1523/JNEUROSCI.08-11-04340.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilgin N, Senol S, Gucuyener K, Gokcora N, Sener S. Is increased D2 receptor availability associated with response to stimulant medication in ADHD. Dev Med Child Neurol. 2001;43:755–760. doi: 10.1017/s0012162201001384. [DOI] [PubMed] [Google Scholar]
- Imperato A, Di Chiara G. Trans-striatal dialysis coupled to reverse phase high performance liquid chromatography with electrochemical detection: a new method for the study of the in vivo release of endogenous dopamine and metabolites. J Neurosci. 1984;4:966–977. doi: 10.1523/JNEUROSCI.04-04-00966.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones MD, Hess EJ. Norepinephrine regulates locomotor hyperactivity in the mouse mutant coloboma. Pharmacol Biochem Behav. 2003;75:209–216. doi: 10.1016/s0091-3057(03)00073-x. [DOI] [PubMed] [Google Scholar]
- Jones MD, Williams ME, Hess EJ. Abnormal presynaptic catecholamine regulation in a hyperactive SNAP-25-deficient mouse mutant. Pharmacol Biochem Behav. 2001a;68:669–676. doi: 10.1016/s0091-3057(01)00481-6. [DOI] [PubMed] [Google Scholar]
- Jones MD, Williams ME, Hess EJ. Expression of catecholaminergic mRNAs in the hyperactive mouse mutant coloboma. Brain Res Mol Brain Res. 2001b;96:114–121. doi: 10.1016/s0169-328x(01)00281-9. [DOI] [PubMed] [Google Scholar]
- Kim DS, Szczypka MS, Palmiter RD. Dopamine-deficient mice are hypersensitive to dopamine receptor agonists. J Neurosci. 2000;20:4405–4413. doi: 10.1523/JNEUROSCI.20-12-04405.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiyatkin EA, Rebec GV. Dopaminergic modulation of glutamate-induced excitations of neurons in the neostriatum and nucleus accumbens of awake, unrestrained rats. J Neurophysiol. 1996;75:142–153. doi: 10.1152/jn.1996.75.1.142. [DOI] [PubMed] [Google Scholar]
- Kostrzewa RM. Dopamine receptor supersensitivity. Neurosci Biobehav Rev. 1995;19:1–17. doi: 10.1016/0149-7634(94)00019-w. [DOI] [PubMed] [Google Scholar]
- Kruzich PJ, Suchland KL, Grandy DK. Dopamine D4 receptor-deficient mice, congenic on the C57BL/6J background, are hypersensitive to amphetamine. Synapse. 2004;53:131–139. doi: 10.1002/syn.20043. [DOI] [PubMed] [Google Scholar]
- Kuczenski R, Segal D. Concomitant characterization of behavioral and striatal neurotransmitter response to amphetamine using in vivo microdialysis. J Neurosci. 1989;9:2051–2065. doi: 10.1523/JNEUROSCI.09-06-02051.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuczenski R, Segal DS. Effects of methylphenidate on extracellular dopamine, serotonin, and norepinephrine: comparison with amphetamine. J Neurochem. 1997;68:2032–2037. doi: 10.1046/j.1471-4159.1997.68052032.x. [DOI] [PubMed] [Google Scholar]
- Kustanovich V, Merriman B, McGough J, McCracken JT, Smalley SL, Nelson SF. Biased paternal transmission of SNAP-25 risk alleles in attention-deficit hyperactivity disorder. Mol Psychiatry. 2003;8:309–315. doi: 10.1038/sj.mp.4001247. [DOI] [PubMed] [Google Scholar]
- Lou HC, Rosa P, Pryds O, Karrebaek H, Lunding J, Cumming P, Gjedde A. ADHD: increased dopamine receptor availability linked to attention deficit and low neonatal cerebral blood flow. Dev Med Child Neurol. 2004;46:179–183. doi: 10.1017/s0012162204000313. [DOI] [PubMed] [Google Scholar]
- McCracken JT. A two-part model of stimulant action on attention-deficit hyperactivity disorder in children. J Neuropsychiatry Clin Neurosci. 1991;3:201–209. doi: 10.1176/jnp.3.2.201. [DOI] [PubMed] [Google Scholar]
- Mill J, Curran S, Kent L, Gould A, Huchkett L, Richards S, Taylor E, Asherson A. Association study of a SNAP-25 microsatellite and attention deficit hyperactivity disorder. Am J Med Genet. 2002;114:269–271. doi: 10.1002/ajmg.10253. [DOI] [PubMed] [Google Scholar]
- Miller KW, Sanders-Bush E, Dingell JV. p-Chloramphetamine--species differences in the rate of disappearance and the lowering of cerebral serotonin. Biochem Pharmacol. 1971;20:500–503. doi: 10.1016/0006-2952(71)90092-x. [DOI] [PubMed] [Google Scholar]
- O’Donnell P, Grace AA. Tonic D2-mediated attenuation of cortical excitation in nucleus accumbens neurons recorded in vitro. Brain Res. 1994;634:105–112. doi: 10.1016/0006-8993(94)90263-1. [DOI] [PubMed] [Google Scholar]
- O’Neill MF, Shaw G. Comparison of dopamine receptor antagonists on hyperlocomotion induced by cocaine, amphetamine, MK-801 and the dopamine D1 agonist C-APB in mice. Psychopharmacology (Berl) 1999;145:237–250. doi: 10.1007/s002130051055. [DOI] [PubMed] [Google Scholar]
- Ogren SO, Archer T, Johansson C. Evidence for a selective brain noradrenergic involvement in the locomotor stimulant effects of amphetamine in the rat. Neurosci Lett. 1983;43:327–331. doi: 10.1016/0304-3940(83)90209-4. [DOI] [PubMed] [Google Scholar]
- Oswald LM, Wong DF, McCaul M, Zhou Y, Kuwabara H, Choi L, Brasic J, Wand GS. Relationships among ventral striatal dopamine release, cortisol secretion, and subjective responses to amphetamine. Neuropsychopharmacology. 2005;30:821–832. doi: 10.1038/sj.npp.1300667. [DOI] [PubMed] [Google Scholar]
- Page ME, Brown K, Lucki I. Simultaneous analyses of the neurochemical and behavioral effects of the norepinephrine reuptake inhibitor reboxetine in a rat model of antidepressant action. Psychopharmacology (Berl) 2003;165:194–201. doi: 10.1007/s00213-002-1269-x. [DOI] [PubMed] [Google Scholar]
- Piercey MF, Hyslop DK, Hoffmann WE. Excitation of type II caudate neurons by systemic administration of dopamine agonists. Brain Res. 1996;706:249–258. doi: 10.1016/0006-8993(95)01151-x. [DOI] [PubMed] [Google Scholar]
- Raber J, Mehta PP, Kreifeldt M, Parsons LH, Weiss F, Bloom FE, Wilson MC. Coloboma hyperactive mutant mice exhibit regional and transmitter-specific deficits in neurotransmission. J Neurochem. 1997;68:176–186. doi: 10.1046/j.1471-4159.1997.68010176.x. [DOI] [PubMed] [Google Scholar]
- Riffee WH, Ludden TM, Wilcox RE, Gerald MC. Brain and plasma concentrations of amphetamine isomers in mice. J Pharmacol Exp Ther. 1978;206:586–594. [PubMed] [Google Scholar]
- Robinson TE, Camp DM. Does amphetamine preferentially increase the extracellular concentration of dopamine in the mesolimbic system of freely moving rats? Neuropsychopharmacology. 1990;3:163–173. [PubMed] [Google Scholar]
- Ruskin DN, Bergstrom DA, Shenker A, Freeman LE, Baek D, Walters JR. Drugs used in the treatment of attention-deficit/hyperactivity disorder affect postsynaptic firing rate and oscillation without preferential dopamine autoreceptor action. Biol Psychiatry. 2001;49:340–350. doi: 10.1016/s0006-3223(00)00987-2. [DOI] [PubMed] [Google Scholar]
- Sahakian BJ, Robbins TW. Are the effects of psychomotor stimulant drugs on hyperactive children really paradoxical? Med Hypotheses. 1977;3:154–158. doi: 10.1016/0306-9877(77)90065-2. [DOI] [PubMed] [Google Scholar]
- Salmi P, Malmgren K, Svensson TH, Ahlenius S. Stimulation of forward locomotion by SCH-23390 and raclopride in d-amphetamine-treated rats. Naunyn Schmiedebergs Arch Pharmacol. 1998;357:593–599. doi: 10.1007/pl00005213. [DOI] [PubMed] [Google Scholar]
- Seeman P, Madras BK. Anti-hyperactivity medication: methylphenidate and amphetamine. Mol Psychiatry. 1998;3:386–396. doi: 10.1038/sj.mp.4000421. [DOI] [PubMed] [Google Scholar]
- Shippenberg TS, He M, Chefer V. The use of microdialysis in the mouse: conventional versus quantitative techniques. Psychopharmacology (Berl) 1999;147:33–34. doi: 10.1007/s002130051138. [DOI] [PubMed] [Google Scholar]
- Skirboll LR, Grace AA, Bunney BS. Dopamine auto- and postsynaptic receptors: electrophysiological evidence for differential sensitivity to dopamine agonists. Science. 1979;206:80–82. doi: 10.1126/science.482929. [DOI] [PubMed] [Google Scholar]
- Smith AD, Justice JB. The effect of inhibition of synthesis, release, metabolism and uptake on the microdialysis extraction fraction of dopamine. J Neurosci Methods. 1994;54:75–82. doi: 10.1016/0165-0270(94)90161-9. [DOI] [PubMed] [Google Scholar]
- Solanto MV. Neuropsychopharmacological mechanisms of stimulant drug action in attention-deficit hyperactivity disorder: a review and integration. Behav Brain Res. 1998;94:127–152. doi: 10.1016/s0166-4328(97)00175-7. [DOI] [PubMed] [Google Scholar]
- Söllner T, Whiteheart SW, Brunner M, Erdjument-Bromage H, Germanos S, Tempst P, Rothman JE. SNAP receptors implicated in vesicle targeting and fusion. Nature. 1993;362:318–324. doi: 10.1038/362318a0. [DOI] [PubMed] [Google Scholar]
- Udo de Haes JI, Kortekaas R, Van Waarde A, Maguire RP, Pruim J, den Boer JA. Assessment of methylphenidate-induced changes in binding of continuously infused [(11)C]-raclopride in healthy human subjects: correlation with subjective effects. Psychopharmacology (Berl) 2005;183:322–330. doi: 10.1007/s00213-005-0193-2. [DOI] [PubMed] [Google Scholar]
- Viggiano D, Ruocco LA, Sadile AG. Dopamine phenotype and behaviour in animal models: in relation to attention deficit hyperactivity disorder. Neurosci Biobehav Rev. 2003;27:623–637. doi: 10.1016/j.neubiorev.2003.08.006. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Wang G, Fowler JS, Logan J, Gerasimov M, Maynard L, Ding Y, Gatley SJ, Gifford A, Franceschi D. Therapeutic doses of oral methylphenidate significantly increase extracellular dopamine in the human brain. J Neurosci. 2001;21:RC121. doi: 10.1523/JNEUROSCI.21-02-j0001.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AR, Grace AA. Opposite influences of endogenous dopamine D1 and D2 receptor activation on activity states and electrophysiological properties of striatal neurons: studies combining in vivo intracellular recordings and reverse microdialysis. J Neurosci. 2002;22:294–304. doi: 10.1523/JNEUROSCI.22-01-00294.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu F, Gainetdinov RR, Wetsel WC, Jones SR, Bohn LM, Miller GW, Wang Y-M, Caron MG. Mice lacking the norepinephrine transporter are supersensitive to psychostimulants. Nature Neuroscience. 2000;3:465–471. doi: 10.1038/74839. [DOI] [PubMed] [Google Scholar]
- Zheng P, Zhang XX, Bunney BS, Shi WX. Opposite modulation of cortical N-methyl-D-aspartate receptor-mediated responses by low and high concentrations of dopamine. Neuroscience. 1999;91:527–535. doi: 10.1016/s0306-4522(98)00604-6. [DOI] [PubMed] [Google Scholar]
- Zhuang X, Oosting RS, Jones SR, Gainetdinov RR, Miller GW, Caron MG, Hen R. Hyperactivity and impaired response habituation in hyperdopaminergic mice. Proc Natl Acad Sci U S A. 2001;98:1982–1987. doi: 10.1073/pnas.98.4.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]