

Summary
New structures of RNA polymerase II (pol II) transcribing complexes reveal a likely key to transcription. The trigger loop swings beneath a correct nucleoside triphosphate (NTP) in the nucleotide addition site, closing off the active center, and forming an extensive network of interactions with the NTP base, sugar, phosphates, and additional pol II residues. A histidine side chain in the trigger loop, precisely positioned by these interactions, may literally “trigger” phosphodiester bond formation. Recognition and catalysis are thus coupled, ensuring the fidelity of transcription.
Introduction
The fundamental mechanism of transcription is conserved among cellular RNA polymerases. Common features include an unwound region, or “transcription bubble,” of about 15 base pairs of the DNA template and some eight residues of the RNA transcript hybridized with the DNA in the center of the bubble. The enzymes involved, bacterial RNA polymerase (RNAP) and eukaryotic RNA polymerases (pol I, II, and III), are capable of both forward and retrograde movement (“backtracking”) on the DNA. Forward movement is favored by the binding of nucleoside triphosphates (NTPs), while backtracking occurs especially when the enzyme encounters an impediment, such as damaged DNA.
Structures of transcribing complexes have been modeled on the basis of protein-DNA cross-linking data for RNAP and determined by X-ray crystallography for pol II. The first pol II transcribing complex for X-ray crystallography was produced by initiation on a “tailed” template and stalling due to the omission of an NTP (Gnatt et al., 2001). The resulting structure revealed the complex in the “pre-translocation” state, with the nucleotide just added to the RNA transcript still occupying the addition or “A” site. A more facile and versatile method was subsequently developed, based on the finding that pol II binds a transcription bubble formed from DNA and RNA oligonucleotides and is able to extend the RNA (Kireeva et al., 2000; Westover et al., 2004). An X-ray structure obtained by this approach revealed the transcribing complex in the “post-translocation” state, with the A site available for entry of a matched NTP. Crystals of the transcribing complex in the post-translocation state were soaked with a matched nucleotide, UTP, and with an unmatched nucleotide, ATP, revealing a second NTP-binding site, termed the entry or “E” site (Figure 1A) (Westover et al., 2004). All NTPs can bind the E site, whereas only an NTP matched for base pairing with the DNA template binds the A site for addition to the growing RNA chain (Westover et al., 2004). The addition step is presumed to involve two Mg2+ ions, one stably associated with the enzyme and the other only transiently, entering with the NTP and leaving upon pyrophosphate release.
Figure 1. A and E Sites in the Pol II Transcribing Complex (A).
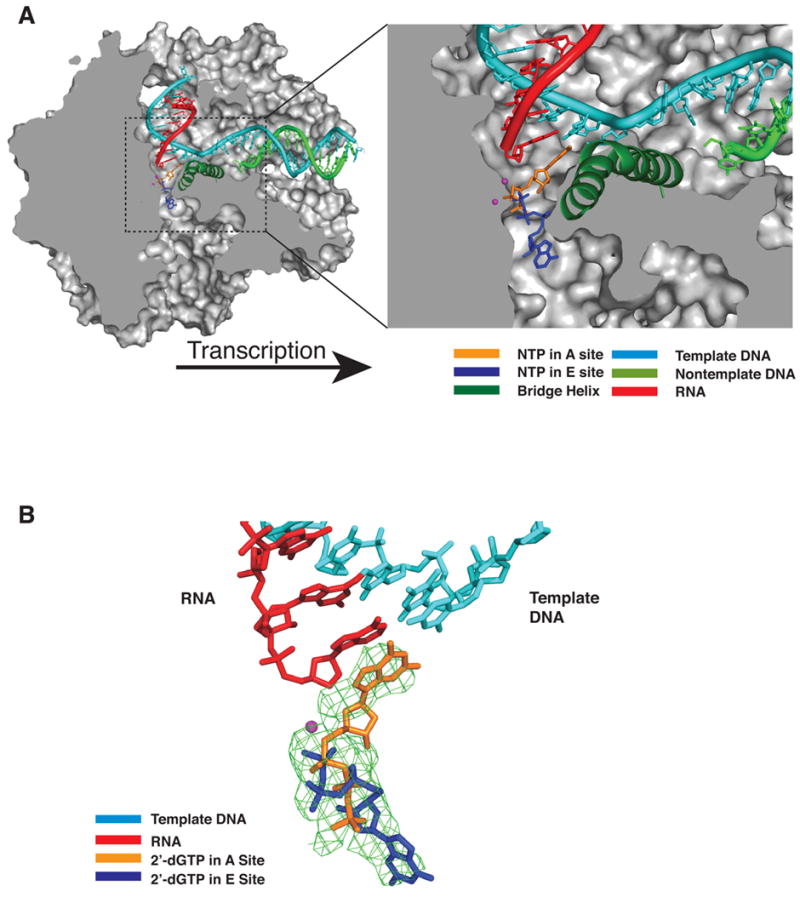
Cutaway view of the pol II transcribing complex. Template DNA, non-template DNA, RNA, GTP in the A site, and ATP in the E site are shown in cyan, green, red, orange, and blue, respectively. The bridge helix (Rpb1 815–848) is in green and Mg2+ ions are shown as magenta spheres. The pol II surface is shown in gray (Westover et al., 2004).
(B) Difference electron density map for transcribing complex crystals soaked with 2’-dGTP. An Fo-Fc omit map contoured at 3.0 sigma is shown in green mesh. Template DNA, RNA, 2’-dGTP (A site) and 2’-dGTP (E site) are shown in cyan, red, orange, and blue, respectively.
Beyond these findings, little is known of the basis for nucleotide selection or catalysis. The way in which the correctly matched and positioned NTP is recognized and how this recognition leads to catalysis remain obscure. The energies of base pairing and stacking are insufficient for base selectivity, and the question arises of why transient occupation of the A site by either incorrect NTP or 2’-dNTP substrates does not lead to erroneous RNA synthesis. Genetic and biochemical studies have implicated two conserved polymerase domains, termed F and G, in the transcription mechanism (Allison et al., 1985; Archambault et al., 1998; Bar-Nahum et al., 2005; Hekmatpanah and Young, 1991; Thuillier et al., 1996; Weilbaecher et al., 1994). Structural studies have identified these two domains with elements adjacent to the polymerase active site, termed the bridge helix (F) and trigger loop (G) (Cramer et al., 2001). In the X-ray structures of transcribing complexes, however, no contact of these structural elements with NTP in the A or E sites has been observed. Here we report a series of pol II transcribing complex structures that reveal such contacts and suggest the roles of these domains in the transcription mechanism. These structures also address matters arising from the literature concerning the multiplicity of NTP binding sites and the locations of active center magnesium ions.
Results
Conformation of the Trigger Loop with NTP in the A Site
A pol II transcribing complex was formed as before (Westover et al., 2004), with a 29-residue template DNA, 10-residue RNA lacking a 3’-OH group, and 14-residue DNA complementary to the template downstream of the RNA. The only difference from the complex prepared previously was the substitution of C for A in the template at the i+1 position (coding base immediately downstream of the RNA). Crystals of the complex were then soaked with GTP for occupation of the A site, and the structure was solved by molecular replacement starting from the previous transcribing complex model (Westover et al., 2004), followed by rigid body and restrained refinement. A difference electron density map, calculated with nucleoside triphosphate removed (Fo-Fc omit map), showed density in the A site corresponding with the structure of an NTP, essentially the same as the previous results for crystals formed from DNA specifying incorporation of UTP at the i+1 position and soaked with UTP (PDB 1R9S)(Westover et al., 2004). The protein structure, however, revealed continuous density beneath that for the NTP not seen in this location previously (Figure 2A). This density was attributable to the trigger loop (Rpb1 residues between about 1070 and 1100) which has been detected in other pol II structures, but in different conformations (Kettenberger et al., 2003; Westover et al., 2004).
Figure 2. Trigger Loop Conformations and Pol II Structural Changes.
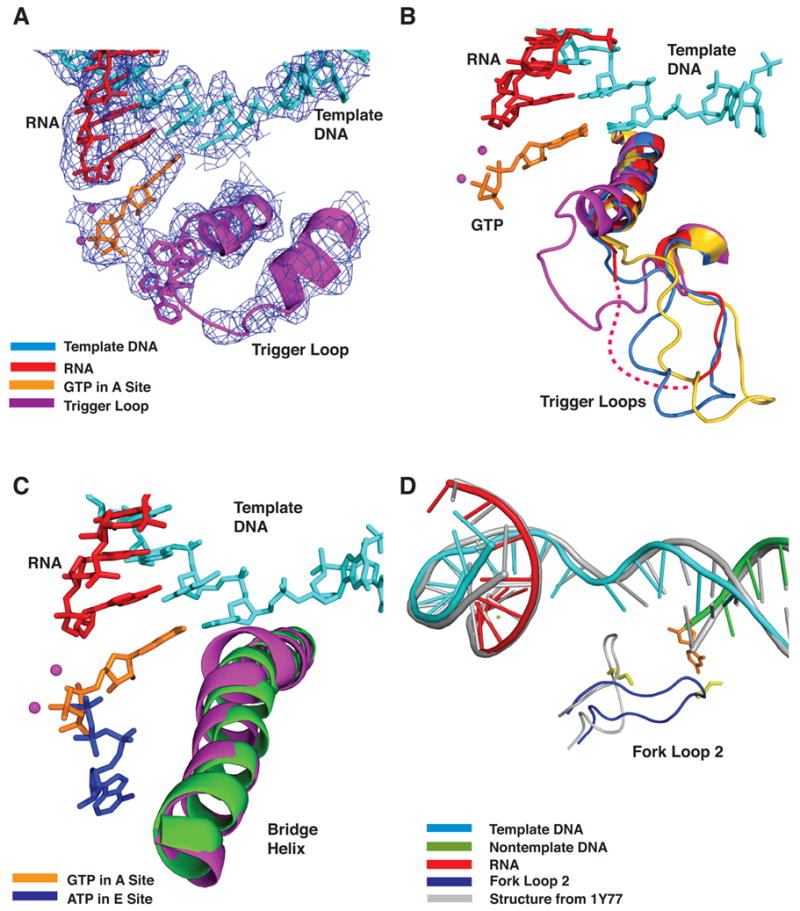
(A) DNA, RNA, trigger loop, and GTP in the A site. The prime_switch map contoured at 1.0 sigma is shown in blue mesh. Template DNA, RNA and GTP are shown in cyan, red, and orange, respectively. The trigger loop is shown in magenta and Mg2+ ions are shown as magenta spheres.
(B) Multiple conformations of the trigger loop. Four Pol II transcribing complexes- GTP (A site, low Mg2+) in magenta, ATP (E site, low Mg2+, 1R9T) in red, UTP (E site, high Mg2+) in marine, and pol II-TFIIS (No nucleotide, high Mg2+, 1Y1V) in yellow are superimposed on Cα atoms in the bridge helix and trigger loop. Other colors as in (A).
(C) Bridge helix movement in transcribing complex with GTP at low Mg2+ concentration. Pol II transcribing complexes with GTP (low Mg2+) in the A site and with ATP in the E site (1R9T, low Mg2+) are superimposed on Cα atoms in the bridge helix. Template DNA, RNA, GTP (A site) and ATP (E site) are shown in cyan, red, orange and blue, respectively. The bridge helices in GTP (low Mg2+) and 1R9T are shown in magenta and green, respectively. Mg2+ ions are shown as magenta spheres.
(D) Fork loop 2 movement. Template DNA, non-template DNA, RNA, and fork loop 2 (Rpb2 502–509) in a pol II transcribing complex with 2’-dGTP at low Mg2+ concentration are cyan, green, red, and blue, respectively. Template DNA, non-template DNA, RNA, and fork loop 2 in a pol II transcribing complex with GMPCPP at high Mg2+ concentration (1Y77) are gray. The side chain of Lys507 is yellow and the nucleotide base at the 5’-end of the non-template strand is orange.
Of fourteen transcribing complex structures in which the trigger loop has been observed - ten from this work (Table 1), three from Westover et al., 2004, and one in the presence of TFIIS (Kettenberger et al., 2003) - only two, with correct NTP in the A site, revealed the trigger loop in proximity to the A site. These two structures are with GTP in the A site (Figure 2A) and, as described (Westover et al., 2004), with UTP in the A site (recalculated here with improved data processing, using REFMAC with TLS with 5 defined TLS domains, revealing the trigger loop in a similar orientation to that with GTP in the A site)(Supplementary Figure 1). Among the remaining twelve complexes, three additional conformations of the trigger loop could be discerned, all remote from the A site (Figure 2B), and in some cases, significantly disordered in center of the loop. Among these complexes were three formed by transcription stalled due to damage in the DNA template (Table 1). The structures of complexes stalled at damage sites did not differ significantly from those stalled by omission of NTPs at the resolution of our analysis (data not shown). The multiple conformations of the trigger loop revealed by crystallography are consistent with the characterization of the trigger loop as a flexible element that oscillates between positions near the active site and positions near downstream DNA (Ederth et al., 2006; Epshtein et al., 2002; Palangat et al., 2004; Vassylyev et al., 2002).
Table 1.
Crystallographic Data and Structure Statistics
| Pol II Transcribing Complex | Pol II | ||||
|---|---|---|---|---|---|
| Crystal Condition | High Mg2+ | High Mg2+ | High Mg2+ | High Mn2+ | |
| Nucleotide Soak | 2’-dUTP | UTP | GMPCPP | None | |
| Space Group | C2 | C2 | C2 | I222 | |
| Unit Cell Dimension | 170.6, 222.8, 195.3, 90.0, 101.3, 90.0 | 170.6, 222.7,196.2, 90.0, 101.9, 90.0 | 170.9, 223.1, 195.4, 90.0, 102.4, 90.0 | 122.3, 222.9, 375.9, 90.0, 90.0, 90.0 | |
| Wavelength(Å) | 1.00 | 1.00 | 0.98 | 1.17 | |
| Resolution(Å) | 48-3.0(3.1–3.0) | 50-3.4(3.5-3.4) | 40-3.4 | 40-3.4 | |
| Unique Reflections | 141,295 | 91,438 | 95,301 | 64,867 | |
| Completeness(%) | 98.9(94.9) | 93.0(76.6) | 97.1(94.8) | 98.1(98.9) | |
| Redundancy | 3.2(2.4) | 2.6(1.4) | 3.0(2.6) | 5.2(5.2) | |
| I/Sigma | 8.1(1.4) | 10.4(1.3) | 8.0(1.4) | 25.6(4.7) | |
| Mosaicity(º) | 0.51 | 0.63 | 0.64 | 0.61 | |
| Rsym (%) | 14.2(74.6) | 9.0(36.1) | 12.5(54.1) | 13.2(52.3) | |
| Rfact/Rfree | 22.2/27.9 | 27.1/33.3 | 28.3/33.3 | 24.9/32.3 | |
| PDB Access Code | 2NVQ | 2NVS | 2NVT | 2NVY | |
| Pol II Transcribing Complex | |||||
|
| |||||
| Crystal Condition | low Mg2+ | low Mg2+ | low Mg2+ | low Mg2+ | low Mg2+ |
| Nucleotide Soak | 2’-dUTP | 2’-dGTP | GMPCPP | GTP | UTP (refine) |
| Space Group | C2 | C2 | C2 | C2 | C2 |
| Unit Cell Dimension | 168.8, 222.5, 193.1, 90.0, 101.3, 90.0 | 168.1, 222.5, 193.1, 90.0,100.4, 90.0 | 170.1, 222.0, 194.7, 90.0, 101.4, 90.0 | 171.1, 222.0, 195.2, 90.0, 102.6, 90.0 | 169.0, 222.0, 194.0, 90.0, 101.0, 90.0 |
| Wavelength(Å) | 1.01 | 0.98 | 1.10 | 0.98 | 0.98 |
| Resolution(Å) | 50-3.6(3.73-3.6) | 50-3.4(3.52-3.4) | 50-3.5(3.63-3.5) | 50-3.95(4.09-3.95) | 40-4.2(4.3-4.2) |
| Unique Reflections | 80,737 | 94,186 | 84,357 | 60,760 | 50,043 |
| Completeness(%) | 93.1(93.9) | 98.3(90.8) | 95.4(81.3) | 98.3(92.2) | 95.8(93.2) |
| Redundancy | 2.8(2.6) | 5.3(4.3) | 8.2(3.8) | 3.4(2.9) | 2.5 |
| I/Sigma | 9.2 (2.0) | 14.6(2.0) | 10.2(1.3) | 10.3(2.3) | 11.8 |
| Mosaicity(º) | 0.53 | 0.64 | 0.67 | 0.34 | 0.89 |
| Rsym (%) | 13.4(72.9) | 14.4(56.2) | 18.6(53.3) | 14.5(46.5) | 15.7(36.1) |
| Rfact/Rfree | 29.8/31.2 | 24.8/29.2 | 24.2/30.3 | 27.2/34.8 | 27.1/33.3 |
| PDB Access Code | 2NVX | 2E2I | 2E2J | 2E2H | 2NVZ |
| Pol II Transcribing Complex with Damaged DNA Template | |||||
|
| |||||
| Crystal Condition | low Mg2+ | low Mg2+ | low Mg2+ | ||
| Damage Type | AAF | CisDDP | CisDDP | ||
| Nucleotide Soak | CTP | GTP | CTP | ||
| Space Group | C2 | C2 | C2 | ||
| Unit Cell Dimension | 169.5, 222.3, 194.3, 90.0, 101.5, 90.0 | 169.0, 222.4, 193.5, 90.0, 101.6, 90.0 | 171.6, 222.7, 196.5, 90.0, 102.3, 90.0 | ||
| Wavelength(Å) | 1.00 | 1.01 | 1.01 | ||
| Resolution(Å) | 46-3.95(4.09-3.95) | 50-3.2(3.31-3.2) | 50-3.4(3.52-3.4) | ||
| Unique Reflections | 62,516 | 111,401 | 91,246 | ||
| Completeness(%) | 95.0(95.0) | 96.8(95.9) | 92.3(87.9) | ||
| Redundancy | 2.7(2.6) | 1.9(1.8) | 2.8(2.3) | ||
| I/Sigma | 15.9(4.4) | 9.5(1.5) | 14.3(1.8) | ||
| Mosaicity(º) | 0.61 | 0.33 | 0.87 | ||
| Rsym (%) | 10.3(26.3) | 8.7(52.8) | 18.7(63.6) | ||
| Rfact/Rfree | 26.6/28.4 | 22.1/27.6 | 24.4/32.6 | ||
Selection of NTPs
The trigger loop engages in a network of interactions with GTP in the A site, with the “bridge helix,” and with other residues of subunits Rpb1 and Rpb2 lining the active center region (Figures 3A, 3B, and Supplementary Figure 2). Trigger loop residue Leu1081 makes hydrophobic contact with the nucleotide base, while Gln1078 interacts through Rpb1 Asn479 with the 3’-OH of the ribose ring (possibly also directly, since the Gln1078 to 3’-OH distance is 3.9 Å, permitting a weak hydrogen bond) and His1085 makes a hydrogen bond or salt bridge with the β-phosphate. His1085 is, in turn, positioned by hydrogen bonding with trigger loop Asn1082 and the Rpb2 Ser1019 main chain carbonyl group. Arg446 lies in close proximity to the 2’-OH of the ribose ring. These interactions serve to recognize all features of the NTP in the A site and to detect its precise location in the site.
Figure 3. Trigger Loop Interactions and Nucleotide Selectivity.
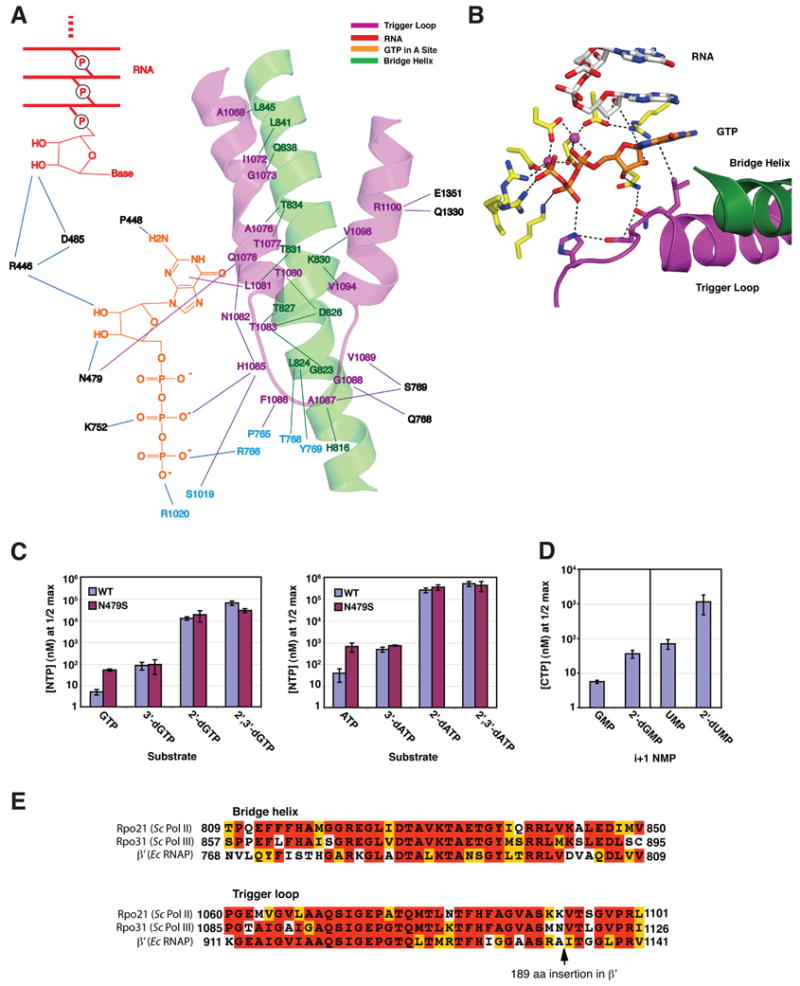
(A) The trigger loop network. Trigger loop and bridge helix are magenta and green, GTP is orange, and the 3’end of the RNA is red. Other residues of Rpb1 and Rpb2 are indicated in black and cyan.
(B) Interactions with GTP in the A site. Trigger loop and bridge helix are magenta and green, GTP is orange, and the 3’-end of the RNA is white, except oxygen and nitrogen atoms are highlighted in red and blue. Other residues of Rpb1 and Rpb2 are shown in yellow.
(C) Role of Rpb1 Asn479 in selection of the ribose sugar: addition of GTP, ATP and derivatives. Transcribing complexes were formed with wild type and Rpb1 N479S pol II as described for crystallography, with templates specifying addition of either G or A, except with a 9 rather than a 10-residue RNA, which was extended to 10 residues with 32P-radiolabeled NTP. After removal of unincorporated label, complexes were challenged with increasing amounts of template-specified NTP or dNTP derivatives and extension to 11 residues was measured after 5 min (Supplementary Figure 3A). These data were plotted and non-linear regression was performed to determine maximal incorporation of each substrate and the concentration that gave 50% maximal incorporation (apparent KM, Supplementary Figure 3B). Apparent KM values for incorporation of GTP and ATP derivatives by wild type and Rpb1 N479S pol II are shown in the bar graphs. Error bars represent mean +/− standard deviation of at least four apparent KM determinations.
(D) Diminished rate of nucleotide addition following incorporation of 2’-dNMP. Apparent KM values for addition of CTP subsequent to incorporation of either GMP, 2’-dGMP, UMP or 2’-dUMP by wild type pol II elongation complexes are shown. Representative gels and data plots are shown in Supplementary Figure 3C,D. Error bars represent mean +/− standard deviation of eight apparent KM determinations.
(E) Conservation of bridge helix and trigger loop residues. The sequences of bridge helix and trigger loop from S.cerevisiae Pol II, Pol III (Sc) and E. coli RNA polymerase (Ec RNAP) were aligned using MUSCLE (Edgar, 2004) and then adjusted by hand to account for the 189 aa insertion in E. coli RNAP. Alignment was colored for conservation in MACBOXSHADE.
Mutation of the RNAP residue analogous to pol II Asn479 leads to a 4.8-fold loss of rG/2’-dG discrimination, suggesting a role for this residue in selection for a 2’-OH group of the ribose ring (Svetlov et al., 2004). Our X-ray structure, however, reveals an interaction of Asn479 with the 3’-OH group of the ring. To investigate this discrepancy, we prepared transcribing complexes as for X-ray analysis from both wild type and Rpb1 N479S mutant pol II and monitored the addition of GTP and derivatives. The rates of incorporation of ribo NTPs were too rapid to measure by our methods, so we determined the NTP concentration at which the addition of a single nucleotide was half maximal during a fixed time (Svetlov et al., 2004)(Figure 3C, Supplementary Figure 3A, 3B). This NTP concentration, or “apparent KM,” depends on both the affinity for substrate and the rate of the nucleotide addition reaction. The Rpb1 N479S mutant pol II showed a 7-fold loss of G/2’-dG discrimination, and the underlying cause was an increase in apparent KM of the mutant for GTP, not a decrease in apparent KM for 2’-dGTP, similar to results for RNAP (Svetlov et al., 2004). The observed increase in apparent KM, however, was due solely to loss of interaction with the 3’-OH group, since a 10-fold difference between wild type and mutant enzymes observed with GTP was abolished with a 3’-deoxy derivative. Similar results were obtained for the addition of ATP and derivatives to transcribing complexes containing the appropriate template DNA (Figure 3C). The role of Rpb1 Arg446 in recognition of the 2’-OH group could not be assessed by this approach, because rpb1-R446A was inviable in our strain background and mutant enzyme could not be purified from a heterozygous strain (data not shown).
The strongest selection for the ribose sugar is observed for wild type pol II between GTP and a 2’-deoxy derivative (Figure 3C). The question arises of whether the 2’-deoxy derivative suffers a diminished affinity for the A site or an impairment of catalysis. To this end, crystals of pol II transcribing complexes were formed as described above and soaked with 2’-dGTP (matched to a template C). The resulting difference map (Fo-Fc omit map) showed election density in both A and E sites (Figure 1B), which fit well with the locations of NTP previously determined for these sites. Occupancy refinement revealed a majority of 2’-dGTP (68%) in the E site. Similarly, a transcribing complex formed as described (Westover et al., 2004) and soaked with 2’-dUTP (matched to a template A) was revealed a preponderance of nucleotide (75%) in the E site (data not shown). Evidently a 2’-deoxy nucleotide has an appreciable affinity for the A site, and the basis for its far slower rate of incorporation lies elsewhere.
The rates of addition of 2’-deoxy nucleotides were so slow they could be measured by conventional methods. The values of Vmax obtained, 0.023 nucleotides per second for 2’-dGTP and 0.052 for 2’-dUTP (Supplementary Figure 4), were at least 400-fold lower than the maximal rate of pol II transcription with ribo NTPs in vitro (Uptain et al., 1997). The values of KM obtained, 46.9 μM for 2’-dGTP and 821 μM for 2’-dUTP, were more nearly comparable to those of 42 μMfor GTP and 12-135 μM for UTP reported for RNAP (Rhodes and Chamberlin, 1974). These findings suggest that selection against 2’-dNTPs occurs primarily at the level of catalysis.
The network of interactions involving the trigger loop and other pol II residues with NTP in the A site (Figures 3A, 3B) also includes the 2’–OH group of the residue at the -1 position in the RNA, as suggested previously (Gnatt et al., 2001; Svetlov et al., 2004). This leads to the expectation that a transcribing complex, once having incorporated a 2’-deoxy nucleotide, will be refractory to further nucleotide addition. Indeed, incorporation of 2’-dUMP or 2’dGMP, depending on the DNA template, increased the apparent KM for subsequent NTP (Figure 3D, Supplementary Figure 3C, 3D). Incorporation of a subsequent 2’-dNTP was virtually undetectable (data not shown).
Trigger Loop Effect on Bridge Helix Conformation
Trigger loop interactions with the bridge helix are extensive and affect its conformation (Figures 2C, 3A). The bridge helix is slightly unwound and bent in the center (residues 826–830), resulting in a movement of 2–2.7 Å of Cα atoms (2.3 Å for Asp826, 2.7 Å for Thr827, and 2.0 Å for Ala828, 1.7 Å for Val829, and 2.1 Å for Lys830) in the direction of the NTP and DNA-RNA hybrid helix. The bend is in a different location from that seen in the structure of bacterial RNAP and proposed to play a role in DNA/RNA translocation during transcription. These observations are consistent with the involvement of the trigger loop in the control of bridge helix motion (Bar-Nahum et al., 2005; Epshtein et al., 2002).
Movement of Fork Loop 2
The structure of transcribing complex with 2’-deoxy nucleotide revealed a movement of a protein element termed fork loop 2 (Rpb2 residues 502–509) that has been suggested to be involved in setting the downstream boundary of the transcription bubble (Gnatt et al., 2001), and whose mutation may reduce the polymerization rate (Trinh et al., 2006). The location of fork loop 2 in a previous transcribing complex structure would clash with the residue at position +3 in the nontemplate DNA strand if this residue were base-paired to the template strand (Kettenberger et al., 2003). In our structure of a transcribing complex soaked with 2’-dGTP, fork loop 2 is rotated by about 90°, accentuating this potential clash (Figure 2D). Nontemplate residue +3 is flipped out, interacting with fork loop 2 through the side chain of Lys507. The movement of fork loop 2, apparently in concert with dissociation of the DNA duplex at position +3, points to a role of this protein element in the unwinding of downstream DNA.
Three Mg Ion Binding Sites
An elevated concentration of Mg2+ concentration caused a shift even of a matched rNTP from A to E site. Crystals of transcribing complexes formed as before (Westover et al., 2004), except in 150 mM rather than 5 mM Mg2+, and soaked with UTP (matched to a template A), showed a preponderance (72%) of nucleotide in the E site (Table 1). This shift to the E site may contribute to the inhibitory effect of a high Mg2+ concentration on transcription ((Chamberlin and Berg, 1962; Fox and Weiss, 1964; Furth et al., 1962; Rhodes and Chamberlin, 1974), data not shown).
Inhibition may also be due to altered location of Mg2+ in the transcribing complex at high Mg2+ concentration. Previous structures determined at low Mg2+ concentration revealed two Mg2+ ions, one also associated with free pol II, designated metal A, and a second found only in the transcribing complex with NTP, designated metal B (Westover et al., 2004). Metal A is coordinated by Rpb1 Asp481, Asp483, Asp485, and by the α-phosphate of the NTP when present; metal B is coordinated by the α, β, and γ phosphates of UTP and by Rpb1 Asp481, Asp483, and Rpb2 Asp837. These two metals are believed to play essential roles in catalysis. They are also present in all transcribing complex structures at low Mg2+ concentration with bound nucleotides, whether in A or E sites, reported here. At high Mg2+ concentration, however, metal B is absent and is replaced by Mg2+ at a new location about 4 Å away, designated metal C (Figure 4A). Some variation in the locations of metals A and B was previously noted (Westover et al., 2004). Superposition of previous and present Mg2+ locations shows a clear division in three groups and the variation within the groups (Figure 4A). Metal C is in the same location as previously reported for a low occupancy Mg2+ site in free pol II (Cramer et al., 2001), and has been confirmed as a metal-binding site by anomalous difference analysis for free pol II at high Mn2+ concentration (Table 1). The occupancy of metal C in transcribing complexes at high Mg2+ concentration is comparable to that of metal A (for example, electron density peaks at 8.7 sigma for metal A and 6.7 sigma for metal C). Since metals B and C are mutually exclusive, and the alignment of metals A and B is likely to be crucial for catalysis, the shift from B to C in high Mg2+ concentration is unavoidably inhibitory.
Figure 4. Three Mg2+ Ion Sites and Binding of GMPCPP in Pol II Transcribing Complexes.
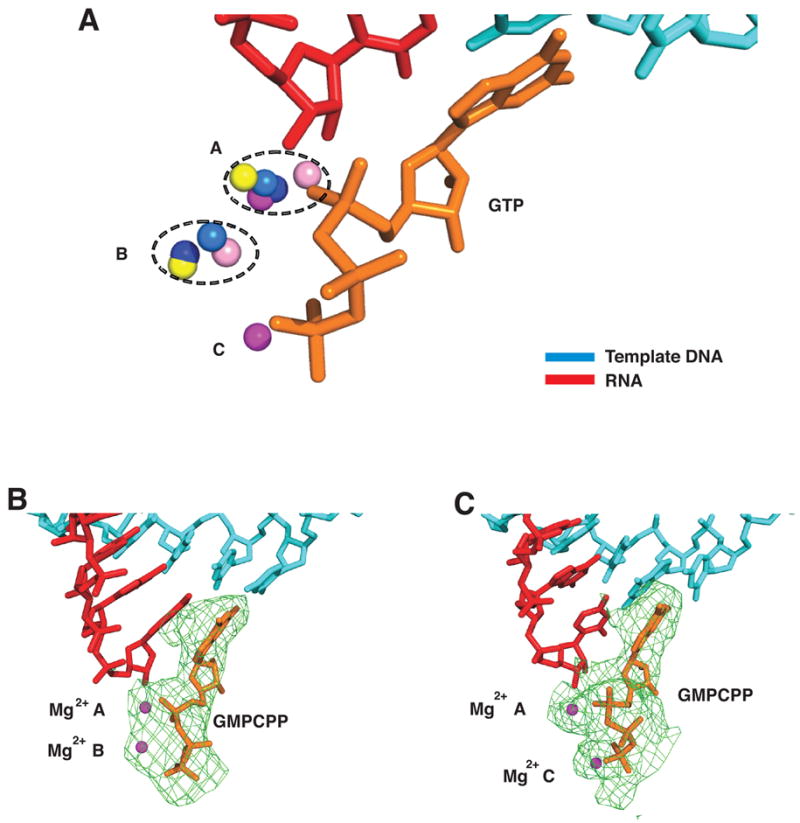
(A) Three Mg2+ ion sites in pol II transcribing complexes. Structures of five pol II transcribing complexes were aligned on phosphates of template DNA and RNA in the active site region. Template DNA, RNA, and GMPCPP at high Mg2+ concentration are cyan, red, and orange, respectively. Mg2+ ions from transcribing complexes (structure, Mg2+ concentration in parentheses) with ATP in the E site (1R9T, low), UTP in the A site (1R9S, low), GTP (Table 1, low), GMPCPP (Table 1, low) and GMPCPP (Table 1, high) are depicted as salmon, blue, yellow, marine, and magenta spheres, respectively. The average distance between metals A and B, B and C, and A and C are 4.0, 4.2, and 5.5 Å, respectively.
(B) GMPCPP (orange) in the A site at low Mg2+ concentration. Color code as in Figure 1B.
(C) GMPCPP (orange) in the A site at high Mg2+ concentration. Color code as in Figure 1B.
No Evidence for a “Pre-insertion” Site
It has been reported that the non-reactive NTP analog GMPCPP binds a transcribing complex in a distinct location between the A and E sites, referred to as a “pre-insertion” site (Kettenberger et al., 2004). The evidence came from crystallographic data 78.4% complete (69.8% in the final shell) at 4.5 Å resolution, obtained with pol II containing two small subunits on the periphery of the enzyme that have no influence upon transcription elongation (Edwards et al., 1991). We repeated the experiment with GMPCPP exactly as described (Kettenberger et al., 2004) except with the use of pol II lacking the two peripheral subunits, to obtain higher resolution, and with the omission of a disordered region of non-template DNA. Data 95–97% complete to 3.4–3.5 Å resolution were collected from crystals in both low and high Mg2+ conditions (Table 1). Difference maps calculated by subtracting the structure with GMPCPP removed (Fo-Fc omit map) revealed electron density only in the A site in both high and low Mg2+ conditions (Figure 4B, C). The fit of GMPCPP to this density was almost identical for the base and sugar in high and low Mg2+ conditions, and only slightly different for the phosphates. Retention of GMPCCP in the A site at high Mg2+ concentration, in contrast with UTP (see above), may reflect a higher affinity of guanine nucleotides for the A site, due to stronger base pairing and stacking interactions.
Discussion
We have extended previous studies of NTP binding to pol II transcribing complexes, in regard to the nucleotide base and sugar, and by screening hundreds of crystals for improved data quality and resolution. The notable finding from this work is the association of the trigger loop with matched rNTP in the A site. We propose this association is the decisive event in NTP recognition and catalysis. Our proposal is almost self-evident from the structure, and is supported by genetic and biochemical work of others, as detailed below.
Role of the Trigger Loop in Nucleotide Recognition and Catalysis
The trigger loop is a mobile element, allowing entry of NTP into the E and A sites in conformations previously observed, and sealing off the A site in the conformation reported here. Located beneath NTP in the A site, the trigger loop directly contacts the base and β-phosphate, and indirectly contacts 2’- and 3’-OH groups of the ribose sugar as well. Numerous interactions with other pol II residues serve to configure and position the trigger loop, so it reads out not only the chemical nature of the NTP but also the parameters of the DNA-RNA hybrid helix in the A site. A well-defined conformation of the trigger loop may be capable of readout to Ångstrom precision. Inasmuch as the hybrid helix differs substantially from B-form DNA (difference of 3 Å in minor groove width and 5.5 Å in root-mean-square phosphorous positions), such readout would readily distinguish ribo from deoxyribo NTPs, as well as providing powerful discrimination against purine-purine and pyrimidine-pyrimidine mispairing.
Two further features of trigger loop interaction may be crucial for transcription. First, the contact of His1085 with the NTP β-phosphate noted above may be key to catalysis. The distance between the imidazole N-H group and β phosphate oxygen is about 3.5 Å, optimal for hydrogen bonding or salt bridge interaction. The protonated imidazole group would be expected to withdraw electron density from the phosphate and facilitate SN2 attack of the RNA 3’-terminal OH group, leading to phosphodiester bond formation (Figure 5). This proposed mechanism is closely analogous to those for protein phosphatase 1 and for ribonuclease A, in which protonated imidazole groups facilitate SN2 attack as proton donors for leaving groups (Barford, 1996; Silverman, 2000; Zhang et al., 1996). Second, trigger loop interaction with NTP in the A site is evidently poised on the verge of stability, since the interaction could only be detected with improved data quality and analysis. If any feature of the NTP or its location is incorrect, the interaction will be lost.
Figure 5. Proposed Role of His1085 in Phosphodiester Bond Formation.
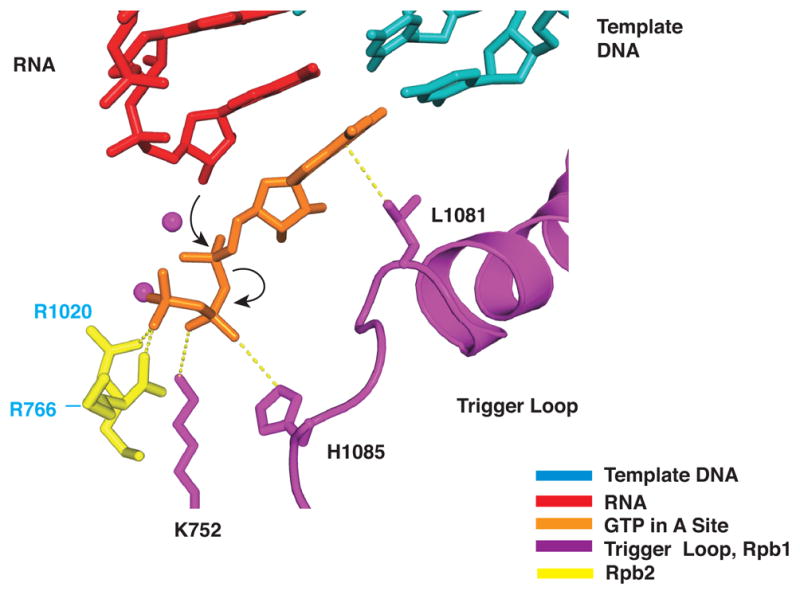
Color code as in Figure 2A, with side chains of Rpb1 Lys752, Rpb1 His1085, Rpb2 Arg1020 (yellow), and Rpb2 Arg766 (yellow). Nucleophilic attack and phosphoanhydride bond breakage are indicated by black arrows.
The trigger loop may therefore couple nucleotide recognition to catalysis. In the presence of matched rNTP in the A site, it will swing into position and literally “trigger” phosphodiester bond formation. An incorrect NTP in the A site will not support trigger loop interaction and so is unlikely to undergo catalysis. When reaction with a correct NTP does occur, the release of pyrophosphate disrupts contact with His1085, likely destabilizing trigger loop interaction and freeing the DNA-RNA hybrid for translocation. Movement of the trigger loop, coupled to that of the bridge helix (Figures 2C, 3A), may contribute to the translocation process (Bar-Nahum et al., 2005; Gnatt et al., 2001).
The proposed coupling of recognition to catalysis through trigger loop interactions resolves an apparent contradiction between two lines of evidence reported here. A 2’-deoxy NTP matched for base pairing to the template DNA exhibits 25–30% occupancy of the A site, so significant incorporation of the deoxy nucleotide might be expected. Such misincorporation is, however, exceedingly rare (Figure 3C); selectivity for a 2’-OH group is even greater, by a factor of 100, than for a 3’-OH group. The energy of a single hydrogen bond to the 2’-OH group cannot account for such selectivity. Rather it may be explained by trigger loop recognition of many features of a ribo/deoxyribo hybrid base pair (see above). In the absence of such recognition, as in the case of a deoxy/deoxy base pair, stable trigger loop interaction with the NTP does not occur and catalysis is much diminished.
Why is selectivity for a 2’-OH group so far greater than that for a 3’-OH? The likely answer is that only discrimination with respect to the 2’-OH is required, since 2’-deoxy NTPs occur in nature but 3’-deoxy NTPs do not. Discrimination with respect to the 2’-OH is achieved in two ways. First, the concentrations of 2’-deoxy NTPs are at least ten-fold lower than those of rNTPs in vivo (Albert and Gudas, 1985; Kornberg and Baker, 1992; Mathews, 1972; Reichard, 1985). Second, pol II achieves over 1000-fold selectivity in incorporation (Figure 3C).
Support for a role of the trigger loop in A site NTP transactions comes from results of mutagenesis of bacterial RNAP. Residues in the RNAP trigger loop, including E. coli β’ Met932 (pol II trigger loop residue Leu1081) can be cross-linked to 8-azido AMP and to 4-thio UMP at the 3’-end of the RNA in a transcribing complex (Borukhov et al., 1991; Epshtein et al., 2002; Markovtsov et al., 1996). Mutation of β’ Thr934 (pol II trigger loop residue Thr1083) to alanine diminished this cross-linking and reduced the affinity for NTP during transcription elongation ~60-fold (Epshtein et al., 2002). Since pol II trigger loop residue Thr1083 is in hydrogen bond contact with bridge helix residues Thr827, Asp826 and Gly823 (Figure 3A), we suppose the mutation to alanine disrupts this contact and destabilizes the trigger loop conformation involved in A-site nucleotide interaction. The observed cross-linking to the RNA 3’-end is attributed to backtracking of the RNA by one residue into the A site (Borukhov et al., 1991; Epshtein et al., 2002; Markovtsov et al., 1996).
Indeed the trigger loop has been suggested to participate in the control of backtracking. Mutants bearing deletions of 40–60 residues in the trigger loop are defective in transcript cleavage induced by the bacterial counterparts of TFIIS (GreA and GreB) and exhibit dramatically reduced transcription elongation rates at sub-saturating substrate concentrations, due to prolonged pausing at sites of transcriptional arrest (Zakharova et al., 1998). Other mutations in the RNAP trigger loop enhance either forward translocation or backtracking, leading to the suggestion that the trigger loop modulates the movement of RNAP (Bar-Nahum et al., 2005). Mutations that alter transcription start site selection may also be explained by effects on pol II movement (Hekmatpanah and Young, 1991). Finally, recessive lethal mutations in the RNAP trigger loop are believed to affect termination through interaction with the 3’-end of the transcript and are defective in elongation (Weilbaecher et al., 1994).
The trigger loop may contribute in yet additional ways to the transcription process. It may not only enhance the affinity of NTP for the A site but also prevent return to the E site, due to a steric clash with NTP in that site (Figure 6A). His1085 may participate not only in phosphodiester bond formation but also in TFIIS-induced cleavage of backtracked RNA.
Figure 6. Trigger Loop Clashes with Nucleotide in the E Site and with α-Amanitin.
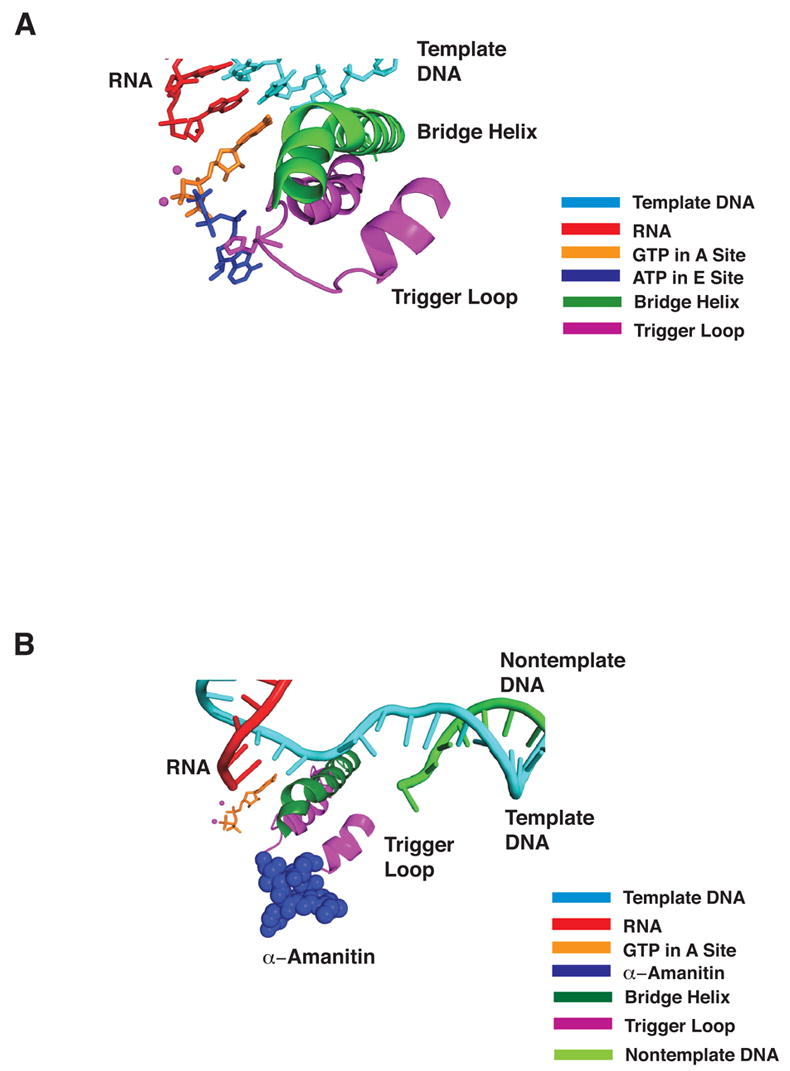
(A) Color code as in Figure 2A, with side chain of trigger loop residue His1085 included and ATP in the E site (1R9T, blue).
(B) As in (A) with E site nucleotide omitted and α-amanitin (CPK model in blue, from 1K83) included.
The Trigger Loop as a Target of Regulatory Factors and Inhibitors
NusA and NusG may modulate RNAP transcription through interactions with the trigger loop (Bar-Nahum et al., 2005; Chlenov et al., 2005; Ito and Nakamura, 1996). DksA and Gfh1 have been also reported to interact with the trigger loop (Laptenko et al., 2003; Perederina et al., 2004; Symersky et al., 2006). Fe-EDTA hydroxyl radical footprinting has suggested that Gre A and Gre B interact with the trigger loop and the bridge helix as well (Laptenko et al., 2003). It is noteworthy that many bacteria harbor insertions of hundreds of amino acids in the middle of the trigger loop (For example, in E.coli RNAP see Figure 3E), which may play species-specific regulatory roles.
The most potent pol II inhibitor, α-amanitin, was previously shown to bind adjacent to the bridge helix beneath the active center (Bushnell et al., 2002). In this location, it will clash with the trigger loop interacting with A site nucleotide (Figure 6B). In all likelihood, α-amanitin prevents the trigger loop from swinging into position beneath A site nucleotide and promoting catalysis.
The RNAP inhibitor streptolydigin also binds in the vicinity of the trigger loop (Temiakov et al., 2005; Tuske et al., 2005), altering its conformation, and disrupting its interaction with the bridge helix. Amino acid replacements in the trigger loop confer streptolydigin resistance (Tuske et al., 2005; Yang and Price, 1995), and such substitutions also confer resistance to Microcin J25 (Delgado et al., 2001; Mukhopadhyay et al., 2004; Yuzenkova et al., 2002). Finally, an antibody that binds to the RNAP trigger loop has a specific inhibitory effect on both polymerization and pyrophosphorylsis (Zakharova et al., 1998) .
Inhibition Through a Third Metal Ion Binding Site in the Transcribing Complex
Two Mg2+ ions associated with the pol II transcribing complex as previously described, designated metals A and B, are believed to be crucial for catalysis. Binding of a third Mg2+ ion, designated metal C, at high Mg2+ concentration as reported here, is incompatible with binding of metal B and is therefore inhibitory to transcription. This mechanism of inhibition is similar to the attenuation of RNase H activity at high Mg2+ concentration, due to recruitment of a third metal ion, compromising coordination of active site Mg2+ ions (Nowotny et al., 2005). The structure of T. thermophilus RNAP-Tagetitoxin and RNAP–ppGpp complexes also revealed a third Mg2+ ion in the vicinity of the active site that interfered with binding of the second Mg2+ ion required for catalysis, induced active site alterations, and stabilized an inactive intermediate (Artsimovitch et al., 2004; Vassylyev et al., 2005).
Only Two NTP Sites in the Active Center
The partial occupancy of A and E sites by dNTP (Figure 1B) is consistent with the idea of equilibration between the sites. None of the structures reported here reveals a site intermediate between A and E as has been previously suggested (Kettenberger et al., 2004). The discrepancy may be attributed to the lower resolution and lower data quality of the previous analysis. In any case, the data upon which the suggestion was based were obtained at high Mg2+ concentration, where metal B shifts to metal C and significant inhibition of transcription is observed ((Chamberlin and Berg, 1962; Fox and Weiss, 1964; Furth et al., 1962; Rhodes and Chamberlin, 1974), and data not shown). The absence of an intermediate or “pre-insertion” site represents an important difference between single and multi-subunit RNA polymerases. The single subunit enzymes also lack any feature corresponding to the trigger loop, further indicative a fundamental difference in the transcription mechanism.
Conclusions
X-ray studies have addressed the longstanding question of how nucleotide selectivity is achieved in transcription by cellular RNA polymerases. Base pairing and base stacking, as well as hydrogen bonding to ribose hydroxyl groups, are insufficient to account for the degree of selectivity observed. For example, it is shown here that a ribonucleotide is incorporated into RNA several hundred-fold more rapidly than a 2’-deoxy ribonucleotide. The basis for this extraordinary specificity lies in a structural element of the polymerase termed the trigger loop, which makes both direct and indirect contact with all features of the nucleotide in the polymerase active center. The trigger loop detects the topology of a correct RNA-DNA hybrid base pair, and thus excludes not only purine-purine and pyrimidine-pyrimidine mismatches, but also a 2’-deoxy ribonucleotide, due to a significant difference in helix parameters between DNA-DNA and RNA-DNA helices.
Experimental Procedures
Crystallization
Ten-subunit yeast S. cerevisiae pol II was purified as described (Cramer et al., 2000). Pol II transcribing complexes were assembled with the use of synthetic oligonucleotides as described (Westover et al., 2004). Briefly, DNA and RNA oligonucleotides were annealed and mixed with pol II and 3’-dATP (except for transcribing complexes to be soaked with GMPCPP, where 3’-dATP was omitted) in 20 mM Tris (pH 7.5), 40 mM KCl, and 5 mM DTT. The final mixture included 2 mM pol II, 10 mM template DNA strand, 20 mM non-template DNA strand and RNA oligonucleotides. The mixture was kept for 1 h at room temperature, and excess oligonucleotides were removed by ultrafiltration. The pol II transcribing complexes were crystallized by hanging drop vapor diffusion as described (Gnatt et al., 2001; Kettenberger et al., 2004; Westover et al., 2004). The crystals were obtained at both low Mg2+ concentration (390 mM (NH4)2HPO4/NaH2PO4, pH 6.5, 50 mM dioxane, 10 mM DTT, and 9–11% PEG6000) and high Mg2+ concentration (200 mM NH4OAc, 150 mM Mg(OAc)2, 50 mM Hepes, pH 7.0, and 5% PEG6000). For pol II transcribing complexes containing damaged DNA, the damaged strands were prepared and purified as described (Lindsley and Fuchs, 1994; Zamble et al., 1996), and transcribing complexes were assembled as above.
The crystals were transferred stepwise to freezing buffer as described (Cramer et al., 2001; Kettenberger et al., 2004; Westover et al., 2004). For the addition of GTP, UTP, 2’-dGTP and 2’-dUTP at low Mg2+ concentration, 10–20 mM nucleotides and 5 mM MgCl2 were added to the freezing buffer (Westover et al., 2004). For the addition of GMPCPP (guanosine-5′-[(α,β)-methyleno]-triphosphate, Jena Biosciences), 3 mM GMPCPP was added to the freezing buffer (5 mM DTT, 35 mM dioxane, 11.2% PEG6K, 70 mM MES (pH 6.3) 245 mM NaCl, 11.9% PEG400) along with 5 mM Mg(OAc)2 (low Mg2+ concentration) or 150 mM Mg(OAc)2 (high Mg2+ concentration).
Data Collection and Processing
Diffraction data were collected at beamlines 11-1 and 9-2 at the Stanford Synchrontron Radiation Laboratory and beamlines 5.0.2 and 8.2.2 at the Advanced Light Source, Lawrence Berkeley National Laboratory. Data were processed in DENZO and SCALEPACK (HKL2000) (Otwinowski and Minor, 1997). Model building was carried with the program Coot (Emsley and Cowtan, 2004), and refinement was done using REFMAC with TLS (CCP4i) (Potterton et al., 2003; Winn et al., 2001). The models were superimposed with phosphate atoms near the active site (+2 to −2) or as indicated in the description of PYMOL (DeLano, 2002).
Nucleotide Addition
For addition of rNTPs subsequent to either rNMP or 2’-dNMP incorporation, transcribing complexes were formed as above with 10 pmol 10-subunit pol II, 120 pmol template, 240 pmol RNA and 240 pmol downstream non-template DNA, in transcription buffer (TB) containing 20 mM Tris pH 8.0, 40 mM KCl, 5 mM MgCl2, and 2 mM DTT. Complexes were incubated with 50 μCi alpha-32P-ATP (3000 Ci/mmol)(Perkin Elmer) to label active elongation complexes for 5 min at room temperature. The rNTP or 2’-dNTP specified for incorporation at the subsequent template position was then added at a final concentration of 200 μM for 5 min at room temperature. Unincorporated NTPs and 2’-dNTPs were removed by passage through a G50 spin column (GE Health Sciences) that had been prequilibrated in TB. Reactions were then diluted to a pol II concentration of approximately 20–30 nM. Aliquots (5 μL) were then added to 5 μL of various concentrations of CTP diluted in TB, the next nucleotide specified for incorporation, and incubated for 5 min at room temperature. Reactions were stopped with 10 μL 2X stop solution (10 M urea, 50 mM EDTA, 1X TBE (90 mM Tris base, 90 mM boric acid, 2 mM EDTA), 0.01% xylene cyanol, 0.01% bromophenol blue). Reaction products were separated in 18% 19:1 acrylamide:bisacrylamide, 7 M urea, 1X TBE gels. Visualization and quantification of products was performed with the use of a PhosphorImager (Molecular Dynamics). The concentration of nucleotide that gave 50% maximal incorporation over the reaction time was determined by plotting incorporation data in GraphPad Prism and non-linear regression analysis (Supplementary Figure 3).
For assessment of wild type and mutant pol II selectivity for rNTP and dNTP substrates, nucleic acids and pol II elongation complexes were formed as above. DNA templates were identical except at the i+1 and i+2 positions. Templates for testing selectivity for G, C, and U substrates in the i+2 position specified ATP in the i+1 position, while the template for testing selectivity for A substrates in the i+2 position specified CTP in the i+1 position. In these experiments, wild type and an Rpb1 N479S mutant were complete, 12-subunit enzymes purified with the use of a TAP-tag on Rpb3. The enzymes were isolated from pre-cleared whole-cell lysate prepared in 2X extract buffer (100 mM Tris pH 7.5, 300 mM KOAc, 10% glycerol, 5 mM DTT, 2X protease inhibitor cocktail) by adsorption on IgG columns, and washing with 20 column vol of 1X extract buffer, 20 column vol of 50 mM Tris pH 7.5, 500 mM (NH4)2SO4,, 5% glycerol, 2 mM DTT, 20 column vol of 1X extract buffer supplemented with 5 mM ATP and 5 mM MgCl2, and finally, 20 column vol of 1X extract buffer without protease inhibitors. Enzymes were eluted from IgG columns with TEV protease, adsorbed on calmodulin affinity columns in the presence of 500 μM CaCl2 and washed and eluted as described except for the reduction of CaCl2 to 500 μM in appropriate buffers (Puig et al., 2001). Complexes were then exchanged into TB by concentration and dilution followed by concentration and storage at –80° C in TB plus 10% glycerol. Reactions were labeled as above with either ATP or CTP, with the same concentrations of components. Reactions were diluted to 5 nM pol II and 5 μL aliquots were added to 5 μL of various concentrations of rNTPs or dNTPs and incubated for 5 min at room temperature. Reactions were stopped and analyzed as above.
Supplementary Material
Acknowledgments
This research was supported by NIH grants GM49985 and GM36559 to R.D.K. C.D.K. was supported by a Fellowship from the Helen Hay Whitney Foundation. Portions of the research were carried out at the Stanford Synchrotron Radiation Laboratory, a national user facility operated by Stanford University on behalf of the U.S. Department of Energy, Office of Basic Energy Sciences. The SSRL Structural Molecular Biology Program is supported by the Department of Energy, Office of Biological and Environmental Research, and by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program, and the National Institute of General Medical Sciences. Portions of this research were conducted at the Advanced Light Source a national user facility operated by Lawrence Berkeley National Laboratory, on behalf of the U.S. Department of Energy, Office of Basic Energy Sciences. The Berkeley Center for Structural Biology is supported in part by the Department of Energy, Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Albert DA, Gudas LJ. Ribonucleotide reductase activity and deoxyribonucleoside triphosphate metabolism during the cell cycle of S49 wild-type and mutant mouse T-lymphoma cells. J Biol Chem. 1985;260:679–684. [PubMed] [Google Scholar]
- Allison LA, Moyle M, Shales M, Ingles CJ. Extensive homology among the largest subunits of eukaryotic and prokaryotic RNA polymerases. Cell. 1985;42:599–610. doi: 10.1016/0092-8674(85)90117-5. [DOI] [PubMed] [Google Scholar]
- Archambault J, Jansma DB, Kawasoe JH, Arndt KT, Greenblatt J, Friesen JD. Stimulation of transcription by mutations affecting conserved regions of RNA polymerase II. J Bacteriol. 1998;180:2590–2598. doi: 10.1128/jb.180.10.2590-2598.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artsimovitch I, Patlan V, Sekine S, Vassylyeva MN, Hosaka T, Ochi K, Yokoyama S, Vassylyev DG. Structural basis for transcription regulation by alarmone ppGpp. Cell. 2004;117:299–310. doi: 10.1016/s0092-8674(04)00401-5. [DOI] [PubMed] [Google Scholar]
- Bar-Nahum G, Epshtein V, Ruckenstein AE, Rafikov R, Mustaev A, Nudler E. A ratchet mechanism of transcription elongation and its control. Cell. 2005;120:183–193. doi: 10.1016/j.cell.2004.11.045. [DOI] [PubMed] [Google Scholar]
- Barford D. Molecular mechanisms of the protein serine/threonine phosphatases. Trends Biochem Sci. 1996;21:407–412. doi: 10.1016/s0968-0004(96)10060-8. [DOI] [PubMed] [Google Scholar]
- Borukhov S, Lee J, Goldfarb A. Mapping of a contact for the RNA 3' terminus in the largest subunit of RNA polymerase. J Biol Chem. 1991;266:23932–23935. [PubMed] [Google Scholar]
- Bushnell DA, Cramer P, Kornberg RD. Structural basis of transcription: alpha-amanitin-RNA polymerase II cocrystal at 2.8 A resolution. Proc Natl Acad Sci USA. 2002;99:1218–1222. doi: 10.1073/pnas.251664698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlin M, Berg P. Deoxyribo ucleic acid-directed synthesis of ribonucleic acid by an enzyme from Escherichia coli. Proc Natl Acad Sci USA. 1962;48:81–94. doi: 10.1073/pnas.48.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chlenov M, Masuda S, Murakami KS, Nikiforov V, Darst SA, Mustaev A. Structure and function of lineage-specific sequence insertions in the bacterial RNA polymerase beta' subunit. J Mol Biol. 2005;353:138–154. doi: 10.1016/j.jmb.2005.07.073. [DOI] [PubMed] [Google Scholar]
- Cramer P, Bushnell DA, Fu J, Gnatt AL, Maier-Davis B, Thompson NE, Burgess RR, Edwards AM, David PR, Kornberg RD. Architecture of RNA polymerase II and implications for the transcription mechanism. Science. 2000;288:640–649. doi: 10.1126/science.288.5466.640. [DOI] [PubMed] [Google Scholar]
- Cramer P, Bushnell DA, Kornberg RD. Structural basis of transcription: RNA polymerase II at 2.8 angstrom resolution. Science. 2001;292:1863–1876. [Google Scholar]
- DeLano WL. The PyMOL Molecular Graphics System. 2002. [Google Scholar]
- Delgado MA, Rintoul MR, Farias RN, Salomon RA. Escherichia coli RNA polymerase is the target of the cyclopeptide antibiotic microcin J25. J Bacteriol. 2001;183:4543–4550. doi: 10.1128/JB.183.15.4543-4550.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ederth J, Mooney RA, Isaksson LA, Landick R. Functional interplay between the jaw domain of bacterial RNA polymerase and allele-specific residues in the product RNA-binding pocket. J Mol Biol. 2006;356:1163–1179. doi: 10.1016/j.jmb.2005.11.080. [DOI] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards AM, Kane CM, Young RA, Kornberg RD. Two dissociable subunits of yeast RNA polymerase II stimulate the initiation of transcription at a promoter in vitro. J Biol Chem. 1991;266:71–75. [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. 2004;D60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Epshtein V, Mustaev A, Markovtsov V, Bereshchenko O, Nikiforov V, Goldfarb A. Swing-gate model of nucleotide entry into the RNA polymerase active center. Mol Cell. 2002;10:623–634. doi: 10.1016/s1097-2765(02)00640-8. [DOI] [PubMed] [Google Scholar]
- Fox CF, Weiss SB. Enzymatic Synthesis Of Ribonucleic Acid. Ii Properties Of The Deoxyribonucleic Acid-Primed Reaction With Micrococcus Lysodeikticus Ribonucleic Acid Polymerase. J Biol Chem. 1964;239:175–185. [PubMed] [Google Scholar]
- Furth JJ, Hurwitz J, Anders M. The role of deoxyribonucleic acid in ribonucleic acid synthesis. I The purification and properties of ribonucleic acid polymerase. J Biol Chem. 1962;237:2611–2619. [PubMed] [Google Scholar]
- Gnatt AL, Cramer P, Fu J, Bushnell DA, Kornberg RD. Structural basis of transcription: an RNA polymerase II elongation complex at 3.3 A resolution. Science. 2001;292:1876–1882. [Google Scholar]
- Hekmatpanah DS, Young RA. Mutations in a conserved region of RNA polymerase II influence the accuracy of mRNA start site selection. Mol Cell Biol. 1991;11:5781–5791. doi: 10.1128/mcb.11.11.5781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Nakamura Y. Localization of nusA-suppressing amino acid substitutions in the conserved regions of the beta' subunit of Escherichia coli RNA polymerase. Mol Gen Genet. 1996;251:699–706. doi: 10.1007/BF02174119. [DOI] [PubMed] [Google Scholar]
- Kettenberger H, Armache KJ, Cramer P. Architecture of the RNA polymerase II-TFIIS complex and implications for mRNA cleavage. Cell. 2003;114:347–357. doi: 10.1016/s0092-8674(03)00598-1. [DOI] [PubMed] [Google Scholar]
- Kettenberger H, Armache KJ, Cramer P. Complete RNA polymerase II elongation complex structure and its interactions with NTP and TFIIS. Mol Cell. 2004;16:955–965. doi: 10.1016/j.molcel.2004.11.040. [DOI] [PubMed] [Google Scholar]
- Kireeva ML, Komissarova N, Waugh DS, Kashlev M. The 8-nucleotide-long RNA:DNA hybrid is a primary stability determinant of the RNA polymerase II elongation complex. J Biol Chem. 2000;275:6530–6536. doi: 10.1074/jbc.275.9.6530. [DOI] [PubMed] [Google Scholar]
- Kornberg A, Baker TA. DNA Replication. 2. New York, New York, USA: W. H. Freeman and Company; 1992. p. 54. [Google Scholar]
- Laptenko O, Lee J, Lomakin I, Borukhov S. Transcript cleavage factors GreA and GreB act as transient catalytic components of RNA polymerase. EMBO J. 2003;22:6322–6334. doi: 10.1093/emboj/cdg610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsley JE, Fuchs RP. Use of single-turnover kinetics to study bulky adduct bypass by T7 DNA polymerase. Biochemistry. 1994;33:764–772. doi: 10.1021/bi00169a018. [DOI] [PubMed] [Google Scholar]
- Markovtsov V, Mustaev A, Goldfarb A. Protein-RNA interactions in the active center of transcription elongation complex. Proc Natl Acad Sci USA. 1996;93:3221–3226. doi: 10.1073/pnas.93.8.3221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews CK. Biochemistry of deoxyribonucleic acid-defective amber mutants of bacteriophage T4.3 Nucleotide pools. J Biol Chem. 1972;247:7430–7438. [PubMed] [Google Scholar]
- Mukhopadhyay J, Sineva E, Knight J, Levy RM, Ebright RH. Antibacterial peptide microcin J25 inhibits transcription by binding within and obstructing the RNA polymerase secondary channel. Mol Cell. 2004;14:739–751. doi: 10.1016/j.molcel.2004.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowotny M, Gaidamakov SA, Crouch RJ, Yang W. Crystal structures of RNase H bound to an RNA/DNA hybrid: substrate specificity and metal-dependent catalysis. Cell. 2005;121:1005–1016. doi: 10.1016/j.cell.2005.04.024. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Macromolecular Crystallography, Part A, Vol 276. New York: Academic Press; 1997. Processing of X-ray diffraction data collected in oscillation mode; pp. 307–326. [DOI] [PubMed] [Google Scholar]
- Palangat M, Hittinger CT, Landick R. Downstream DNA selectively affects a paused conformation of human RNA polymerase II. J Mol Biol. 2004;341:429–442. doi: 10.1016/j.jmb.2004.06.009. [DOI] [PubMed] [Google Scholar]
- Perederina A, Svetlov V, Vassylyeva MN, Tahirov TH, Yokoyama S, Artsimovitch I, Vassylyev DG. Regulation through the secondary channel--structural framework for ppGpp-DksA synergism during transcription. Cell. 2004;118:297–309. doi: 10.1016/j.cell.2004.06.030. [DOI] [PubMed] [Google Scholar]
- Potterton E, Briggs P, Turkenburg M, Dodson E. A graphical user interface to the CCP4 program suite. Acta Crystallogr D Biol Crystallogr. 2003;59:1131–1137. doi: 10.1107/s0907444903008126. [DOI] [PubMed] [Google Scholar]
- Puig O, Caspary F, Rigaut G, Rutz B, Bouveret E, Bragado-Nilsson E, Wilm M, Seraphin B. The tandem affinity purification (TAP) method: a general procedure of protein complex purification. Methods. 2001;24:218–229. doi: 10.1006/meth.2001.1183. [DOI] [PubMed] [Google Scholar]
- Reichard P. Ribonucleotide reductase and deoxyribonucleotide pools. Basic Life Sci. 1985;31:33–45. doi: 10.1007/978-1-4613-2449-2_3. [DOI] [PubMed] [Google Scholar]
- Rhodes G, Chamberlin MJ. Ribonucleic acid chain elongation by Escherichia coli ribonucleic acid polymerase. I Isolation of ternary complexes and the kinetics of elongation. J Biol Chem. 1974;249:6675–6683. [PubMed] [Google Scholar]
- Silverman RB. The Organic Chemistry of Enzyme-Catalyzed Reactions. San Diego, CA, USA: Academic Press; 2000. pp. 85–88. [Google Scholar]
- Svetlov V, Vassylyev DG, Artsimovitch I. Discrimination against deoxyribonucleotide substrates by bacterial RNA polymerase. J Biol Chem. 2004;279:38087–38090. doi: 10.1074/jbc.C400316200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symersky J, Perederina A, Vassylyeva MN, Svetlov V, Artsimovitch I, Vassylyev DG. Regulation through the RNA polymerase secondary channel. Structural and functional variability of the coiled-coil transcription factors. J Biol Chem. 2006;281:1309–1312. doi: 10.1074/jbc.C500405200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temiakov D, Zenkin N, Vassylyeva MN, Perederina A, Tahirov TH, Kashkina E, Savkina M, Zorov S, Nikiforov V, Igarashi N, et al. Structural basis of transcription inhibition by antibiotic streptolydigin. Mol Cell. 2005;19:655–666. doi: 10.1016/j.molcel.2005.07.020. [DOI] [PubMed] [Google Scholar]
- Thuillier V, Brun I, Sentenac A, Werner M. Mutations in the alpha-amanitin conserved domain of the largest subunit of yeast RNA polymerase III affect pausing, RNA cleavage and transcriptional transitions. EMBO J. 1996;15:618–629. [PMC free article] [PubMed] [Google Scholar]
- Trinh V, Langelier MF, Archambault J, Coulombe B. Structural perspective on mutations affecting the function of multisubunit RNA polymerases. Microbiol Mol Biol Rev. 2006;70:12–36. doi: 10.1128/MMBR.70.1.12-36.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuske S, Sarafianos SG, Wang X, Hudson B, Sineva E, Mukhopadhyay J, Birktoft JJ, Leroy O, Ismail S, Clark AD, Jr, et al. Inhibition of bacterial RNA polymerase by streptolydigin: stabilization of a straight-bridge-helix active-center conformation. Cell. 2005;122:541–552. doi: 10.1016/j.cell.2005.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uptain SM, Kane CM, Chamberlin MJ. Basic mechanisms of transcript elongation and its regulation. Annu Rev Biochem. 1997;66:117–172. doi: 10.1146/annurev.biochem.66.1.117. [DOI] [PubMed] [Google Scholar]
- Vassylyev DG, Sekine S, Laptenko O, Lee J, Vassylyeva MN, Borukhov S, Yokoyama S. Crystal structure of a bacterial RNA polymerase holoenzyme at 2.6 A resolution. Nature. 2002;417:712–719. doi: 10.1038/nature752. [DOI] [PubMed] [Google Scholar]
- Vassylyev DG, Svetlov V, Vassylyeva MN, Perederina A, Igarashi N, Matsugaki N, Wakatsuki S, Artsimovitch I. Structural basis for transcription inhibition by tagetitoxin. Nat Struct Mol Biol. 2005;12:1086–1093. doi: 10.1038/nsmb1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weilbaecher R, Hebron C, Feng G, Landick R. Termination-altering amino acid substitutions in the beta' subunit of Escherichia coli RNA polymerase identify regions involved in RNA chain elongation. Genes Dev. 1994;8:2913–2927. doi: 10.1101/gad.8.23.2913. [DOI] [PubMed] [Google Scholar]
- Westover KD, Bushnell DA, Kornberg RD. Structural basis of transcription: nucleotide selection by rotation in the RNA polymerase II active center. Cell. 2004;119:481–489. doi: 10.1016/j.cell.2004.10.016. [DOI] [PubMed] [Google Scholar]
- Winn MD, Isupov MN, Murshudov GN. Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr D Biol Crystallogr. 2001;57:122–133. doi: 10.1107/s0907444900014736. [DOI] [PubMed] [Google Scholar]
- Yang X, Price CW. Streptolydigin resistance can be conferred by alterations to either the beta or beta' subunits of Bacillus subtilis RNA polymerase. J Biol Chem. 1995;270:23930–23933. doi: 10.1074/jbc.270.41.23930. [DOI] [PubMed] [Google Scholar]
- Yuzenkova J, Delgado M, Nechaev S, Savalia D, Epshtein V, Artsimovitch I, Mooney RA, Landick R, Farias RN, Salomon R, Severinov K. Mutations of bacterial RNA polymerase leading to resistance to microcin J25. J Biol Chem. 2002;277:50867–50875. doi: 10.1074/jbc.M209425200. [DOI] [PubMed] [Google Scholar]
- Zakharova N, Bass I, Arsenieva E, Nikiforov V, Severinov K. Mutations in and monoclonal antibody binding to evolutionary hypervariable region of Escherichia coli RNA polymerase beta' subunit inhibit transcript cleavage and transcript elongation. J Biol Chem. 1998;273:24912–24920. doi: 10.1074/jbc.273.38.24912. [DOI] [PubMed] [Google Scholar]
- Zamble DB, Mu D, Reardon JT, Sancar A, Lippard SJ. Repair of cisplatin--DNA adducts by the mammalian excision nuclease. Biochemistry. 1996;35:10004–10013. doi: 10.1021/bi960453+. [DOI] [PubMed] [Google Scholar]
- Zhang J, Zhang Z, Brew K, Lee EY. Mutational analysis of the catalytic subunit of muscle protein phosphatase-1. Biochemistry. 1996;35:6276–6282. doi: 10.1021/bi952954l. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.