

Abstract
Transgenic rat models of amyotrophic lateral sclerosis (ALS) have recently been developed. Most assays of ALS-symptoms in these models monitor disease onset accurately, but do not identify individuals that will develop these symptoms before the motor deficits become apparent. Peak bodyweight has recently been shown to indicate affected individuals before motor deficits become apparent. However, it must be determined retrospectively due to weight fluctuation. Here, we report that exploratory activities detected by a Photobeam Activity System (PAS) and wire mesh ascending test can be used to detect slight motor deficits in the early phase of ALS. Thus, these tests may be used in addition to peak bodyweight to monitor early disease progression and to assay efficacy of new therapeutic interventions.
Keywords: ALS (Amyotrophic Lateral Sclerosis), transgenic rat, motor function, superoxide dismutase 1
Amyotrophic lateral sclerosis (ALS) or Lou Gehrig’s disease is a devastating neurological disease characterized by selective degeneration of motor neurons. Approximately 10% of patients inherit the disease (familial or FALS), but the rest are sporadic (SALS) with no known genetic link [3]. Most ALS patients develop limb weakness initially, which progresses quickly to generalized muscle atrophy and paralysis. Death ultimately occurs within 5 years due to respiratory paralysis.
In 1993, a mutated gene coding for copper/zinc superoxide dismutase 1 (Cu/Zn-SOD1) was first associated with ALS, marking a major breakthrough in our understanding of ALS pathogenesis. It is now known that approximately 20–25% of the familial cases or 1–2% of total ALS cases are due to dominantly inherited mutations in Cu/Zn-SOD1 [5,14]. Due to the clinical and pathological similarities to human FALS and SALS, our understanding of ALS mechanisms has been enhanced by studies using hemizygous transgenic rodent animal models that express mutant human Cu/Zn-SOD1 genes [7,8,13].
Behavioral analyses have previously been used to evaluate potential therapies for transgenic ALS mice or rats. RotaRod tests were the most frequently used method to evaluate motor performance after ALS therapies in mice [1,4,9,10,18–20]. Other assays included grip strength and wire hanging tests [2,6,12]. Also, an automated motion analyses system (SCANET) and inclined plane test have been used to evaluate disease progression in ALS rats [11]. Unfortunately, these tests in rats only identified individuals at or near symptomatic disease onset (obvious limb weakness defined as limping or paralysis), but failed to detect slight motor deficits during the early disease stage.
Thus, it was a break-through when Matsumoto et al. showed that peak bodyweight was a reliable index of “pre-symptomatic” disease onset, identifying individuals approximately 13 days prior to symptomatic onset [11]. However, due to fluctuations in weight, bodyweight peak must be determined retrospectively [11]. Thus, other assays to identify affected individuals before apparent symptoms develop would be desirable. Here, we report that measurements of exploratory activities in a Photobeam Activity System (PAS) improve early diagnosis by identifying early motor deficits. In addition, a simple wire mesh ascending test monitors both early and late disease progression.
Male hemizygous NTac:SD-TgN(SOD1G93A)L26H rats (Taconic, Hudson, NY), originally developed by Howland et al., were bred locally by crossing with wild-type female NTac:SD rats [8]. Male SOD1G93A progenies were used for further breeding to maintain the line. Human mutant SOD1-negative littermates were not used as breeders to ensure the consistency of the colony phenotype. The breeding protocol was approved by the Institutional Animal Care and Use Committee (IACUC) at The University of Texas Medical Branch. Rats were housed in a centralized Animal Care Facility with a 12-hr light-dark cycle (light 8AM – 8 PM). Food and water were provided ad libitum. SOD1G93A progenies were verified by PCR genotyping. Chromosomal DNAs were extracted from tails of 3-week old pups and subjected to PCR amplification with the following primers: sodi3-f primer (GTGGCATCAGCCCTAATCCA) and sodE4-r primer (CACCAGTGT GCGGCCAATGA).
Rats were weighed once per week beginning at 70 days of age, then twice per week at 120 days of age until symptomatic disease onset (determined by motor score, see below) and finally, once per day until disease end-stage, as defined by a lack of righting reflexes, i.e. inability to right oneself from both sides within 30 sec.
Rats were tested once per week beginning at 70 days of age, then twice per week beginning at 120 days of age until disease end-stage. This test was conducted according to a previous description with modifications [17]. A 10 mm wire mesh (H and W: 45 cm × 15 cm) was placed at an angle of 70° in contact with a cardboard box at the top and the edge of a lab bench at the bottom. Rats were placed on the bottom of the wire mesh and motivated to ascend by placing their littermates in the box at the top. Each rat was pre-trained 3 times per day for 3 consecutive days to adapt to the system. Time to reach the top of the wire mesh was recorded for up to 5 seconds. If a rat was unable to ascend or fell off the wire mesh, a time of 5 sec. was recorded. Three runs were recorded for each rat on each testing day.
Exploratory activity was measured by a PAS once per week beginning at 17 weeks of age. Two activity chambers (40 × 40 × 40 cm) collected movement data analyzed by a PAS (San Diego Instruments, Inc., CA) according to our previous description [16]. The exploratory activity of each rat was assessed over three consecutive 5-minute intervals (total 900 seconds) by the time of photobeam obstruction in an axis-oriented grid system. There were 16 photobeams on each horizontal axis located 4 cm above the chamber floor. Interruption of these photobeams recorded the distance traveled. Obstruction of another set of photobeams located 12 cm above the chamber floor was recorded as rearing events. Resting time occurred when the rats were stationary for at least 1 second.
Symptomatic disease onset and post-symptomatic disease progression were assessed using a modified 5-point motor score system derived from the Matsumoto et al. motor score [11]. This assay was performed once per week beginning at 70 days of age, then twice per week at 120 days of age until disease onset and finally, once per day until the disease end-stage. Rats were allowed to move freely in an open field and were scored blindly by an observer. A non-parametrical scoring system was used following these criteria: 5, normal movement; 4, loss of some weight bearing in the hindlimbs, or limping or dragging of any limb, but still able to stand on hindlimbs; 3, loss of most weight bearing in hindlimbs, dragging of lower body and inability to stand on hindlimbs; 2, complete loss of weight bearing in hindlimbs accompanied by forelimb weakness, unable to drag the lower body, but righting reflexes present from both sides; 1, a righting reflex from only one side; 0, absent righting reflexes from both sides within 30 seconds (defined as the end-stage). Symptomatic disease onset was identified when the rats showed a score of 4. Lifespan was determined by the age of the rats at end-stage (score of 0), at which time the rats were euthanized.
The two-tailed unpaired Student’s t-test with Welch’s correction was used to compare the transgenic ALS and wild-type littermate data from the motor score assessments, wire mesh ascending test times and Photobeam Activity System. A p value less than 0.05 was considered statistically significant. Data were expressed as means ± S.E.M. Lifespan and disease onset were depicted using Kaplan-Meier survival curves. Statistical analyses were done using GraphPad Prism Version 4 software (GraphPad Software, Inc., San Diego, CA).
Bodyweight measurements monitor the extent of muscle wasting in ALS rats. The bodyweights of ALS rats peaked at 143.5 ± 12.9 days (Table 1) and began to decrease approximately 24 days prior to the average age of symptomatic disease onset (Fig. 1a). The wire mesh ascending test monitors muscle strength and limb coordination, both of which are affected in ALS. Transgenic ALS rats significantly increased the time spent climbing a distance of 45 cm as early as 145 days of age compared to their wild-type littermates (Fig. 1b). Note that a dramatic increase of ascending time occurred close to the average age of symptomatic disease onset.
Table 1.
Summary of methods for identifying pre-symptomatic disease onset, symptomatic disease onset and lifespan.
| Evaluation methods | Age in days (range) |
|---|---|
| Pre-symptomatic disease onset | |
| Peak bodyweighta | 143.5 ± 12.9 (114–163) |
| Distance traveled in PASb | 151.3 ± 19.0 (118–176) |
| Ascending timec | 159.6 ± 13.3 (138–173) |
| Symptomatic disease onset | |
| Observable motor deficitsd | 167.6 ± 15.8 (138–187) |
| Rearing events in PASe | 165.0 ± 15.5 (138–186) |
| Resting time in PASf | 174.3 ± 12.5 (156–191) |
| End-stage disease | |
| Lifespang | 188.7 ± 15.2 (165–207) |
Mean ± SD. ALS transgenic rats (n=10). Male:female ratio is 1:1.
Max weight prior to a constant decline
Total distance traveled ≤ 6500 cm
Average climbing time ≥ 1.8 sec.
Motor score of 4
Total rearing events ≤ 60 events
Total resting time ≥ 450 sec.
Motor score of 0
Fig. 1.
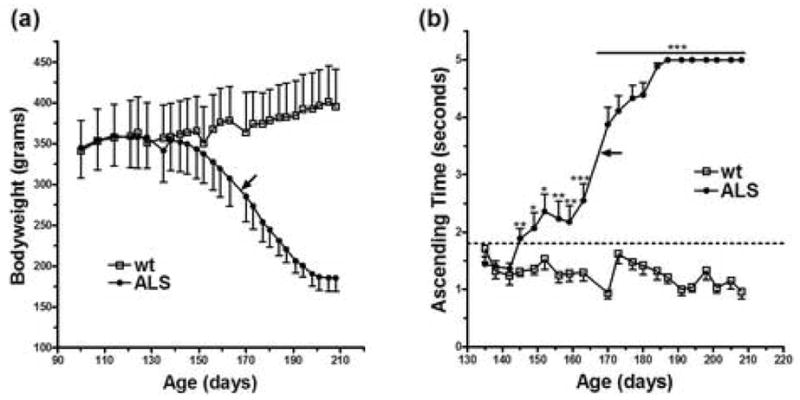
Bodyweight measurements and time recordings on the wire mesh ascending test monitor pre-symptomatic disease progression. Bodyweight measurements (a) and wire mesh ascending test time recordings (b). wt, wild-type littermates (n=6). ALS, transgenic ALS rats (n=10). Male:female ratio 1:1 in both groups. Black arrows represent the average age of symptomatic disease onset (167.6 days). Dot line depicts the first value in ALS rats that is significantly different from wild-type littermates. *p < 0.05. ** p < 0.01. ***p < 0.001.
ALS pathogenesis causes a rapid decline in motor strength and activity. Several key parameters of motor function, including walking distance, rearing capability and resting time were evaluated using a Photobeam Activity System (PAS). The total distance traveled in the PAS showed a consistent and significant decrease in the transgenic ALS group compared to the wild-type littermates at 21 weeks of age (Fig. 2a). The number of rearing events significantly declined in consecutive weeks beginning at 23 weeks of age (Fig. 2b). However, the resting time did not significantly increase until 24 weeks of age (Fig. 2c), which was near symptomatic disease onset determined by motor scoring. All three parameters showed a trend of pre-symptomatic disease progression, but only walking distance and rearing capability significantly declined prior to symptomatic disease onset (Table 1).
Fig. 2.
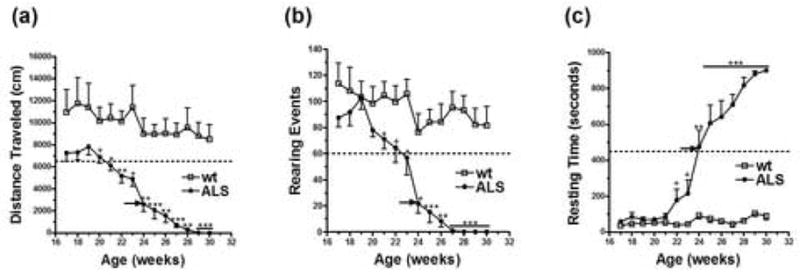
Distance traveled and rearing capability, but not resting time, as measured in a Photobeam Activity System (PAS) identify pre-symptomatic motor deficits. Total distance traveled (a), total number of rearing events (b) and total resting time (c) as determined over a 15-min time period in a PAS. wt, wild-type littermates (n=6). ALS, transgenic ALS rats (n=10). Male: female ratio 1:1 in both groups. Black arrows represent the average age of symptomatic disease onset (167.6 days). Dot line depicts the first value in ALS rats that is significantly different from wild-type littermates. *p < 0.05. ** p < 0.01. ***p < 0.001. + 0.05 < p < 0.06.
Symptomatic disease onset was subjectively determined through motor score assessment. The day of symptomatic disease onset was defined as the day when the motor score of a rat decreased from 5 to 4. Post-symptomatic disease progression was subsequently evaluated through further assessments of motor score until each rat reached a motor score of 0. The day when the motor score reached 0 was considered the ‘end-stage’ of the lifespan for each rat. When averaging motor scores from all ALS rats on each assessment day, ALS rats first showed a significant decrease in motor score at 170 days of age, which was comparable to the average age of symptomatic disease onset (Fig. 3a). The Kaplan-Meier survival curves for symptomatic disease onset (DO) and end-stage (ES) are depicted in Fig. 3b. The interval between DO and ES was about 21 days (Table 1).
Fig. 3.
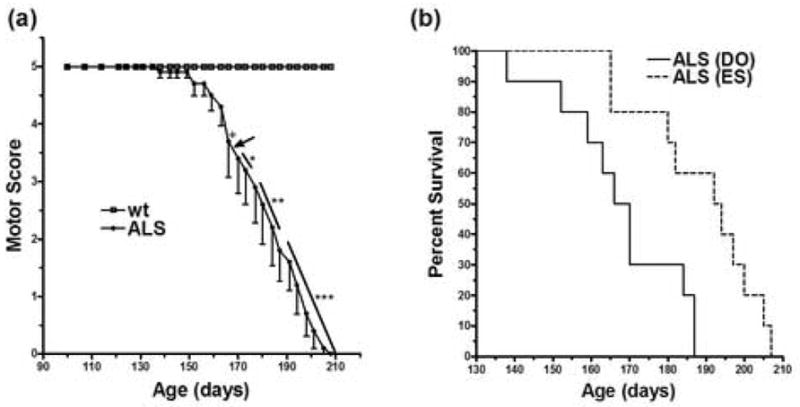
Transgenic ALS rats show a delayed symptomatic disease onset and lifespan compared to previous reports. Post-symptomatic disease progression based on a modified 5-point motor score (a). Kaplan-Meier survival curves depict the symptomatic disease onset (DO) and the end-stage (ES) (b). wt, wild-type littermates (n=6). ALS, transgenic ALS rats (n=10). Male: female ratio is 1:1 in both groups. Black arrow represents the average age of symptomatic disease onset (167.6 days). *p < 0.05. ** p < 0.01. ***p < 0.001. + p = 0.0703.
The major thesis of the present study is that the earlier one can identify individuals that will develop ALS or ALS-like symptoms, the more effective therapy is likely to be. Accordingly, early diagnosis, particularly before substantial motor deficits can be recognized, is a matter of considerable importance. Most previously used tests, such as the motor score test used in this study, do an excellent job of monitoring the disease process, but they only indicate ALS-like individuals after motor symptoms become apparent (limping and paralysis). A real advance has been made recently in demonstrating that the peak weight of ALS rats identified individuals that will develop motor deficits with precision well before the onset of observable symptoms [11]. The difficulty with this assay is that rat weights vary enough that one cannot be sure when the peak is reached until a marked weight loss is seen, which is later than the time of peak weight. Therefore, further tests to identify individuals that will develop motor deficits are desirable.
In the present study, we tested the ability of a PAS and wire mesh ascending test to detect subtle motor abnormalities in transgenic ALS rats. These tests, in combination with bodyweight measurements, allowed us to objectively monitor pre-symptomatic disease onset more precisely than the use of peak bodyweight alone. Accordingly, the above tests should be combined with determinations of peak weight to obtain the most objective and earliest assessment of the onset of symptoms in ALS rats.
In more detail, the wire mesh ascending test first detected a significant difference between the transgenic ALS rats and their wild-type littermates at 145 days of age. Using the threshold cutoff value of a 1.8-sec climbing time, the average age of the ALS rats was approximately 160 days. Thus, this test could detect motor deficits about 1 week prior to symptomatic disease onset (Table 1). Furthermore, the day each rat recorded an average climbing time of 3 seconds or more correlated well (r2 = 0.8490) with the day of symptomatic disease onset. Better correlative values may have been obtained if this test was performed on a daily basis rather than twice per week. The conclusion is that the wire mesh ascending test was an adequate behavioral test to identify subtle pre-symptomatic motor deficits.
All three PAS parameters also indicated onset and monitored disease progression to detect signs of motor weakness too subtle to be visually observed in ALS rats, but total distance traveled was the best indicator of pre-symptomatic motor abnormalities. Using a threshold value of 6500 cm walking distance, this test detected motor abnormalities approximately 16 days prior to symptomatic disease onset. Rearing capability also dramatically decreased in the ALS rats. However, when a threshold of 60 events was used, rearing deficits were only detected 2–3 days prior to symptomatic disease onset. Similarly, total resting time drastically increased in the transgenic ALS rats, but resting time did not identify a pre-symptomatic deficit in motor activity using a cutoff value of 450 seconds. It is likely, however, that increasing the number of rats in the study would increase the capability of these three parameters to detect pre-symptomatic motor abnormalities.
In order to minimize litter and gender variations, the wild-type and transgenic groups each consisted of an assortment of rats from five and six litters, respectively, in a 1:1 ratio of males to females. However, a careful examination of potential effects of gender matching and littermate pairing on wire mesh ascending time and PAS activity remains to be conducted in future studies with larger sample sizes. Furthermore, due to the large variation in disease progression in the ALS rodent model, trial experiments and a subsequent power analysis must be performed in order to determine the number of transgenic rats required to distinguish a significant treatment effect between groups when evaluating ascending time and PAS activities.
Finally, the symptomatic disease onset (168 days) and lifespan (189 days) in our colony as determined by a modified motor score (Table 1) were considerably delayed compared to what were earlier described, 115 days for disease onset and 126 days for lifespan, in the original transgenic line [8]. The cause of these differences remains to be determined, but may indicate a decrease in mutant SOD1 expression in our colony or may be due to large variations in disease onset that characterize this model [15].
In summary, we discovered that the exploratory activity measured in a PAS and the climbing time measured by a wire mesh ascending test, in combination with bodyweight measurements, can be used to better define early signs of disease progression in transgenic ALS rats. This is particularly useful in order to properly evaluate how potential therapies such as cell transplantation, intrathecal drug administration or gene therapy act to delay ALS disease progression.
Acknowledgments
The authors thank the technical assistance from Tiffany J. Dunn, the critical review by Richard Coggeshall and the support from the National Institute of Neurological Disorders and Stroke (NS046025) and the John S. Dunn Research Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Andreassen OA, Dedeoglu A, Klivenyi P, Beal MF, Bush AI. N-acetyl-L-cysteine improves survival and preserves motor performance in an animal model of familial amyotrophic lateral sclerosis. Neuroreport. 2000;11:2491–2493. doi: 10.1097/00001756-200008030-00029. [DOI] [PubMed] [Google Scholar]
- 2.Azari MF, Lopes EC, Stubna C, Turner BJ, Zang DW, Nicola NA, Kurek JB, Cheema SS. Behavioural and anatomical effects of systemically administered leukemia inhibitory factor in the SOD1(G93A) (G1H) mouse model of familial amyotrophic lateral sclerosis. Brain Res. 2003;982:92–97. doi: 10.1016/s0006-8993(03)02989-5. [DOI] [PubMed] [Google Scholar]
- 3.Bruijn LI, Miller TM, Cleveland DW. Unraveling the mechanisms involved in motor neuron degeneration in ALS. Annu Rev Neurosci. 2004;27:723–749. doi: 10.1146/annurev.neuro.27.070203.144244. [DOI] [PubMed] [Google Scholar]
- 4.Chiba T, Yamada M, Sasabe J, Terashita K, Aiso S, Matsuoka M, Nishimoto I. Colivelin prolongs survival of an ALS model mouse. Biochem Biophys Res Commun. 2006;343:793–798. doi: 10.1016/j.bbrc.2006.02.184. [DOI] [PubMed] [Google Scholar]
- 5.Gaudette M, Hirano M, Siddique T. Current status of SOD1 mutations in familial amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1:83–89. doi: 10.1080/14660820050515377. [DOI] [PubMed] [Google Scholar]
- 6.Guo H, Lai LC, Butchbach MER, Stockinger MP, Shan X, Bishop GA, Lin CLG. Increased expression of the glial glutamate transporter EAAT2 modulates excitotoxicity and delays the onset but not the outcome of ALS in mice. Hum Mol Genet. 2003;12:2519–2532. doi: 10.1093/hmg/ddg267. [DOI] [PubMed] [Google Scholar]
- 7.Gurney ME, Pu HF, Chiu AY, Dalcanto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX, Chen WJ, Zhai P, Sufit RL, Siddique T. Motor-Neuron Degeneration in Mice That Express A Human Cu,Zn Superoxide-Dismutase Mutation. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- 8.Howland DS, Liu J, She YJ, Goad B, Maragakis NJ, Kim B, Erickson J, Kulik J, DeVito L, Psaltis G, DeGennaro LJ, Cleveland DW, Rothstein JD. Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS) Proc Natl Acad Sci USA. 2002;99:1604–1609. doi: 10.1073/pnas.032539299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kiaei M, Petri S, Kipiani K, Gardian G, Choi DK, Chen JY, Calingasan NY, Schafer P, Muller GW, Stewart C, Hensley K, Beal MF. Thalidomide and lenalidomide extend survival in a transgenic mouse model of amyotrophic lateral sclerosis. J Neurosci. 2006;26:2467–2473. doi: 10.1523/JNEUROSCI.5253-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Klivenyi P, Ferrante RJ, Matthews RT, Bogdanov MB, Klein AM, Andreassen OA, Mueller G, Wermer M, Kaddurah-Daouk R, Beal MF. Neuroprotective effects of creatine in a transgenic animal model of amyotrophic lateral sclerosis. Nat Med. 1999;5:347–350. doi: 10.1038/6568. [DOI] [PubMed] [Google Scholar]
- 11.Matsumoto A, Okada Y, Nakamichi M, Nakamura M, Toyama Y, Sobue G, Nagai M, Aoki M, Itoyama Y, Okano H. Disease progression of human SOD1 (G93A) transgenic ALS model rats. J Neurosci Res. 2006;83:119–133. doi: 10.1002/jnr.20708. [DOI] [PubMed] [Google Scholar]
- 12.Miana-Mena FJ, Munos MJ, Yague G, Mendez M, Moreno M, Ciriza J, Zaragoza P, Osta R. Optimal methods to characterize the G93A mouse model of ALS. Amyotroph Lateral Scler Other Motor Neuron Disord. 2005;6:55–62. doi: 10.1080/14660820510026162. [DOI] [PubMed] [Google Scholar]
- 13.Nagai M, Aoki M, Miyoshi I, Kato M, Pasinelli P, Kasai N, Brown RH, Itoyama Y. Rats expressing human cytosolic copper-zinc superoxide dismutase transgenes with amyotrophic lateral sclerosis: Associated mutations develop motor neuron disease. J Neurosci. 2001;21:9246–9254. doi: 10.1523/JNEUROSCI.21-23-09246.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, Oregan JP, Deng HX, Rahmani Z, Krizus A, Mckennayasek D, Cayabyab A, Gaston SM, Berger R, Tanzi RE, Halperin JJ, Herzfeldt B, Vandenbergh R, Hung WY, Bird T, Deng G, Mulder DW, Smyth C, Laing NG, Soriano E, Pericakvance MA, Haines J, Rouleau GA, Gusella JS, Horvitz HR, Brown RH. Mutations in Cu/Zn Superoxide-Dismutase Gene Are Associated with Familial Amyotrophic-Lateral-Sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- 15.Storkebaum E, Lambrechts D, Dewerchin M, Moreno-Murciano MP, Appelmans S, Oh H, Van Damme P, Rutten B, Man WY, De Mol M, Wyns S, Manka D, Vermeulen K, Van den Bosch L, Mertens N, Schmitz C, Robberecht W, Conway EM, Collen D, Moons L, Carmeliet P. Treatment of motoneuron degeneration by intracerebroventricular delivery of VEGF in a rat model of ALS. Nat Neurosci. 2005;8:85–92. doi: 10.1038/nn1360. [DOI] [PubMed] [Google Scholar]
- 16.Tarasenko YI, Nie L, McAdoo D, Johnson K, Hulsebosch C, Grady J, Wu P. Survival and differentiation of human neural stem cells in contusion-injured rat spinal cord. J Neurosci Res. doi: 10.1002/jnr.21098. in press. [DOI] [PubMed] [Google Scholar]
- 17.Tchekalarova J, Kubova H, Mares P. Postnatal caffeine exposure: effects on motor skills and locomotor activity during ontogenesis. Behav Brain Res. 2005;160:99–106. doi: 10.1016/j.bbr.2004.11.018. [DOI] [PubMed] [Google Scholar]
- 18.Van den Bosch L, Tilkin P, Lemmens G, Robberecht W. Minocycline delays disease onset and mortality in a transgenic model of ALS. Neuroreport. 2002;13:1067–1070. doi: 10.1097/00001756-200206120-00018. [DOI] [PubMed] [Google Scholar]
- 19.Wang LJ, Lu YY, Muramatsu S, Ikeguchi K, Fujimoto K, Okada T, Mizukami H, Matsushita T, Hanazono Y, Kume A, Nagatsu T, Ozawa K, Nakano I. Neuroprotective effects of glial cell line-derived neurotrophic factor mediated by an adeno-associated virus vector in a transgenic animal model of amyotrophic lateral sclerosis. J Neurosci. 2002;22:6920–6928. doi: 10.1523/JNEUROSCI.22-16-06920.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhu S, Stavrovskaya IG, Drozda M, Kim BYS, Ona V, Li MW, Sarang S, Liu AS, Hartley DM, Du CW, Gullans S, Ferrante RJ, Przedborski S, Kristal BS, Friedlander RM. Minocycline inhibits cytochrome c release and delays progression of amyotrophic lateral sclerosis in mice. Nature. 2002;417:74–78. doi: 10.1038/417074a. [DOI] [PubMed] [Google Scholar]