

Abstract
The cellular mechanisms underlying Purkinje neuron death in various neurodegenerative disorders of the cerebellum are poorly understood. Here we investigate an in vitro model of cerebellar neuronal death. We report that cerebellar Purkinje neurons, deprived of trophic factors, die by a form of programmed cell death distinct from the apoptotic death of neighboring granule neurons. Purkinje neuron death was characterized by excessive autophagic-lysosomal vacuolation. Autophagy and death of Purkinje neurons were inhibited by nerve growth factor (NGF) and were activated by NGF-neutralizing antibodies. Although treatment with antisense oligonucleotides to the p75 neurotrophin receptor (p75ntr) decreased basal survival of cultured cerebellar neurons, p75ntr-antisense decreased autophagy and completely inhibited death of Purkinje neurons induced by trophic factor withdrawal. Moreover, adenoviral expression of a p75ntr mutant lacking the ligand-binding domain induced vacuolation and death of Purkinje neurons. These results suggest that p75ntr is required for Purkinje neuron survival in the presence of trophic support; however, during trophic factor withdrawal, p75ntr contributes to Purkinje neuron autophagy and death. The autophagic morphology resembles that found in neurodegenerative disorders, suggesting a potential role for this pathway in neurological disease.
Keywords: autophagy, Purkinje neuron, p75ntr, cell death, neurotrophin, vacuoles
Introduction
Chronic neurodegenerative diseases are characterized by a selective loss of specific neuronal populations over a period of years or even decades. Although the underlying causes of most neurodegenerative diseases are unclear, the loss of neurons and neuronal contacts is a key feature of disease pathology. Elucidation of the cellular mechanisms regulating neuronal cell death is critical for developing new therapeutic strategies to slow or halt the progressive neurodegeneration in these disorders.
Purkinje neurons in the cerebellum integrate input to the cerebellar cortex from the mossy fibers and climbing fibers and subsequently generate inhibitory output to the deep cerebellar nuclei (Ghez and Thach, 2000). In addition to their essential role in proper cerebellar function, Purkinje neurons provide critical trophic support to developing cerebellar granule neurons and inferior olivary neurons (Torres-Aleman et al., 1994; Zanjani et al., 1994; Linseman et al., 2002a). Purkinje neurons are specifically lost in various neurodegenerative conditions such as spinocerebellar ataxias (Koeppen, 1998; Watase et al., 2002), ataxia telangectasia (Gatti and Vinters, 1985; Borghesani et al., 2000), autism (Ritvo et al., 1986; Bailey et al., 1998), and certain prion encephalopathies (Ferrer et al., 1991; Watanabe and Duchen, 1993; Lasmezas et al., 1997). Although Purkinje cell loss is a critical feature of disease pathology, the molecular mechanisms underlying Purkinje cell death remain poorly understood.
The Lurcher (Lc) mouse has been extensively used as an in vivo model of Purkinje neuron degeneration. The cell degeneration in the Lurcher cerebellum results from a single point mutation in the δ2 glutamate receptor (GluRδ2), whose expression is restricted to Purkinje neurons (Zuo et al., 1997). The degeneration and loss of Purkinje neurons in Lurcher cerebellum is followed by a secondary death of cerebellar granule neurons and inferior olivary neurons attributable to loss of trophic support normally provided by their afferent target Purkinje neurons (Wetts and Herrup, 1982). Because GluRδ2Lc causes a constitutive depolarization of Purkinje neurons, it was originally thought that the death of Purkinje neurons in the Lc cerebellum was akin to excitotoxicity mediated by excessive calcium influx. However, a recent report suggested that the mechanism of GluRδ2Lc-induced Purkinje cell degeneration can be dissociated from depolarization (Selimi et al., 2003). Instead, Lc Purkinje cell death is hypothesized to involve interactions between the mutant GluRδ2 receptor and the proteins nPIST and Beclin 1, because these protein-protein interactions can lead to cell death and increase autophagy when overexpressed in heterologous cells (Yue et al., 2002). Furthermore, both groups were able to demonstrate the appearance of autophagic vacuoles in Lc Purkinje neurons before degeneration, suggesting that upregulated autophagy is an early feature of dying Purkinje neurons (Yue et al., 2002; Selimi et al., 2003).
Autophagy is a degradative pathway responsible for the bulk of proteolysis in normal cells. Autophagy is an evolutionarily conserved pathway that leads to the degradation of proteins and entire organelles in cells undergoing stress; however, in extreme cases it can result in cellular dysfunction and cell death (Klionsky and Emr, 2000). Autophagy begins with the formation of double-membrane vesicles that sequester cytoplasm and organelles in autophagosomes (autophagic vacuoles in mammalian cells). The autophagosomes fuse with lysosomes, forming autophagolysosomes. The contents of autophagolysosomes are degraded by lysosomal enzymes into basic macromolecules, which are then recycled for use in essential cellular functions (Stromhaug and Klionsky, 2001).
Dysregulation of autophagolysosomal activity can lead to cell death and is implicated in several neurodegenerative conditions, including Parkinson's disease (Anglade et al., 1997), Alzheimer's disease (Cataldo et al., 1996; Nixon et al., 2000), Lewy body dementias (Zhu et al., 2003), Huntington's disease (Kegel et al., 2000; Petersen et al., 2001), and prion encephalopathies (Boellaard et al., 1991; Liberski et al., 2002). Nutrient deprivation, including withdrawal of serum (Mitchener et al., 1976), is one stimulus known to induce autophagy. In this report, we describe an in vitro model to investigate signaling pathways that regulate autophagy and survival in cerebellar Purkinje neurons. We used cerebellar cultures from early postnatal rats to investigate Purkinje neuron death in response to trophic factor withdrawal. In this model, Purkinje cell loss was characterized by extensive cytoplasmic vacuolation and a marked absence of nuclear condensation or fragmentation. The vacuoles stained with markers for autophagic vacuoles and lysosomes and the presence of abundant, enlarged autophago(lyso)somes was confirmed by transmission electron microscopy. The autophagy inhibitor 3-methyladenine diminished cytoplasmic vacuolation and moderately increased the survival of Purkinje neurons. Nerve growth factor (NGF) also inhibited autophagy and death of Purkinje neurons. The protective effects of NGF were mediated by the low-affinity p75 neurotrophin receptor (p75ntr). Our data support the hypothesis that p75ntr can regulate autophagy and death in Purkinje neurons.
Materials and Methods
Materials. Polyclonal antibodies to calbindin-D28k and p75ntr, monoclonal antibodies to NGF, and purified NGF were obtained from Chemicon (Temecula, CA). Polyclonal antibodies to NGF and monoclonal antibodies to phospho-TrkA were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). The monoclonal phosphotyrosine antibody (Ab) was obtained from Upstate Cell Signaling Solutions (Lake Placid, NY). A polyclonal antibody to the intracellular domain of rat p75ntr was obtained from Covance (Berkeley, CA). Cy3- and FITC-conjugated secondary antibodies for immunocytochemistry were purchased from Jackson ImmunoResearch (West Grove, PA). Horseradish peroxidase-linked secondary antibodies and reagents for enhanced chemiluminescence detection were obtained from Amersham Biosciences (Piscataway, NJ). Lysosensor blue was obtained from Molecular Probes (Eugene, OR). Monodansylcadaverine, 3-methyladenine, and 4,6-diamidino-2-phenylindole (DAPI) were from Sigma (St. Louis, MO). Adenoviral cytomegalovirus (CMV; negative control adenovirus) was from Dr. Jerry Schaack (University of Colorado Health Sciences Center, Denver, CO). The adenoviral rat p75ntr myristylated intracellular domain (p75mICD) has been described previously (Roux et al., 2001).
Cell culture. Rat cerebellar granule neurons were isolated from 7-d-old Sprague Dawley rat pups as described previously (D'Mello et al., 1993). Briefly, neurons were plated at a density of 2.0 × 106 cells/ml in basal modified Eagle's medium (BME) containing 10% fetal bovine serum, 25 mm KCl, 2 mm l-glutamine, and penicillin (100 U/ml)-streptomycin (100 μg/ml; Invitrogen, Gaithersburg, MD) Cytosine arabinoside (10 μm) was added to the culture medium 24 hr after plating to limit the growth of non-neuronal cells. Experiments were performed after 7 d in culture. Death was induced by removing the serum- and high-potassium-containing media and replacing it with serum-free and 5 mm potassium BME.
Adenoviral infection. Five days after plating, neuronal cultures were infected with either control adenovirus (adenoviral CMV) or adenoviral p75ntr, full-length or truncated, mICD, each at the indicated multiplicity of infection. After infection, cells were returned to the incubator for 48 hr at 37°C and 10% CO2. On day 7, neurons were processed for live cell imaging or fixed for immunocytochemistry.
Antisense. The p75ntr antisense and missense oligonucleotides (both at 5 μm) were added to the cultures at the time of plating [day in vitro (DIV) 0] by repeatedly triturating the cells in the presence of the oligonucleotides before seeding. The oligonucleotides were present throughout the culture. Trophic factor withdrawal was performed on day 5 or 6 in culture (DIV 5 or 6) for either 24 or 48 hr. After this treatment, the cells were processed for live cell lysosensor experiments or fixed for immunocytochemistry to count Purkinje neurons. In all cases, the antisense was taken up by neurons with nearly 100% efficiency as assayed by visualization of the fluorescently labeled oligonucleotides. HPLC-purified phosphorothioate oligonucleotides were purchased from Integrated DNA Technologies (Coralville, IA). Sequences used were as follows: rat p75ntr 5′ antisense, 5′-ACCTGCCCTCCTCATTGCA-3′; and rat p75ntr 5′ missense, 5′-CTCCCACTCGTCATTCGAC-3′. The rat 5′ p75ntr antisense was also purchased with a 5′ 56-FAM fluorescent label so that uptake by the neurons could be monitored. The antisense sequence used has been characterized previously and has been shown to be effective at inhibiting p75ntr-mediated cell death both in vitro and in vivo (Barrett and Bartlett, 1994; Cheema et al., 1996; Lowry et al., 2001).
Immunocytochemistry. Neuronal cultures were plated on polyethyleneimine-coated glass coverslips. Neurons were infected and induced to undergo death as indicated previously. After treatment, the neurons were fixed with 4% paraformaldehyde and then permeabilized and blocked with PBS, pH 7.4, containing 0.2% Triton X-100 and 5% BSA. Cells were then incubated with polyclonal antibodies against calbindin (1:250), active caspase-3 (1:500), or p75ntr (1:250) overnight at 4°C diluted in PBS containing 0.1% Triton X-100 and 2% BSA. Primary antibodies were then removed, and the cells were washed at least six times with PBS at room temperature. The neurons were then incubated with Cy3-conjugated donkey anti-rabbit secondary antibodies (1:500) and DAPI (1 μg/ml) for 1 hr at room temperature. The cells were then washed at least six more times with PBS, and coverslips were adhered to glass slides with mounting medium (0.1% p-phenylenediamine in 75% glycerol in PBS). Imaging was performed on a Zeiss (Thornwood, NY) Axioplan 2 microscope equipped with a Cooke Sensicam deep-cooled CCD camera, and images were analyzed with the Slidebook software program (Intelligent Imaging Innovations Inc., Denver, CO).
Purkinje cell counts. Purkinje neuron numbers were measured by counting the number of calbindin-positive cells in 152 fields under 63× oil that were randomly selected by following a fixed grid pattern over the coverslip. The total area counted per coverslip was 14.6 mm2 or ∼13% of the coverslip. In addition to calbindin staining, cells were also judged on their morphology. Cells counted as Purkinje neurons had large rounded cell bodies, elaborate processes, and larger, more oval nuclei (in comparison with the smaller, rounder nuclei of granule neurons). Some very bright calbindin-positive cells that were either as small as the granule neurons, having bipolar processes, or very large flattened cells, lacking elaborate processes, were not counted because previous studies on in vitro differentiation of Purkinje neurons suggest that these cells may represent examples of delayed or aberrant development of Purkinje neurons in culture (Baptista et al., 1994). At least three coverslips were counted per experimental condition. The numbers were averaged and expressed as a percentage of the average number counted in the appropriate controls. This was repeated for at least three independent experiments.
Live cell imaging and vacuolation measurements. Lysosensor blue and monodansylcadaverine (MDC) were added to the cultures at the end of the indicated treatments and returned to the incubator for 20 min. The cells were then washed three times with 37°C phenol red-free DMEM to remove nonspecifically bound dyes. The coverslips were mounted onto glass slides on ∼10 μl of phenol red-free DMEM; excess media were aspirated; and the coverslips were sealed and imaged immediately. Purkinje neurons were identified on the basis of their morphology (large, oval nuclei, large cell body, and multiple processes) by scanning the coverslips under bright-field differential interference contrast with a 63× oil objective. Images of the lysosensor blue and MDC fluorescence were captured on the DAPI and FITC channels, respectively, with the 100× oil objective. The lysosensor blue images were used to measure the diameters of all the visible lysosomes in at least six Purkinje neurons per condition. This was usually between 100 and 200 lysosomes. This was repeated for at least three independent experiments.
Transmission electron microscopy. Cultured cells were fixed in 2% paraformaldehyde and 2.5% glutaraldehyde for 1 hr, postfixed in 1% osmium tetroxide for 1 hr at 4°C, and processed for embedding in the culture dish. Cells were then gently scraped and embedded in blocks of Eponaraldite. Thin sections were stained with 4% aqueous uranyl acetate and lead citrate and examined on a Philips CM-12 electron microscope.
Lysate preparation. After incubation for the indicated times and with the reagents specified above, the culture medium was aspirated, cells were washed once with 2 ml of ice-cold PBS (PBS, pH 7.4), placed on ice, and scraped into lysis buffer (200 μl/35 mm well) containing 20 mm HEPES, pH 7.4, 1% Triton X-100, 50 mm NaCl, 1 mm EGTA, 5 mm β-glycerophosphate, 30 mm sodium pyrophosphate, 100 μm sodium orthovanadate, 1 mm phenylmethylsulfonyl fluoride, 10 μg/ml leupeptin, and 10 μg/ml aprotinin. Cell debris was removed by centrifugation at 6000 × g for 3 min and the protein concentration of the supernatant was determined using a commercially available protein assay kit (Pierce, Rockford, IL). Equal amounts of supernatant protein were diluted to a final concentration of 1× SDS-PAGE sample buffer, boiled for 5 min, and electrophoresed through 7.5% polyacrylamide gels. Proteins were transferred to polyvinylidene membranes (Millipore, Bedford, MA) and processed for immunoblot analysis.
Immunoblot analysis. Nonspecific binding sites were blocked in PBS, pH 7.4, containing 0.1% Tween 20 (PBS-T) and 1% BSA for 1 hr at room temperature. Primary antibodies were diluted in blocking solution and incubated with the membranes for 1 hr. Excess primary Ab was removed by washing the membranes three times in PBS-T. The blots were then incubated with the appropriate horseradish peroxidase-conjugated secondary Ab diluted in PBS-T for 1 hr and were subsequently washed three times in PBS-T. Immunoreactive proteins were detected by enhanced chemiluminescence. In some experiments, membranes were reprobed after stripping in 0.1 m Tris-HCl, pH 8.0, 2% SDS, and 100 mm β-mercaptoethanol for 30 min at 52°C. The blots were rinsed twice in PBS-T and processed as above with a different primary Ab. Autoluminograms shown are representative of at least three independent experiments.
Data analysis. Results shown represent the means ± SEM for the number of independent experiments performed. Statistical differences between the means of unpaired sets of data were evaluated using one-way ANOVA followed by post hoc Tukey's test; p < 0.05 was considered statistically significant.
Results
Trophic factor withdrawal results in a loss of Purkinje neurons that is morphologically distinct from apoptosis
We have investigated Purkinje neuron death induced by trophic factor withdrawal using primary cerebellar neuronal cultures. Although these cultures have been extensively used to study signaling pathways that regulate survival of cerebellar granule neurons (D'Mello et al., 1993; Linseman et al., 2002b; Vaudry et al., 2003), they also provide a model system for studying differentiated Purkinje neurons (Baptista et al., 1994). Primary neuronal cultures are derived from dissociated cerebella of early postnatal rats. These neuronal cultures survive and differentiate when maintained in medium containing 10% fetal calf serum and a depolarizing concentration of potassium (25 mm). Granule neurons, which constitute ∼98% of the cell population, undergo classical apoptosis, characterized by chromatin condensation and caspase activation, when they are deprived of serum and depolarizing potassium (trophic factor withdrawal; (D'Mello et al., 1993; Linseman et al., 2002b).
In contrast, Purkinje neurons, which make up ∼2% of the culture, undergo a distinct nonapoptotic form of cell death in response to trophic factor withdrawal. Quantification of the number of Purkinje neurons after trophic factor withdrawal is shown in Figure 1A. Purkinje neurons were identified by staining with antibodies to the Purkinje marker calbindin-D28k. After 24 or 48 hr of trophic factor withdrawal, Purkinje numbers were 50.4 ± 1.4 and 22.3 ± 7.0% of controls, respectively. The remaining Purkinje neurons showed markedly different morphology from that of control cells, characterized by extensive cytoplasmic vacuolation (Fig. 1, compare control cells, B, C, with trophic factor-deprived cells, E, F). In contrast to granule neurons deprived of trophic support, which demonstrated substantial nuclear condensation and fragmentation characteristic of apoptosis (Fig. 1, compare D, G), Purkinje neurons showed no obvious signs of nuclear condensation or fragmentation (Fig. 1, compare nuclei indicated in D, G, arrows). The degenerating Purkinje neurons did not stain with propidium iodide, indicating that they retained membrane integrity throughout the death process (data not shown). The maintenance of membrane integrity suggested that Purkinje death did not occur by necrosis. Vacuole formation and subsequent loss of Purkinje neurons required new RNA synthesis because addition of the transcriptional inhibitor actinomycin D (ActD) decreased vacuolation and significantly increased the numbers of Purkinje cells to 78.7 ± 1.5% compared with 50.4 ± 1.4% (p < 0.05) after 24 hr of trophic factor withdrawal. The ability of ActD to protect Purkinje neurons further suggested that the mechanism of death was not necrotic but was rather a regulated program of cell suicide.
Figure 1.

Trophic factor withdrawal induces a nonapoptotic death pathway in Purkinje neurons. Cultured cerebellar neurons were allowed to differentiate in medium containing 10% fetal calf serum and 25 mm potassium. On day 7, neurons were either maintained in control medium (25K+Ser) or subjected to trophic factor withdrawal medium (5K-Ser) for 24-48 hr. A, Purkinje number was quantified by counting the total number of calbindin-positive Purkinje cells in randomly chosen, equally sized areas per condition (total area, 14.6 mm2). Numbers are plotted as a percentage of control. Values represent the mean ± SEM of three independent experiments each performed in triplicate. **Significant difference from 25K+S control at p < 0.01, one-way ANOVA, Tukey's post hoc test. B-G, Cells incubated in either control medium (B-D) or deprived of trophic factors (E-G) were fixed and stained with polyclonal antibodies against calbindin-D28k (a specific marker of Purkinje neurons; red) and the nuclear dye DAPI (blue). B, C, Purkinje neurons maintained in control medium demonstrate differentiated morphology. D, DAPI staining reveals the nuclei of healthy granule neurons and a Purkinje neuron (arrow). E, F, Trophic factor withdrawal induced extensive cytoplasmic vacuolation of Purkinje neurons (see magnified insets). G, DAPI staining reveals increased nuclear condensation and fragmentation in granule neurons, whereas there is a notable lack of nuclear condensation in Purkinje nuclei (arrow).
Purkinje neurodegeneration is associated with increased autophagy
Extensive cytoplasmic vacuolation was the most prominent morphological feature distinguishing healthy from degenerating Purkinje neurons in response to trophic factor withdrawal. To determine the mechanism underlying the loss of Purkinje neurons, we first characterized the nature of the vacuoles formed during withdrawal conditions. The formation of extensive cytoplasmic vacuoles is consistent with the upregulation of autophagy. The autofluorescent drug MDC has been shown to specifically accumulate in autophagic vacuoles (Biederbick et al., 1995; Munafo and Colombo, 2001). To determine whether autophagy was activated in Purkinje neurons subjected to trophic factor withdrawal, we incubated the cerebellar cultures with MDC and then visualized the MDC staining using live cell imaging techniques. In addition to MDC, the neurons were stained with lysosensor blue, a pH-sensitive dye that fluoresces in acidic compartments (pKa, 5.2), thus specifically revealing lysosomes. These experiments demonstrated that control Purkinje neurons contained both lysosomal and autophagic vacuoles. However, in healthy Purkinje neurons these vacuoles were on average very small (Fig. 2A-D). In contrast, 24 hr of trophic factor withdrawal induced a marked increase in the sizes of autophagic and lysosomal vacuoles, as detected by MDC and lysosensor blue staining, respectively (Fig. 2E-H).
Figure 2.
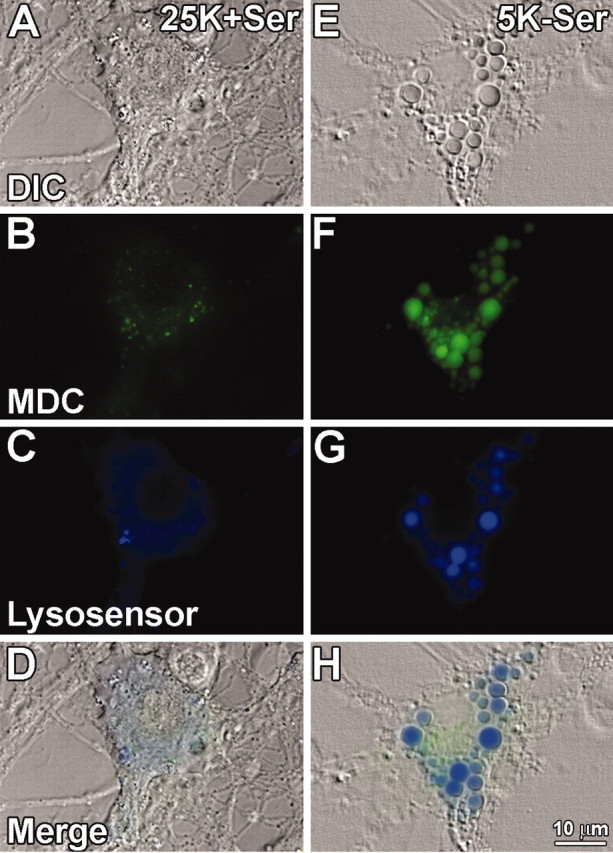
Autophagic and lysosomal markers stain vacuoles that form during Purkinje neuron degeneration. Purkinje neurons maintained in either control (25K+Ser) or trophic factor withdrawal (5K-Ser) media for 24 hr were stained with the autophagic marker MDC (green), and lysosensor blue, a pH-sensitive dye that fluoresces blue in acidic environments. Live cell imaging of control (A-D) and trophic factor-deprived (E-H) Purkinje neurons was performed. A, E, Bright-field images of cells identified as Purkinje neurons based on their morphology, characterized by a large cytoplasm (compared with granule neurons) and extensive neuronal processes. B, F, MDC staining demonstrating increases in autophagosomes in Purkinje neurons after trophic factor withdrawal. C, G, Lysosensor blue staining reveals acidic organelles (i.e., lysosomes) and demonstrates a marked increase in the size of lysosomes in Purkinje neurons after withdrawal of trophic factors. D, H, All three fields merged, demonstrating colabeling of some cytoplasmic vacuoles with both MDC and lysosensor blue indicative of autophagolysosomal fusion.
To determine whether augmented autophagy contributed to Purkinje neuron death, we incubated the cultures with 5 mm 3-methyladenine (3MA), a drug known to inhibit the formation of autophagosomes (Seglen and Gordon, 1982). Quantitative analysis revealed that trophic factor withdrawal significantly increased the size of vacuoles in Purkinje neurons (Fig. 3A). In healthy Purkinje neurons, ∼89% of the lysosensor blue-positive vacuoles were <0.75 μm in diameter. After 24 hr of trophic factor withdrawal, most (∼47%) of the lysosensor blue-positive vacuoles in the dying Purkinje neurons were much larger with a diameter, between 0.75 and 1.5 μm, and a significant percentage (∼32%) were >1.5 μm in diameter. Addition of 5 mm 3MA to the cerebellar cultures almost completely maintained the lysosensor size profile of vacuoles in the range observed in control Purkinje neurons. The inhibition of autophagic activity by 3-methyladenine correlated with significantly increased numbers of Purkinje neurons that remained after 24 hr of trophic factor deprivation (Fig. 3B).
Figure 3.
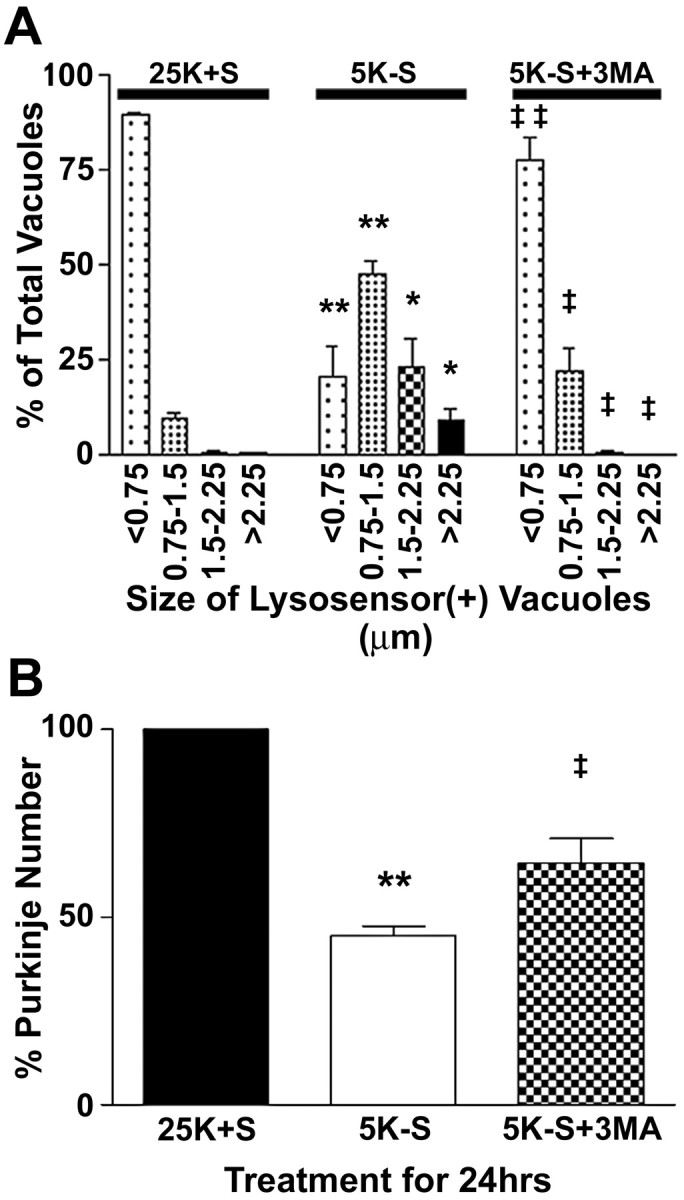
The autophagy inhibitor 3MA, blocks the increased vacuolation and loss of Purkinje neurons. Purkinje neurons were maintained in either control medium (25K+S) or trophic factor withdrawal medium (5K-S) in the absence or presence of 5 mm 3MA (5K-S+3MA) for 24 hr. A, The effects of the various treatments on vacuole size were quantified by measuring the diameters of all visible lysosensor blue-positive vacuoles in 6-12 Purkinje neurons per treatment condition in at least three independent experiments. Usually, the total number of vacuoles measured per condition was between 100 and 200. The size distribution was graphed as a percentage of total vacuoles that were within the indicated size ranges. B, Quantitation of the effects of 3MA on Purkinje neuron numbers demonstrate that addition of 5 mm 3MA to trophic withdrawal medium partially rescues Purkinje neurons from death after 24 hr of trophic factor withdrawal, compared with healthy controls. Numbers are mean ± SEM values of seven independent experiments, each performed in triplicate. **, *Significant difference from 25K+S (**p < 0.01; *p < 0.05). ‡‡,‡Significant difference from 5K-S (‡‡p < 0.01; ‡p < 0.05).
Electron microscopic analysis of autophagic vacuoles in trophic factor-deprived Purkinje neurons
Taken together, the above data suggested that autophagy is upregulated in Purkinje neurons subjected to trophic factor withdrawal. Electron microscopy was performed on cerebellar cultures to confirm that the enlarged MDC- and lysosensor-positive vacuoles observed after trophic factor withdrawal were indeed autophagic vacuoles. The electron micrographs revealed that the cytoplasm of identified Purkinje cells showed multiple autophagosomes, multivesicular bodies, and other secondary lysosomes ranging in size from 0.7 to 3.0 μm after 24 hr of trophic factor withdrawal (Fig. 4). Some of these autophagic structures contained ribosomes, glycogen, and disorganized membranous organelles (Fig. 4A,B, asterisks, C). Occasionally, a double membrane was observed extending around cytoplasmic contents (Fig. 4C, white arrows), indicative of earlier stages of autophagy (Shelburne et al., 1973). The cytoplasm of Purkinje neurons also contained lipid droplets (Fig. 4A). In Purkinje neurons, the nuclear chromatin was uniformly dispersed (Fig. 4A). In contrast to the Purkinje neurons, large autophagolysosomes were absent from the granule neurons, which instead showed mild cytoplasmic condensation and peripheral chromatin clumping (Fig. 4A, black arrow).
Figure 4.
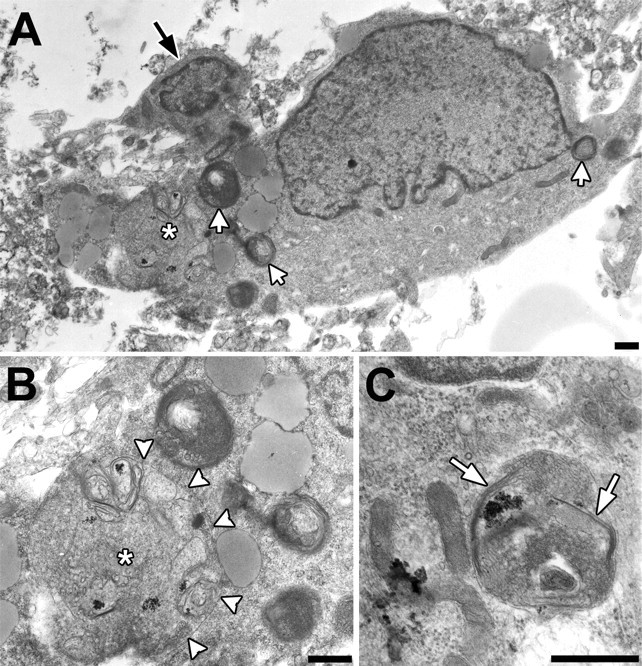
Transmission electron microscopy of Purkinje neurons after 24 hr of trophic factor withdrawal demonstrates ultra-structural features of autophagy. A, Low-power micrograph of a large Purkinje cell with multiple secondary lysosomes (white arrows) and a large autophagic vacuole (asterisk). In contrast, the smaller cerebellar granule neuron (black arrow) does not show these changes. B, Detail of the large autophagic vacuole indicated by the asterisk. There are disorganized membranous structures consistent with endoplasmic reticulum, ribosomes, and glycogen, delimited predominantly by a single membrane (white arrowheads). C, Micrograph showing an autophagosome delimited by a double membrane (white arrows). Note the similarity of the contents to adjacent mitochondria and cytoplasmic glycogen (lower left). Scale bars, 500 nm.
Purkinje neuron autophagy and death are caspase-independent
Previous studies have shown that trophic factor withdrawal and other death-inducing stimuli can simultaneously induce both autophagy and apoptosis (Jia et al., 1997; Xue et al., 1999; Uchiyama, 2001). In these models, caspase activation and apoptosis occurred downstream of autophagy and could be blocked by 3MA. To determine whether the autophagic death pathway in Purkinje neurons acted in concert with caspase activation, we added the broad-spectrum caspase inhibitor zVAD-FMK to Purkinje neurons undergoing trophic factor withdrawal. Addition of zVAD-FMK (100 μm), which effectively inhibits caspase activity in cerebellar granule neurons, was unable to block Purkinje vacuolation or death. The numbers of Purkinje neurons remaining after 24 hr of trophic factor withdrawal in the absence and presence of zVAD-FMK were 55.7 ± 2.8 and 54.7 ± 3.5, respectively. Furthermore, we were unable to detect increases in activated caspase-3 by immunocytochemical techniques in Purkinje neurons undergoing death induced by trophic factor withdrawal (data not shown). In contrast, activated caspase-3 immunoreactivity increases markedly in the granule neurons after trophic factor withdrawal (Linseman et al., 2003). These results suggest that neither autophagy nor death of Purkinje neurons required caspase activation.
NGF promotes survival and decreases autophagy in Purkinje neurons
Having established that trophic factor withdrawal induced autophagy and death of cerebellar Purkinje neurons, we next examined whether neurotrophin signaling could protect Purkinje neurons in this model. NGF is a neurotrophin known to promote the survival and differentiation of many types of neurons both in vitro and in vivo, including Purkinje neurons (Legrand and Clos, 1991; Cohen-Cory et al., 1993; Mount et al., 1998). We added NGF at various concentrations to the cultures at the time of trophic factor withdrawal and determined its effect on Purkinje survival. High concentrations of NGF (>50 ng/ml) significantly increased Purkinje numbers compared with trophic factor withdrawal alone (Fig. 5E). To determine whether NGF could prevent extensive autophagic vacuolation, cultures were subjected to trophic factor withdrawal in the absence or presence of NGF, and quantitative analysis of vacuolation was performed using live cell lysosensor measurements (Fig. 5G). Again, in the presence of trophic support, the majority of Purkinje neurons contained small (<0.75 μm) lysosensor-positive vacuoles. After 24 hr of trophic factor withdrawal, most Purkinje neurons contained very large vacuoles, with 49% of vacuoles between 0.75 and 1.5 μm, ∼21% of vacuoles between 1.5 and 2.25 μm, and ∼13% of vacuoles >2.25 μm. The addition of NGF (2.5 μg/ml) to the trophic factor withdrawal media significantly decreased the overall vacuolation of Purkinje neurons. The lysosome size profile was shifted, with the majority of vacuoles (∼76%) being <0.75 μm in diameter, ∼19% of vacuoles between 0.75 and 1.5 μm, ∼4% of vacuoles between 1.5 and 2.25 μm, and <1% of vacuoles >2.25 μm. Images shown are representative of the overall effects of the various treatments (Fig. 5A-C).
Figure 5.
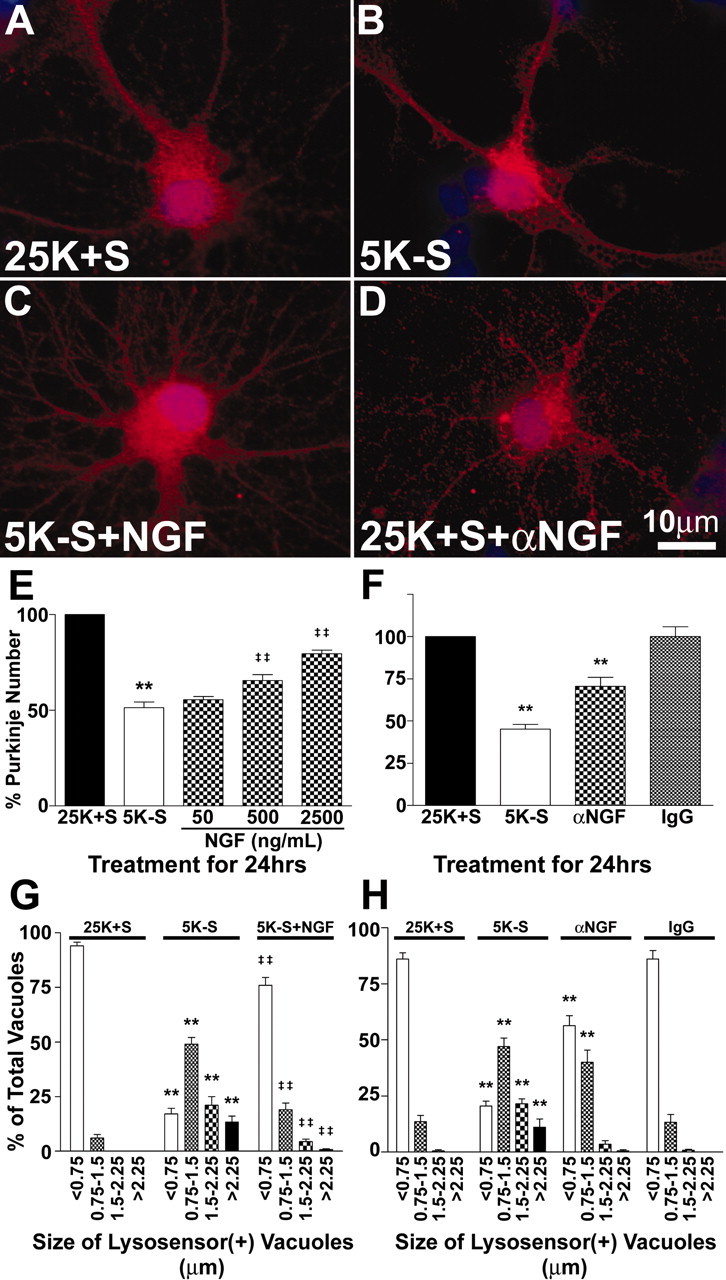
The neurotrophin NGF blocks the loss and the increased vacuolation of Purkinje neurons. A-D, Purkinje neurons maintained in various conditions were fixed and stained with antibodies to calbindin-D28k (a specific marker of Purkinje neurons; red) and the nuclear dye DAPI (blue). Images shown are representative of the effects of the various treatments. Purkinje neurons were maintained in control medium (A, 25K+S), trophic factor withdrawal medium (B, 5K-S), trophic factor withdrawal medium with 2500 ng/ml NGF (C, 5K-S+NGF), and control medium with 2 μg/ml NGF-neutralizing antibodies (D, 25K+S+αNGF) for 24 hr. E, Quantitation of the effects of NGF on Purkinje neuron numbers demonstrates that NGF partially rescues Purkinje neurons from death in a concentration-dependent manner after 24 hr of trophic factor withdrawal, compared with healthy controls. F, Quantitation of the effects of NGF-neutralizing antibodies on Purkinje neuron numbers demonstrates that depleting NGF from control media results in a loss of Purkinje neurons, whereas control antibodies had no effect on Purkinje survival. Numbers are mean ± SEM values of at least three independent experiments, each performed in triplicate. G, H, The effects of the various treatments on vacuole size were quantified by measuring the diameters of all visible lysosensor blue-positive vacuoles in 6-12 Purkinje neurons per treatment condition in at least three independent experiments. Usually, the total number of vacuoles measured per condition was between 100 and 200. The size distribution was graphed as a percentage of total vacuoles that were within the indicated size ranges. G, A high concentration of NGF (2500 ng/ml) significantly decreased the autophagic vacuolation of Purkinje neurons induced by 24 hr of trophic factor withdrawal. H, NGF-neutralizing antibodies significantly increased the vacuolation of Purkinje neurons in control media. E-H, **, *Significant difference from 25K+S (**p < 0.01; *p < 0.05). ‡‡,‡Significant difference from 5K-S (‡‡p < 0.01; ‡p < 0.05).
To determine whether decreasing NGF in the cerebellar cultures would be sufficient to induce autophagy and death of Purkinje neurons, we added NGF-neutralizing antibodies to the media and determined the effects on Purkinje survival and vacuolation. Addition of NGF-neutralizing antibodies to the cultures for 24 hr reduced the numbers of Purkinje neurons to 70.6 ± 5.3% of controls (Fig. 5F). Control IgG had no effect on the number of Purkinje neurons. The effect of the NGF-neutralizing antibodies was less than the effect of complete trophic factor withdrawal, which reduced Purkinje numbers to 45.2 ± 2.7% of control (Fig. 5F). To determine whether the NGF-neutralizing antibodies could induce autophagic vacuolation in Purkinje neurons, we performed live cell imaging experiments to measure the sizes of Purkinje lysosomes (Fig. 5H). Again, control Purkinje neurons contained mostly small lysosensor-positive vacuoles, whereas 24 hr of trophic factor withdrawal markedly induced the appearance of much larger vacuoles. Addition of NGF-neutralizing antibodies similarly increased the appearance of larger vacuoles in Purkinje neurons, although not to the same extent as trophic factor withdrawal. Control IgG had no effect on the size distribution of the vacuoles. The ability of NGF-neutralizing antibodies to induce the formation of vacuoles was also observed by calbindin staining (Fig. 5, compare A, D).
The protective effects of NGF on Purkinje neurons are independent of TrkA signaling
NGF exerts its effects by binding to two distinct receptors, the TrkA receptor and p75ntr. The high concentrations of NGF required to promote Purkinje survival suggested that the p75ntr receptor mediated the neuroprotective effects of NGF in these cultures. Further data supporting this hypothesis come from experiments showing that TrkA receptor tyrosine phosphorylation is high under basal conditions and does not change on trophic factor withdrawal (up to 6 hr) or on addition of high doses of NGF (Fig. 6A,B). Furthermore, Purkinje neurons demonstrated significantly increased autophagic vacuolation (within 3–4 hr of trophic factor withdrawal, as assessed by live cell lysosensor measurements of vacuole size; Fig. 6C) before any significant decrease in TrkA activation.
Figure 6.

The protective effects of NGF are not mediated by TrkA. Immunoblot analysis was performed to determine the effects of trophic factor withdrawal and various doses of NGF on TrkA activation. A, Immunoblot of samples from cultures subjected to 0, 1, 2, 4, and 6 hr of trophic factor withdrawal with a phosphotyrosine-specific Ab. B, Immunoblot of samples from cultures subjected to acid wash, followed by 2 hr of trophic factor withdrawal and then incubation with 0, 5, 50, 500, or 1000 ng/ml of NGF for 10 min. C, The effects of a short time course of trophic factor withdrawal on vacuole size were quantified by measuring the diameters of all visible lysosensor blue-positive vacuoles in 6-12 Purkinje neurons per treatment condition in at least three independent experiments. Usually, the total number of vacuoles measured per condition was between 100 and 200. The size distribution was graphed as a percentage of total vacuoles that were within the indicated size ranges. **, *Significant difference from 25K+S (**p < 0.01; *p < 0.05).
In the presence of trophic factors, the p75ntr supports Purkinje neuron survival
The p75 neurotrophin receptor is expressed throughout the developing cerebellum and demonstrates specific spatial and temporal regulation (Yan and Johnson, 1988; Carter et al., 2003). In particular, p75ntr is expressed in the developing Purkinje neurons by postnatal day 7, the time point at which we obtained our cerebellar cultures. p75ntr immunoreactivity gradually decreases until it becomes absent from adult cerebellum; however, injury such as axotomy of Purkinje neurons results in marked re-expression of p75ntr in the injured neurons (Martinez-Murillo et al., 1993). These data suggest that p75ntr is involved in both the development of the cerebellum and injury responses in Purkinje neurons.
To determine whether p75ntr was involved in mediating autophagy and death of Purkinje neurons, cerebellar cultures were incubated with antisense oligonucleotides directed against a 5′ region spanning the initiation codon of the rat p75ntr mRNA. This antisense sequence has been shown to be effective at preventing the NGF withdrawal or axotomy-induced death of dorsal root ganglion neurons (Barrett and Bartlett, 1994; Cheema et al., 1996) and the axotomy-induced death of spinal motor neurons (Lowry et al., 2001), which are mediated by p75ntr. Dissociated cerebellar tissue was triturated with 5 μm 56-FAM-labeled antisense or missense phosphorothioate oligonucleotides at the time of plating. The antisense entered both granule neurons and Purkinje neurons in the culture, with nearly 100% efficiency as assessed by fluorescence microscopy. The p75ntr antisense had two distinct effects on the neurons in the culture. Inclusion of the antisense in the culture medium for 7 d resulted in a significant decrease in the basal survival of both cerebellar granule and Purkinje neurons. Purkinje neuron number with the p75ntr antisense was 35 ± 2.3% of control cultures. The effect of the p75ntr antisense on cell numbers was specific because cultures treated with scrambled missense oligonucleotides did not demonstrate diminished survival. Also, the loss of cells induced by p75ntr antisense was not apparent until approximately day 4 or 5 in culture (as assed by cell density by bright-field examination of the cultures). This corresponds to the time in culture when NGF production is first detectable by Western blot analysis (data not shown). These findings suggest that p75ntr mediates survival in response to autocrine and paracrine neurotrophic factors such as NGF and possibly other neurotrophins such as brain-derived neurotrophic factor (BDNF) or neurotrophin-3 (NT-3), which are known to be secreted by cells in these cerebellar cultures (Favaron et al., 1993; Leingartner et al., 1994; Condorelli et al., 1998; Marini et al., 1998; Bhave et al., 1999).
p75ntr promotes autophagy and death of Purkinje neurons in the absence of trophic stimuli
In contrast to its effects on basal survival, the p75ntr antisense completely blocked the death of Purkinje neurons induced by 48 hr of trophic factor withdrawal (Fig. 7A). Moreover, 5 μm antisense decreased the autophagic vacuolation of Purkinje neurons elicited by 24 hr of trophic factor withdrawal (Fig. 7B). Again, in the presence of trophic support, most (∼90%) of the Purkinje neuron lysosensor-positive vacuoles were <0.75 μm, and only ∼10% of vacuoles were between 0.75 and 1.5 μm, with no vacuoles >1.5 μm in diameter After 24 hr of trophic factor withdrawal, most Purkinje neurons contained very large vacuoles, with ∼49% of vacuoles between 0.75 and 1.5 μm, ∼13% of vacuoles between 1.5 and 2.25 μm, and ∼11% of vacuoles >2.25 μm. Cultures that were preincubated with p75ntr antisense oligonucleotides demonstrated a shifted lysosome size profile, with ∼70% of vacuoles <0.75 μm in diameter, ∼26% of vacuoles between 0.75 and 1.5 μm, ∼4% of vacuoles between 1.5 and 2.25 μm, and <1% of vacuoles >2.25 μm. In contrast, missense oligonucleotides had no effect on the lysosome size profile of Purkinje neurons deprived of trophic support (Fig. 7B). Collectively, the above results indicate that antisense-mediated depletion of p75ntr decreases the basal survival of Purkinje neurons in the presence of trophic support. However, under conditions of trophic factor withdrawal, decreasing p75ntr provides a survival advantage to the remaining Purkinje neurons. The ability of p75ntr to mediate both the survival and death in the same neuron depending on cellular context is consistent with previous reports of its effects on dorsal root ganglion neuron survival (Barrett and Bartlett, 1994; Sorensen et al., 2003).
Figure 7.
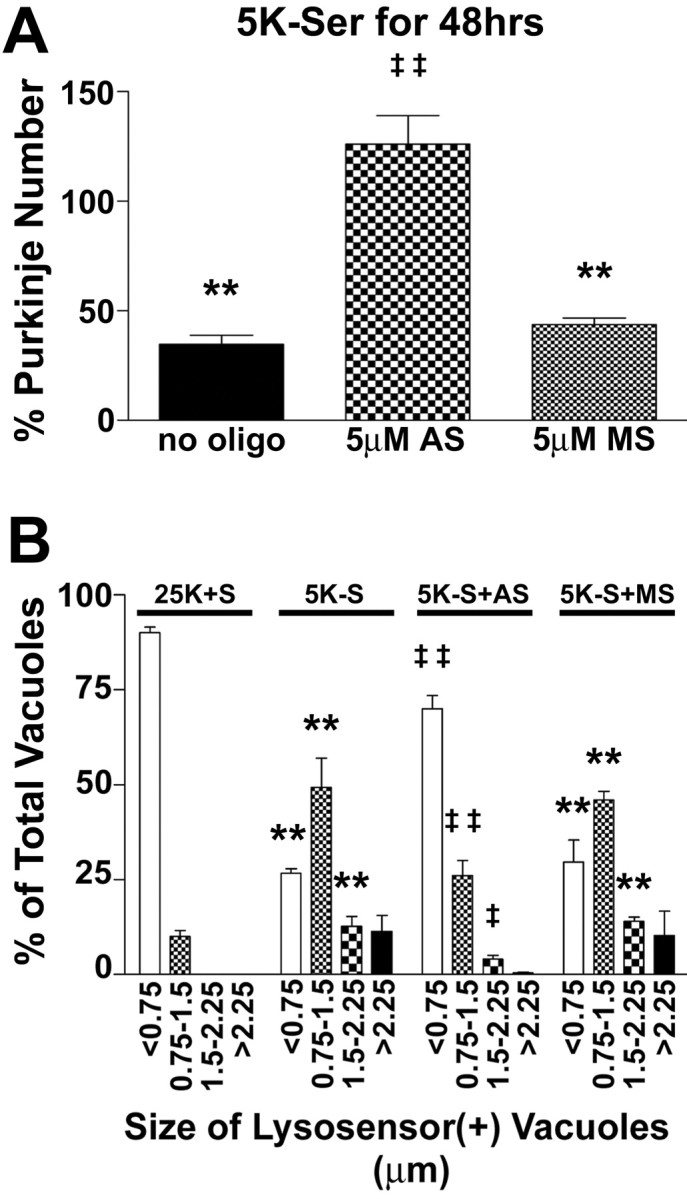
p75ntr antisense inhibits the loss and vacuolation of Purkinje neurons. Purkinje neurons maintained in various conditions were fixed and stained with antibodies to calbindin-D28k. A, Quantitation of the effects p75ntr antisense (AS) and missense (MS) oligonucleotides (oligo) on Purkinje neuron numbers expressed as the percentage of the appropriate control demonstrates that p75ntr antisense completely blocks the loss of Purkinje neurons induced by trophic factor withdrawal, whereas missense oligos had no effect on Purkinje survival. Numbers are mean ± SEM values of at least three independent experiments, each performed in triplicate. B, The effects of the various treatments on vacuole size were quantified by measuring the diameters of all visible lysosensor blue-positive vacuoles in 6-12 Purkinje neurons per treatment condition in at least three independent experiments. Usually, the total number of vacuoles measured per condition was between 100 and 200. The size distribution was graphed as a percentage of total vacuoles that were within the indicated size ranges. **, *Significant difference from 25K+S (**p < 0.01; *p < 0.05). ‡‡,‡Significant difference from 5K-S (‡‡p < 0.01; ‡p < 0.05).
Overexpression of a truncated p75ntr lacking the extracellular domain induces Purkinje neuron autophagy and death in the presence of trophic support
To provide direct evidence that the p75ntr can induce autophagy and death in Purkinje neurons, we investigated the effects of adenoviral-mediated overexpression of a myristoylated rat p75ntr intracellular domain (p75mICD) protein. This p75ntr truncation mutant was used because it lacks the ligand-binding domain, which would complicate the interpretation of results because of the presence of endogenous neurotrophins in these cultures. It has been shown that relatively low expression levels of this p75ntr mutant mediate the survival of PC12 cells. In contrast, high levels of p75mICD expression induce efficient PC12 cell death (Roux et al., 2001). Finally, it has been demonstrated that the intracellular domain possesses the potent cell death-inducing activity of p75ntr (Majdan et al., 1997; Coulson et al., 2000; Rabizadeh et al., 2000; Murray et al., 2003). Cultures were infected with recombinant control CMV or p75mICD adenoviruses at various multiplicities of infection (moi) on day 5 in culture. After incubation for 48 hr, the cultures were fixed and stained with polyclonal antibodies raised against the intracellular domain of rat p75ntr (Majdan et al., 1997), followed by a Cy3-conjugated secondary antibody. Increases in p75ntr immunoreactivity after infection with the p75mICD adenovirus were primarily and consistently observed in cells with morphologies typical of Purkinje neurons. Semiquantitative analysis of fluorescence images indicated that p75ntr expression in p75mICD-infected Purkinje neurons was increased by 3.4-fold compared with uninfected controls or cells infected with control CMV adenovirus. To determine whether overexpression of p75mICD could induce Purkinje neuron death, infected neurons were fixed and stained with calbindin antibodies to quantify Purkinje numbers. Overexpression of p75mICD resulted in a dose-dependent loss of Purkinje neurons, with infection at 50 moi yielding similar levels of Purkinje cell loss, as was observed with trophic factor withdrawal (Fig. 8A). Infection with a control adenovirus at 50 moi did not induce significant Purkinje neuron death (Fig. 8A). To determine whether overexpression of p75mICD could induce autophagic vacuolation in Purkinje neurons, live cell quantification of lysosome size was performed (Fig. 8B). Purkinje neurons that were infected with 50 moi of p75mICD adenovirus demonstrated significantly increased autophagic vacuolation, with only ∼58% of vacuoles <0.75 μm, ∼32% of vacuoles between 0.75 and 1.5 μm, ∼7% of vacuoles between 1.5 and 2.25 μm, and ∼4% of vacuoles >2.25 μm. Infection with 50 moi of control CMV adenovirus did not significantly increase Purkinje neuron vacuolation. In addition to increasing autophagic vacuolation, p75mICD infection caused notable membrane blebbing and, in some rare cases, nuclear condensation and fragmentation in Purkinje neurons. Our observations of increased autophagy with p75mICD expression are consistent with a previous report of p75ntr intracellular domain-induced death being morphologically characterized by increased vacuolation (Coulson et al., 2000), in addition to the regularly reported morphological features of apoptosis.
Figure 8.
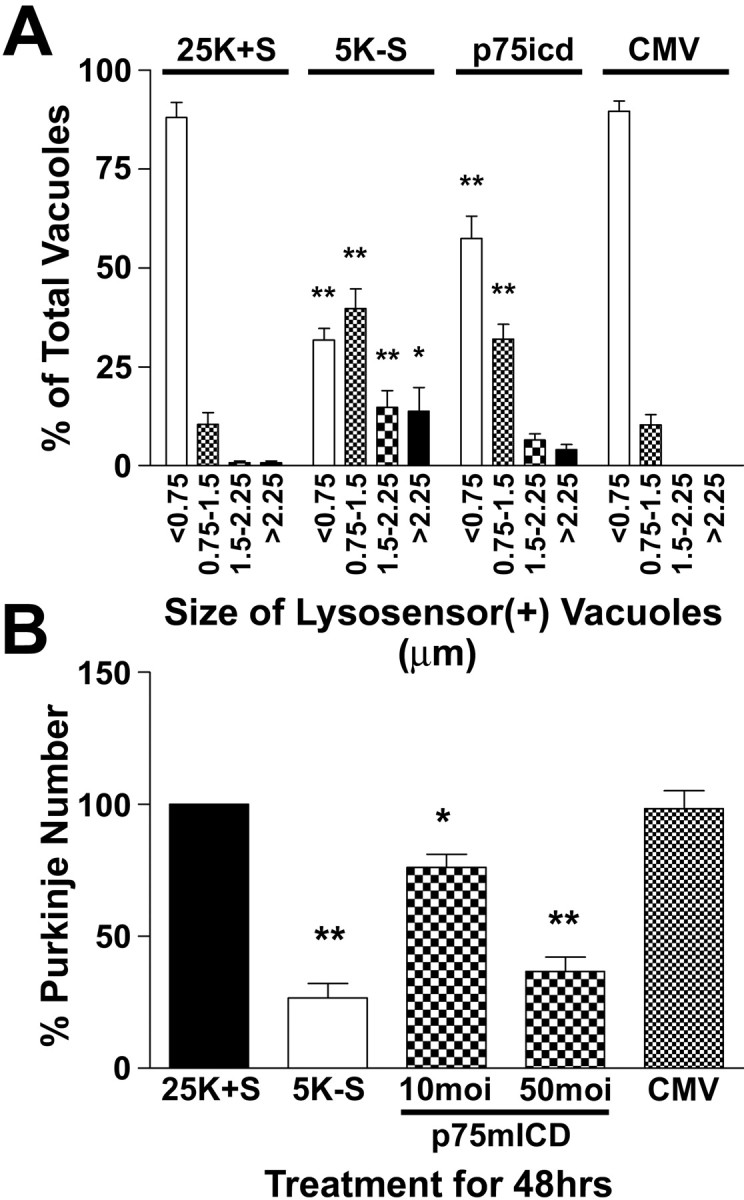
Overexpression of a truncated p75ntr receptor lacking the ligand-binding domain induces the loss and vacuolation of Purkinje neurons. Cultures were subjected to trophic factor withdrawal or adenoviral infection with either control CMV (at 50 moi) or myristoylated intracellular domain p75ntr (at both 10 and 50 moi) adenoviruses on DIV 5. A, The cells were fixed and stained with calbindin antibodies to determine Purkinje neuron numbers after the various treatments. B, The effects of p75mICD and CMV infection at 50 moi on vacuole size were quantified by measuring the diameters of all visible lysosensor blue-positive vacuoles in 6-12 Purkinje neurons per treatment condition in at least three independent experiments. Usually, the total number of vacuoles measured per condition was between 100 and 200. The size distribution was graphed as a percentage of total vacuoles that were within the indicated size ranges. **, *Significant difference from 25K+S (**p < 0.01; *p < 0.05).
Discussion
Morphological studies of diseased human postmortem brain consistently reveal signs of increased autophagic activity in degenerating neuronal populations. There are multiple reports in the literature that describe autophagic vacuoles, as well as disturbances in the lysosomal degradative system in neurodegenerative conditions such as Alzheimer's disease (Cataldo et al., 1996; Nixon et al., 2000), Parkinson's disease (Anglade et al., 1997), Huntington's disease (Kegel et al., 2000; Petersen et al., 2001), prion encephalopathies (Boellaard et al., 1991; Jeffrey et al., 1992), and diffuse Lewy body disease (Zhu et al., 2003). Extensive cytoplasmic vacuole formation, consistent with autophagy, also has been described in degenerating cerebellar Purkinje neurons in models of ischemia (Fessatidis et al., 1993; Barenberg et al., 2001) and in spinocerebellar ataxia 1 transgenic mice (Skinner et al., 2001). Recently, autophagy has been implicated in the death of cerebellar Purkinje neurons in the Lurcher mouse, which suffers from extensive Purkinje neuron degeneration caused by a point mutation in the δ2 glutamate receptor (Zuo et al., 1997). Two reports have also shown that Lurcher Purkinje neurons demonstrate ultrastructural features of autophagy at a time when they begin to degenerate (Yue et al., 2002; Selimi et al., 2003). In addition, overexpression of the mutant glutamate receptor induced autophagy and death in human embryonic kidney 293 cells (Yue et al., 2002). In the current study, we have shown that autophagy is greatly enhanced in Purkinje neurons that are dying as a result of trophic factor deprivation, and treatments that decrease autophagy are associated with increased survival, lending further support to the hypothesis that enhanced autophagy is involved in Purkinje neuron degeneration. Moreover, we have identified a role for p75ntr as a possible mediator of autophagy and death in Purkinje neurons.
It is generally accepted that the neurotrophins NGF, BDNF, NT-3, and NT-4/5 promote survival and differentiation by binding to their cognate Trk receptors (Ibanez et al., 1992). In contrast, deciphering the role of p75ntr signaling in mediating neurotrophin effects has been complicated by several factors. First, p75ntr shares structural homologies with members of the tumor necrosis factor receptor family of death receptors and lacks intrinsic tyrosine kinase activity. Second, all four neurotrophins are able to bind to p75ntr with similar affinity but display selectivity when binding to the Trk receptors (Klein et al., 1991a,b; Lamballe et al., 1991). Third, p75ntr is often coexpressed with Trk receptors, and neurotrophin effects are dependent on the relative complements of Trk and p75 neurotrophin receptors (for review, see Rabizadeh and Bredesen, 2003). In this context, it has been shown that the presence of p75ntr is required for high-affinity binding of neurotrophins to Trk receptors (Hempstead et al., 1991). Yet another layer of complexity is added when one considers that unprocessed proneurotrophins demonstrate higher-affinity binding to p75ntr but have negligible binding to Trk receptors (Lee et al., 2001). Finally, p75ntr may also function as a coreceptor with the Nogo receptor (Wang et al., 2002) or as a receptor for prion (Della-Bianca et al., 2001) and β-amyloid proteins (Yaar et al., 1997). These complexities in neurotrophin signaling may provide some explanation for the paradoxical roles ascribed to p75ntr in the literature. Expression of p75ntr is capable of inducing death, especially when neurotrophin concentrations are limited (Rabizadeh et al., 1993; Barrett and Georgiou, 1996). In addition, p75ntr signaling can result in death in response to neurotrophin binding (Casaccia-Bonnefil et al., 1996; Frade et al., 1996; Kuner and Hertel, 1998; Friedman, 2000). In contrast, other reports suggest that p75ntr plays an important and necessary role in mediating survival in response to neurotrophins (Barrett and Bartlett, 1994; Barrett et al., 1998; DeFreitas et al., 2001; Bui et al., 2002).
In the current report, we provide evidence that p75ntr plays an important role in mediating autophagy and death of cerebellar Purkinje neurons induced by trophic factor withdrawal. The promotion of survival and the reduction of autophagic vacuolation required high concentrations of NGF, suggesting the involvement of p75ntr. TrkA phosphorylation was not correlated with the autophagic vacuolation induced by trophic factor withdrawal or with neuroprotection mediated by NGF. Decreasing available neurotrophin with NGF-neutralizing antibodies induced autophagic vacuolation and death of Purkinje neurons. Antisense to p75ntr decreased autophagy and completely inhibited the loss of Purkinje neurons in response to trophic factor withdrawal. Moreover, adenoviral-mediated overexpression of a myristoylated rat p75ntr intracellular domain protein (p75mICD) resulted in significantly increased autophagic vacuolation that was also accompanied by a significant loss of Purkinje neurons. Finally, our results directly show that Purkinje neuron death induced by either trophic factor withdrawal or p75mICD overexpression is associated with increased autophagic vacuolation, suggesting an important role for autophagy during Purkinje neuron degeneration.
Our results with p75ntr antisense suggest a dual role for p75ntr in mediating both the survival and death of Purkinje neurons. Antisense to p75ntr decreased the survival of Purkinje neurons maintained in healthy conditions. The loss of neurons induced with p75ntr antisense occurred at a time when NGF synthesis becomes detectable, suggesting that p75ntr is required for the basal survival of the cerebellar neurons that is mediated by autocrine and paracrine neurotrophin production in theses cultures. In contrast, Purkinje neurons pretreated with p75ntr antisense were completely resistant to death and demonstrated decreased vacuolation in response to trophic factor withdrawal. Collectively, our results suggest that neurotrophin signaling through p75ntr can regulate the survival and death of Purkinje neurons. One can speculate that in the presence of appropriate neurotrophin, signaling from p75ntr can promote Purkinje survival, and decreasing neurotrophin levels can lead to a loss of prosurvival p75ntr signals. However, it is also possible that a loss of appropriate neurotrophins not only leads to a loss of prosurvival signals but also may result in the generation of prodeath signals from p75ntr. This concept of p75ntr as a dependence receptor has been hypothesized by others to explain the paradoxical nature of p75ntr function (Rabizadeh et al., 2000). In addition, our data have not formally ruled out the possibility that trophic factor withdrawal results in an increase of some ligand that promotes prodeath p75ntr signals because there is evidence in the literature that p75ntr may have other ligands or may also function as a coreceptor with other proteins (Yaar et al., 1997; Della-Bianca et al., 2001; Wang et al., 2002).
Despite the evidence suggesting that autophagy is upregulated in neurodegeneration, very little is known about the cellular mechanisms that regulate autophagy in neurons (for review, see Yuan et al., 2003). Several reports suggest that the activation of autophagy contributes to the death of neurons (Xue et al., 1999; Uchiyama, 2001; Borsello et al., 2003; Weeks, 2003). In contrast, other studies indicate a neuroprotective role for autophagy in Huntington's disease and α-synucleopathies (Qin et al., 2003; Webb et al., 2003). These studies demonstrate that autophagy may be a mechanism for mutant protein degradation and the prevention of plaque formation. Similarly, in cases in which mitochondria are extensively damaged, autophagy may be protective by sequestering and degrading defective mitochondria before they can release death-inducing proteins (Lemasters et al., 2002; Tolkovsky et al., 2002). One can speculate that the degree and specificity of autophagy may determine cell fate. By elucidating the molecular pathways that regulate autophagy, it may be possible to determine which components of autophagy may contribute to neurodegeneration and which may be neuroprotective. In addition to the questions regarding autophagy as a survival or death mechanism, it will be of great importance to understand the effects of autophagy on neuronal function, such as the formation and maintenance of synapses, the transduction of electrical impulses, and neurotransmitter release.
Footnotes
This work was supported by a Department of Veterans Affairs merit award (K.A.H.), Department of Defense Grant DAMD17-99-1-9481 (K.A.H.), National Institutes of Health Grants NS38619-01A1 (K.A.H.) and NS40817 (C.T.C.), a Department of Veterans Affairs Research Enhancement Program award (K.A.H., D.A.L.), and a National Research Service award (M.L.F.-M.). We thank Ardith Ries of the University of Pittsburgh electron microscopy laboratory for technical assistance and Dr. John Shelburne of the Duke University and Veterans Affairs Medical Centers (Durham, NC) for helpful discussion.
Correspondence should be addressed to Kim A. Heidenreich, Department of Pharmacology, C236, University of Colorado Health Sciences Center, 4200 East Ninth Avenue, Denver, CO 80262. E-mail: kim.heidenreich@uchsc.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/244498-12$15.00/0
References
- Anglade P, Vyas S, Javoy-Agid F, Herrero MT, Michel PP, Marquez J, Mouatt-Prigent A, Ruberg M, Hirsch EC, Agid Y (1997) Apoptosis and autophagy in nigral neurons of patients with Parkinson's disease. Histol Histopathol 12: 25-31. [PubMed] [Google Scholar]
- Bailey A, Luthert P, Dean A, Harding B, Janota I, Montgomery M, Rutter M, Lantos P (1998) A clinicopathological study of autism. Brain 121: 889-905. [DOI] [PubMed] [Google Scholar]
- Baptista CA, Hatten ME, Blazeski R, Mason CA (1994) Cell-cell interactions influence survival and differentiation of purified Purkinje cells in vitro. Neuron 12: 243-260. [DOI] [PubMed] [Google Scholar]
- Barenberg P, Strahlendorf H, Strahlendorf J (2001) Hypoxia induces an excitotoxic-type of dark cell degeneration in cerebellar Purkinje neurons. Neurosci Res 40: 245-254. [DOI] [PubMed] [Google Scholar]
- Barrett GL, Bartlett PF (1994) The p75 nerve growth factor receptor mediates survival or death depending on the stage of sensory neuron development. Proc Natl Acad Sci USA 91: 6501-6505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett GL, Georgiou A (1996) The low-affinity nerve growth factor receptor p75NGFR mediates death of PC12 cells after nerve growth factor withdrawal. J Neurosci Res 45: 117-128. [DOI] [PubMed] [Google Scholar]
- Barrett GL, Georgiou A, Reid K, Bartlett PF, Leung D (1998) Rescue of dorsal root sensory neurons by nerve growth factor and neurotrophin-3, but not brain-derived neurotrophic factor or neurotrophin-4, is dependent on the level of the p75 neurotrophin receptor. Neuroscience 85: 1321-1328. [DOI] [PubMed] [Google Scholar]
- Bhave SV, Ghoda L, Hoffman PL (1999) Brain-derived neurotrophic factor mediates the anti-apoptotic effect of NMDA in cerebellar granule neurons: signal transduction cascades and site of ethanol action. J Neurosci 19: 3277-3286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biederbick A, Kern HF, Elsasser HP (1995) Monodansylcadaverine (MDC) is a specific in vivo marker for autophagic vacuoles. Eur J Cell Biol 66: 3-14. [PubMed] [Google Scholar]
- Boellaard JW, Kao M, Schlote W, Diringer H (1991) Neuronal autophagy in experimental scrapie. Acta Neuropathol (Berl) 82: 225-228. [DOI] [PubMed] [Google Scholar]
- Borghesani PR, Alt FW, Bottaro A, Davidson L, Aksoy S, Rathbun GA, Roberts TM, Swat W, Segal RA, Gu Y (2000) Abnormal development of Purkinje cells and lymphocytes in Atm mutant mice. Proc Natl Acad Sci USA 97: 3336-3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borsello T, Croquelois K, Hornung JP, Clarke PG (2003) N-methyl-d-aspartate-triggered neuronal death in organotypic hippocampal cultures is endocytic, autophagic and mediated by the c-Jun N-terminal kinase pathway. Eur J Neurosci 18: 473-485. [DOI] [PubMed] [Google Scholar]
- Bui NT, Konig HG, Culmsee C, Bauerbach E, Poppe M, Krieglstein J, Prehn JH (2002) p75 neurotrophin receptor is required for constitutive and NGF-induced survival signalling in PC12 cells and rat hippocampal neurones. J Neurochem 81: 594-605. [DOI] [PubMed] [Google Scholar]
- Carter AR, Berry EM, Segal RA (2003) Regional expression of p75NTR contributes to neurotrophin regulation of cerebellar patterning. Mol Cell Neurosci 22: 1-13. [DOI] [PubMed] [Google Scholar]
- Casaccia-Bonnefil P, Carter BD, Dobrowsky RT, Chao MV (1996) Death of oligodendrocytes mediated by the interaction of nerve growth factor with its receptor p75. Nature 383: 716-719. [DOI] [PubMed] [Google Scholar]
- Cataldo AM, Hamilton DJ, Barnett JL, Paskevich PA, Nixon RA (1996) Properties of the endosomal-lysosomal system in the human central nervous system: disturbances mark most neurons in populations at risk to degenerate in Alzheimer's disease. J Neurosci 16: 186-199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheema SS, Barrett GL, Bartlett PF (1996) Reducing p75 nerve growth factor receptor levels using antisense oligonucleotides prevents the loss of axotomized sensory neurons in the dorsal root ganglia of newborn rats. J Neurosci Res 46: 239-245. [DOI] [PubMed] [Google Scholar]
- Cohen-Cory S, Elliott RC, Dreyfus CF, Black IB (1993) Depolarizing influences increase low-affinity NGF receptor gene expression in cultured Purkinje neurons. Exp Neurol 119: 165-175. [DOI] [PubMed] [Google Scholar]
- Condorelli DF, Dell'Albani P, Timmusk T, Mudo G, Belluardo N (1998) Differential regulation of BDNF and NT-3 mRNA levels in primary cultures of rat cerebellar neurons. Neurochem Int 32: 87-91. [DOI] [PubMed] [Google Scholar]
- Coulson EJ, Reid K, Baca M, Shipham KA, Hulett SM, Kilpatrick TJ, Bartlett PF (2000) Chopper, a new death domain of the p75 neurotrophin receptor that mediates rapid neuronal cell death. J Biol Chem 275: 30537-30545. [DOI] [PubMed] [Google Scholar]
- DeFreitas MF, McQuillen PS, Shatz CJ (2001) A novel p75NTR signaling pathway promotes survival, not death, of immunopurified neocortical subplate neurons. J Neurosci 21: 5121-5129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Della-Bianca V, Rossi F, Armato U, Dal-Pra I, Costantini C, Perini G, Politi V, Della Valle G (2001) Neurotrophin p75 receptor is involved in neuronal damage by prion peptide-(106-126). J Biol Chem 276: 38929-38933. [DOI] [PubMed] [Google Scholar]
- D'Mello SR, Galli C, Ciotti T, Calissano P (1993) Induction of apoptosis in cerebellar granule neurons by low potassium: inhibition of death by insulin-like growth factor I and cAMP. Proc Natl Acad Sci USA 90: 10989-10993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favaron M, Manev RM, Rimland JM, Candeo P, Beccaro M, Manev H (1993) NMDA-stimulated expression of BDNF mRNA in cultured cerebellar granule neurones. NeuroReport 4: 1171-1174. [PubMed] [Google Scholar]
- Ferrer I, Saracibar N, Gonzalez G (1991) Spongiform encephalopathy and multisystemic degeneration. Neurologia 6: 29-33. [PubMed] [Google Scholar]
- Fessatidis IT, Thomas VL, Shore DF, Hunt RH, Weller RO (1993) Neuropathological features of profoundly hypothermic circulatory arrest: an experimental study in the pig. Cardiovasc Surg 1: 155-160. [PubMed] [Google Scholar]
- Frade JM, Rodriguez-Tebar A, Barde YA (1996) Induction of cell death by endogenous nerve growth factor through its p75 receptor. Nature 383: 166-168. [DOI] [PubMed] [Google Scholar]
- Friedman WJ (2000) Neurotrophins induce death of hippocampal neurons via the p75 receptor. J Neurosci 20: 6340-6346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatti RA, Vinters HV (1985) Cerebellar pathology in ataxia-telangiectasia: the significance of basket cells. Kroc Found Ser 19: 225-232. [PubMed] [Google Scholar]
- Ghez C, Thach WT (2000) The cerebellum. In: Principles of neuroscience (Kandel ER, Schwartz JH, Jessell TM, eds), pp 832-852. New York: McGraw-Hill.
- Hempstead BL, Martin-Zanca D, Kaplan DR, Parada LF, Chao MV (1991) High-affinity NGF binding requires coexpression of the trk proto-oncogene and the low-affinity NGF receptor. Nature 350: 678-683. [DOI] [PubMed] [Google Scholar]
- Ibanez CF, Ebendal T, Barbany G, Murray-Rust J, Blundell TL, Persson H (1992) Disruption of the low affinity receptor-binding site in NGF allows neuronal survival and differentiation by binding to the trk gene product. Cell 69: 329-341. [DOI] [PubMed] [Google Scholar]
- Jeffrey M, Scott JR, Williams A, Fraser H (1992) Ultrastructural features of spongiform encephalopathy transmitted to mice from three species of bovidae. Acta Neuropathol (Berl) 84: 559-569. [DOI] [PubMed] [Google Scholar]
- Jia L, Dourmashkin RR, Allen PD, Gray AB, Newland AC, Kelsey SM (1997) Inhibition of autophagy abrogates tumour necrosis factor alpha induced apoptosis in human T-lymphoblastic leukaemic cells. Br J Haematol 98: 673-685. [DOI] [PubMed] [Google Scholar]
- Kegel KB, Kim M, Sapp E, McIntyre C, Castano JG, Aronin N, DiFiglia M (2000) Huntingtin expression stimulates endosomal-lysosomal activity, endosome tubulation, and autophagy. J Neurosci 20: 7268-7278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein R, Jing SQ, Nanduri V, O'Rourke E, Barbacid M (1991a) The trk proto-oncogene encodes a receptor for nerve growth factor. Cell 65: 189-197. [DOI] [PubMed] [Google Scholar]
- Klein R, Nanduri V, Jing SA, Lamballe F, Tapley P, Bryant S, Cordon-Cardo C, Jones KR, Reichardt LF, Barbacid M (1991b) The trkB tyrosine protein kinase is a receptor for brain-derived neurotrophic factor and neurotrophin-3. Cell 66: 395-403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ, Emr SD (2000) Autophagy as a regulated pathway of cellular degradation. Science 290: 1717-1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koeppen AH (1998) The hereditary ataxias. J Neuropathol Exp Neurol 57: 531-543. [DOI] [PubMed] [Google Scholar]
- Kuner P, Hertel C (1998) NGF induces apoptosis in a human neuroblastoma cell line expressing the neurotrophin receptor p75NTR. J Neurosci Res 54: 465-474. [DOI] [PubMed] [Google Scholar]
- Lamballe F, Klein R, Barbacid M (1991) trkC, a new member of the trk family of tyrosine protein kinases, is a receptor for neurotrophin-3. Cell 66: 967-979. [DOI] [PubMed] [Google Scholar]
- Lasmezas CI, Deslys JP, Robain O, Jaegly A, Beringue V, Peyrin JM, Fournier JG, Hauw JJ, Rossier J, Dormont D (1997) Transmission of the BSE agent to mice in the absence of detectable abnormal prion protein. Science 275: 402-405. [DOI] [PubMed] [Google Scholar]
- Lee R, Kermani P, Teng KK, Hempstead BL (2001) Regulation of cell survival by secreted proneurotrophins. Science 294: 1945-1948. [DOI] [PubMed] [Google Scholar]
- Legrand C, Clos J (1991) Biochemical, immunocytochemical and morphological evidence for an interaction between thyroid hormone and nerve growth factor in the developing cerebellum of normal and hypothyroid rats. Dev Neurosci 13: 382-396. [DOI] [PubMed] [Google Scholar]
- Leingartner A, Heisenberg CP, Kolbeck R, Thoenen H, Lindholm D (1994) Brain-derived neurotrophic factor increases neurotrophin-3 expression in cerebellar granule neurons. J Biol Chem 269: 828-830. [PubMed] [Google Scholar]
- Lemasters JJ, Qian T, He L, Kim JS, Elmore SP, Cascio WE, Brenner DA (2002) Role of mitochondrial inner membrane permeabilization in necrotic cell death, apoptosis, and autophagy. Antioxid Redox Signal 4: 769-781. [DOI] [PubMed] [Google Scholar]
- Liberski PP, Gajdusek DC, Brown P (2002) How do neurons degenerate in prion diseases or transmissible spongiform encephalopathies (TSEs): neuronal autophagy revisited. Acta Neurobiol Exp (Warsz) 62: 141-147. [DOI] [PubMed] [Google Scholar]
- Linseman DA, McClure ML, Bouchard RJ, Laessig TA, Ahmadi FA, Heidenreich KA (2002a) Suppression of death receptor signaling in cerebellar Purkinje neurons protects neighboring granule neurons from apoptosis via an insulin-like growth factor I-dependent mechanism. J Biol Chem 277: 24546-24553. [DOI] [PubMed] [Google Scholar]
- Linseman DA, Phelps RA, Bouchard RJ, Le SS, Laessig TA, McClure ML, Heidenreich KA (2002b) Insulin-like growth factor-I blocks Bcl-2 interacting mediator of cell death (Bim) induction and intrinsic death signaling in cerebellar granule neurons. J Neurosci 22: 9287-9297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linseman DA, Bartley CM, Le SS, Laessig TA, Bouchard RJ, Meintzer MK, Li M, Heidenreich KA (2003) Inactivation of the myocyte enhancer factor-2 repressor histone deacetylase-5 by endogenous Ca2+/calmodulin-dependent kinase II promotes depolarization-mediated cerebellar granule neuron survival. J Biol Chem 278: 41472-41481. [DOI] [PubMed] [Google Scholar]
- Lowry KS, Murray SS, Coulson EJ, Epa R, Bartlett PF, Barrett G, Cheema SS (2001) Systemic administration of antisense p75(NTR) oligodeoxynucleotides rescues axotomised spinal motor neurons. J Neurosci Res 64: 11-17. [DOI] [PubMed] [Google Scholar]
- Majdan M, Lachance C, Gloster A, Aloyz R, Zeindler C, Bamji S, Bhakar A, Belliveau D, Fawcett J, Miller FD, Barker PA (1997) Transgenic mice expressing the intracellular domain of the p75 neurotrophin receptor undergo neuronal apoptosis. J Neurosci 17: 6988-6998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marini AM, Rabin SJ, Lipsky RH, Mocchetti I (1998) Activity-dependent release of brain-derived neurotrophic factor underlies the neuroprotective effect of N-methyl-d-aspartate. J Biol Chem 273: 29394-29399. [DOI] [PubMed] [Google Scholar]
- Martinez-Murillo R, Caro L, Nieto-Sampedro M (1993) Lesion-induced expression of low-affinity nerve growth factor receptor-immunoreactive protein in Purkinje cells of the adult rat. Neuroscience 52: 587-593. [DOI] [PubMed] [Google Scholar]
- Mitchener JS, Shelburne JD, Bradford WD, Hawkins HK (1976) Cellular autophagocytosis induced by deprivation of serum and amino acids in HeLa cells. Am J Pathol 83: 485-491. [PMC free article] [PubMed] [Google Scholar]
- Mount HT, Elkabes S, Dreyfus CF, Black IB (1998) Differential involvement of metabotropic and p75 neurotrophin receptors in effects of nerve growth factor and neurotrophin-3 on cultured Purkinje cell survival. J Neurochem 70: 1045-1053. [DOI] [PubMed] [Google Scholar]
- Munafo DB, Colombo MI (2001) A novel assay to study autophagy: regulation of autophagosome vacuole size by amino acid deprivation. J Cell Sci 114: 3619-3629. [DOI] [PubMed] [Google Scholar]
- Murray SS, Bartlett PF, Lopes EC, Coulson EJ, Greferath U, Cheema SS (2003) Low-affinity neurotrophin receptor with targeted mutation of exon 3 is capable of mediating the death of axotomized neurons. Clin Exp Pharmacol Physiol 30: 217-222. [DOI] [PubMed] [Google Scholar]
- Nixon RA, Cataldo AM, Mathews PM (2000) The endosomal-lysosomal system of neurons in Alzheimer's disease pathogenesis: a review. Neurochem Res 25: 1161-1172. [DOI] [PubMed] [Google Scholar]
- Petersen A, Larsen KE, Behr GG, Romero N, Przedborski S, Brundin P, Sulzer D (2001) Expanded CAG repeats in exon 1 of the Huntington's disease gene stimulate dopamine-mediated striatal neuron autophagy and degeneration. Hum Mol Genet 10: 1243-1254. [DOI] [PubMed] [Google Scholar]
- Qin ZH, Wang Y, Kegel KB, Kazantsev A, Apostol BL, Thompson LM, Yoder J, Aronin N, DiFiglia M (2003) Autophagy regulates the processing of amino terminal Huntingtin fragments. Hum Mol Genet 12: 3231-3244. [DOI] [PubMed] [Google Scholar]
- Rabizadeh S, Bredesen DE (2003) Ten years on: mediation of cell death by the common neurotrophin receptor p75(NTR). Cytokine Growth Factor Rev 14: 225-239. [DOI] [PubMed] [Google Scholar]
- Rabizadeh S, Oh J, Zhong LT, Yang J, Bitler CM, Butcher LL, Bredesen DE (1993) Induction of apoptosis by the low-affinity NGF receptor. Science 261: 345-348. [DOI] [PubMed] [Google Scholar]
- Rabizadeh S, Ye X, Sperandio S, Wang JJ, Ellerby HM, Ellerby LM, Giza C, Andrusiak RL, Frankowski H, Yaron Y, Moayeri NN, Rovelli G, Evans CJ, Butcher LL, Nolan GP, Assa-Munt N, Bredesen DE (2000) Neurotrophin dependence domain: a domain required for the mediation of apoptosis by the p75 neurotrophin receptor. J Mol Neurosci 15: 215-229. [DOI] [PubMed] [Google Scholar]
- Ritvo ER, Freeman BJ, Scheibel AB, Duong T, Robinson H, Guthrie D, Ritvo A (1986) Lower Purkinje cell counts in the cerebella of four autistic subjects: initial findings of the UCLA-NSAC Autopsy Research Report. Am J Psychiatry 143: 862-866. [DOI] [PubMed] [Google Scholar]
- Roux PP, Bhakar AL, Kennedy TE, Barker PA (2001) The p75 neurotrophin receptor activates Akt (protein kinase B) through a phosphatidylinositol 3-kinase-dependent pathway. J Biol Chem 276: 23097-23104. [DOI] [PubMed] [Google Scholar]
- Seglen PO, Gordon PB (1982) 3-Methyladenine: specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc Natl Acad Sci USA 79: 1889-1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selimi F, Lohof AM, Heitz S, Lalouette A, Jarvis CI, Bailly Y, Mariani J (2003) Lurcher GRID2-induced death and depolarization can be dissociated in cerebellar Purkinje cells. Neuron 37: 813-819. [DOI] [PubMed] [Google Scholar]
- Shelburne JD, Arstila AU, Trump BF (1973) Studies on cellular autophagocytosis: cyclic AMP- and dibutyryl cyclic AMP-stimulated autophagy in rat liver. Am J Pathol 72: 521-540. [PMC free article] [PubMed] [Google Scholar]
- Skinner PJ, Vierra-Green CA, Clark HB, Zoghbi HY, Orr HT (2001) Altered trafficking of membrane proteins in purkinje cells of SCA1 transgenic mice. Am J Pathol 159: 905-913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorensen B, Tandrup T, Koltzenburg M, Jakobsen J (2003) No further loss of dorsal root ganglion cells after axotomy in p75 neurotrophin receptor knockout mice. J Comp Neurol 459: 242-250. [DOI] [PubMed] [Google Scholar]
- Stromhaug PE, Klionsky DJ (2001) Approaching the molecular mechanism of autophagy. Traffic 2: 524-531. [DOI] [PubMed] [Google Scholar]
- Tolkovsky AM, Xue L, Fletcher GC, Borutaite V (2002) Mitochondrial disappearance from cells: a clue to the role of autophagy in programmed cell death and disease? Biochimie 84: 233-240. [DOI] [PubMed] [Google Scholar]
- Torres-Aleman I, Pons S, Arevalo MA (1994) The insulin-like growth factor I system in the rat cerebellum: developmental regulation and role in neuronal survival and differentiation. J Neurosci Res 39: 117-126. [DOI] [PubMed] [Google Scholar]
- Uchiyama Y (2001) Autophagic cell death and its execution by lysosomal cathepsins. Arch Histol Cytol 64: 233-246. [DOI] [PubMed] [Google Scholar]
- Vaudry D, Falluel-Morel A, Leuillet S, Vaudry H, Gonzalez BJ (2003) Regulators of cerebellar granule cell development act through specific signaling pathways. Science 300: 1532-1534. [DOI] [PubMed] [Google Scholar]
- Wang KC, Kim JA, Sivasankaran R, Segal R, He Z (2002) P75 interacts with the Nogo receptor as a co-receptor for Nogo, MAG and OMgp. Nature 420: 74-78. [DOI] [PubMed] [Google Scholar]
- Watanabe R, Duchen LW (1993) Cerebral amyloid in human prion disease. Neuropathol Appl Neurobiol 19: 253-260. [DOI] [PubMed] [Google Scholar]
- Watase K, Weeber EJ, Xu B, Antalffy B, Yuva-Paylor L, Hashimoto K, Kano M, Atkinson R, Sun Y, Armstrong DL, Sweatt JD, Orr HT, Paylor R, Zoghbi HY (2002) A long CAG repeat in the mouse Sca1 locus replicates SCA1 features and reveals the impact of protein solubility on selective neurodegeneration. Neuron 34: 905-919. [DOI] [PubMed] [Google Scholar]
- Webb JL, Ravikumar B, Atkins J, Skepper JN, Rubinsztein DC (2003) α-Synuclein is degraded by both autophagy and the proteasome. J Biol Chem 278: 25009-25013. [DOI] [PubMed] [Google Scholar]
- Weeks JC (2003) Thinking globally, acting locally: steroid hormone regulation of the dendritic architecture, synaptic connectivity and death of an individual neuron. Prog Neurobiol 70: 421-442. [DOI] [PubMed] [Google Scholar]
- Wetts R, Herrup K (1982) Interaction of granule, Purkinje and inferior olivary neurons in lurcher chimaeric mice. I. Qualitative studies. J Embryol Exp Morphol 68: 87-98. [PubMed] [Google Scholar]
- Xue L, Fletcher GC, Tolkovsky AM (1999) Autophagy is activated by apoptotic signalling in sympathetic neurons: an alternative mechanism of death execution. Mol Cell Neurosci 14: 180-198. [DOI] [PubMed] [Google Scholar]
- Yaar M, Zhai S, Pilch PF, Doyle SM, Eisenhauer PB, Fine RE, Gilchrest BA (1997) Binding of beta-amyloid to the p75 neurotrophin receptor induces apoptosis: A possible mechanism for Alzheimer's disease. J Clin Invest 100: 2333-2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Q, Johnson Jr EM (1988) An immunohistochemical study of the nerve growth factor receptor in developing rats. J Neurosci 8: 3481-3498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J, Lipinski M, Degterev A (2003) Diversity in the mechanisms of neuronal cell death. Neuron 40: 401-413. [DOI] [PubMed] [Google Scholar]
- Yue Z, Horton A, Bravin M, DeJager PL, Selimi F, Heintz N (2002) A novel protein complex linking the delta 2 glutamate receptor and autophagy: implications for neurodegeneration in lurcher mice. Neuron 35: 921-933. [DOI] [PubMed] [Google Scholar]
- Zanjani HS, Herrup K, Guastavino JM, Delhaye-Bouchaud N, Mariani J (1994) Developmental studies of the inferior olivary nucleus in staggerer mutant mice. Brain Res Dev Brain Res 82: 18-28. [DOI] [PubMed] [Google Scholar]
- Zhu JH, Guo F, Shelburne J, Watkins S, Chu CT (2003) Localization of phosphorylated ERK/MAP kinases to mitochondria and autophagosomes in Lewy body diseases. Brain Pathol 13: 473-481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo J, De Jager PL, Takahashi KA, Jiang W, Linden DJ, Heintz N (1997) Neurodegeneration in Lurcher mice caused by mutation in delta2 glutamate receptor gene. Nature 388: 769-773. [DOI] [PubMed] [Google Scholar]