

Abstract
Fibroblast growth factor 2 (FGF2) inhibits oligodendrocyte progenitor cell (OPC) differentiation during development and limits remyelination following chronic demyelination. The current study examines the mechanism underlying this effect of FGF2 expression on OPC differentiation. Retroviral lineage tracing demonstrates a direct in vivo effect of FGF receptor (FGFR) signaling on OPC differentiation. Retrovirus expressing a dominant negative FGFR construct (FGFRdn) and green fluorescent protein (GFP) was injected into the dorsal columns of postnatal day 7 (P7) mice followed by perfusion at P28. Among the GFP-labeled cells, FGFRdn retrovirus generated a higher proportion of oligodendrocytes than did control infections. This result from FGFRdn expression in OPCs was similar to the result obtained in our previous study using control retrovirus in FGF2 null mice. Further, in vitro retroviral siRNA expression distinguishes the function of specific FGFR isoforms in OPC responses to FGF2. FGF2 inhibition of OPC differentiation was effectively blocked by siRNA targeted to FGFR1, but not FGFR2 or FGFR3. We propose a model of direct FGF2 activation of FGFR1 leading to inhibition of OPC differentiation. This signaling pathway may be an important regulator of oligodendrocyte generation during myelination in development and may perturb OPC generation of remyelinating oligodendrocytes in demyelinating disease. Published 2006 Wiley-Liss, Inc.†
Keywords: differentiation, oligodendrocyte, fibroblast growth factor 2 (FGF2), FGF receptor (FGFR), siRNA
Abbreviations: bp Base pair; CFU Clone forming units; CNS Central nervous system; DMEM Dulbecco Modified Eagle Medium; FBS Fetal bovine serum; FGF2 Fibroblast growth factor 2; FGFR1,2,3 Fibroblast growth factor receptor 1,2,3; GFP Green fluorescent protein; nt Nucleotide; PDGF Platelet derived growth factor-AA; PLP Proteolipid protein; shRNA Short hairpin RNA; siRNA Small interfering RNA
INTRODUCTION
The generation of oligodendrocytes is a critical process for central nervous system (CNS) development and repair. In the embryonic and postnatal CNS, neural stem cells and oligodendrocyte progenitor cells (OPCs) respond to a hierarchy of environmental signals to generate oligodendrocytes that myelinate fiber tracts according to specific temporal and spatial patterns. OPCs and premyelinating oligodendrocytes are abundant in the early postnatal CNS and then are maintained at reduced levels in adults. In demyelinating diseases, such as multiple sclerosis (MS), oligodendrocyte regeneration is a prerequisite for remyelination and the recovery of nerve conduction. OPCs and premyelinating oligodendrocytes may be increased in number in MS lesions yet still fail to differentiate into myelinating oligodendrocytes to effectively remyelinate areas of repeated or chronic demyelination (Chang et al., 2002; Wolswijk, 1998, 2002). Elucidating mechanisms by which environmental signals inhibit OPC generation of myelinating oligodendrocytes may lead to therapeutic targets for treating remyelination failure.
Previous studies indicate that fibroblast growth factor 2 (FGF2) is an important regulator of oligodendrocyte generation during postnatal development and remyelination in rodent models. Expression of FGF2 ligand and FGF receptor (FGFR) isoforms increases coincident with postnatal myelination and in response to demyelination (Armstrong et al., 2002; Bansal et al., 2003; Messersmith et al., 2000; Murtie et al., 2005a). The predominant in vivo effect of FGF2 on oligodendrocyte lineage cells during both myelination and remyelination is inhibition of OPC differentiation (Murtie et al., 2005a,b). Importantly, genetic deletion of endogenous FGF2 results in enhanced oligodendrocyte repopulation of lesions and more extensive remyelination (Armstrong et al., 2002, 2006).
These in vivo studies demonstrate a significant role for endogenous FGF2 in regulating oligodendrocyte lineage development and regeneration. FGF2 can act through three high-affinity FGFR types (FGFR1–3) that are expressed in oligodendrocyte lineage cells as wells as neurons, astrocytes, and microglia (Asai et al., 1993; Ballabriga et al., 1997; Bansal et al., 1996; Belluardo et al., 1997; Miyake et al., 1996). Therefore, it is now important to determine whether FGF2 acts directly through FGFRs on OPCs to regulate OPC differentiation in an in vivo context. To further understand this mechanism of FGF2 action, we must also identify which FGFR type(s) predominantly regulate FGF2 inhibition of OPC differentiation.
In vitro studies of neonatal oligodendrocyte lineage cells have shown that FGF2 inhibits terminal differentiation and induces OPC proliferation (Bansal et al., 1996; Fortin et al., 2005; McKinnon et al., 1990). Within the oligodendrocyte lineage, FGFRs are expressed in a developmentally regulated manner that can be modulated by FGF2 (Bansal et al., 1996; Fortin et al., 2005). FGFR1 is expressed by cells at all stages of the lineage and levels increase with maturation. FGFR2 expression is low in early stages of the lineage and highest in mature oligodendrocytes. FGFR3 expression levels peak at the late progenitor stage and then decline with further maturation. In vitro, FGF2 treatment increases expression of FGFR1 in OPCs but decreases FGFR2 expression in mature oligodendrocytes.
The mechanism by which these FGFR isoforms mediate FGF2 inhibition of OPC differentiation during the generation of myelinating oligodendrocytes is not clear. Treatment of neonatal OPC cultures with neutralizing antibodies to specific FGFR types indicated that simultaneous inactivation of FGFR1 and FGFR3 was required to block FGF2 inhibition of OPC differentiation (Fortin et al., 2005). However, studies of postnatal oligodendrogenesis in FGFR3 knockout mice interpreted FGFR3 signaling as promoting OPC differentiation (Oh et al., 2003). Further analysis is required to more directly assess OPC differentiation and the regulatory contributions of specific FGFR isoforms upon activation by FGF2.
The current study uses retroviral lineage tracing to label proliferating OPCs by green fluorescent protein (GFP) expression and monitor differentiation of subsequently generated cells into mature oligodendrocytes. The retroviral constructs were designed to also express either a dominant negative form of FGFR (FGFRdn) or an isoform-specific short hairpin RNA (shRNA) sequence. The FGFRdn can inactivate all endogenous FGFR types simultaneously to simplify detection of a direct FGFR signaling effect, which otherwise might be obscured by potential redundancy among FGFR isoforms. In vivo retroviral infection of OPCs with FGFRdn in postnatal day 7 (P7) spinal cord demonstrates a direct effect of FGFR signaling in OPCs as inhibiting differentiation during myelination. The effect of FGFRdn inactivation of FGFR signaling is similar to the effect of FGF2 removal, as observed by lineage tracing with control retrovirus in FGF2 null mice (Murtie et al., 2005a). For analysis of the FGFR isoforms regulating OPC differentiation, the shRNA expression was designed for RNA interference to individually inactivate each of the three relevant FGFR isoforms. In vitro retroviral infection of OPCs with constructs expressing each shRNA showed that expression of FGFR1, but not FGFR2 or FGFR3, was required for FGF2 inhibition of OPC differentiation. These experiments implicate FGF2 acting through FGFR1 on OPCs as key components mediating FGF2 inhibition of OPC differentiation. This signaling pathway may be an important regulator of oligodendrocyte generation during myelination in development and remyelination in demyelinating disease.
MATERIALS AND METHODS
All animals were handled in accordance with procedures approved by the USUHS Institutional Animal Care and Use Committee.
Glial Cell Cultures
Primary cultures from neonatal rat brains were prepared as previously described (Armstrong et al., 1995; Armstrong, 1998). Briefly, postnatal day 2 rat brains were dissociated, plated in tissue culture flasks, and allowed to grow for 7–10 days. The flasks were placed on a rotary shaker to dislodge OPCs, which were then plated onto poly-d-lysine-coated chamber slides for proteolipid protein (PLP) mRNA detection or 24-well plates for all other experiments. OPCs were grown in defined medium (Eccleston and Silberberg, 1984) containing high insulin (5 μg/mL), T3 (Triiodothyronine; 30 nM), and 0.5% fetal bovine serum (FBS) (all from Sigma, St. Louis, MO). Under these conditions, the majority of OPCs differentiate into mature oligodendrocytes over 48 h in culture. In contrast, the majority of OPCs proliferate without differentiating when grown in defined medium with mitogens, platelet-derived growth factor AA (PDGF; 10 ng/mL), and FGF2 (10 ng/mL) (both from R and D Systems, Minneapolis, MN). After plating, OPCs were cultured overnight in defined medium with mitogens before incubating in retrovirus stock medium for 6 h. Then the cultures were washed and returned to defined medium with mitogens. At 48 h post-infection, the medium was changed to fresh defined medium with 0.5% FBS and with or without PDGF and FGF2, as noted for specific experiments. At 96 h postinfection the cells were fixed with 4% paraformaldehyde. Assays were repeated from at least three independent cell preparations.
FGFRdn Retroviral Vector Construction and Infections
A truncated FGFR2 sequence was used to generate a dominant negative FGFR construct (FGFRdn; nucleotides 607–2016 encoding the extracellular and transmembrane domains of murine FGFR2 variant IIIc). FGFRdn was expressed either as a direct fusion with a FLAG epitope tag in the pLP-LNCX retroviral vector (Clontech, Palo Alto, CA) or followed by an internal ribosome entry site (IRES) and GFP in the pMXs-IG retroviral plasmid (from T. Kitamura, Tokyo, Japan). Both retroviral plasmids are replication-incompetent. The FGFRdn fragment was generated from the FGFR2 cDNA (Mansukhani et al., 1992) truncated downstream of the transmembrane domain by cutting with EcoRI and BamHI and ligating into the pBluescript II SK (Stratagene; La Jolla, CA) to form FGFRdn-SK. SalI and Bam HI sites were then used to transfer the FGFRdn fragment from FGFRdn-SK into a donor vector with a direct FLAG epitope tag sequence (generated from Clontech pDNR3 donor vector; Nielsen et al., 2004). This pDNR3-FGFRdn-FLAG donor vector was combined with the pLP-LNCX retroviral expression vector, in the presence of cre recombinase to transfer the FGFRdn-FLAG cassette into the retroviral vector according to the manufacturer’s instructions (Clontech; Palo Atlo, CA) resulting in the FGFRdn-FLAG retroviral plasmid. Alternatively, the FGFRdn fragment was isolated from FGFRdn-SK using NotI sites for insertion into the pMXs-IG retroviral vector resulting in the PMXs-FGFRdn retroviral plasmid with GFP as the reporter.
To generate retrovirus stocks, GP2-293 packaging cells (Clontech; Palo Atlo, CA) were co-transfected with each replication-incompetent retroviral plasmid along with pVSVG, which expresses the VSV glycoprotein as a pantropic retrovirus receptor. Cells were transfected at roughly 80% confluency with FuGene6 (Roche Applied Science, Indianapolis, IN). Each 60-mm dish was transfected with 2 μg of retroviral plasmid DNA and 2 μg of pVSVG DNA. At 72 h post-transfection, the supernatant containing the packaged virions was filtered and concentrated by centrifugation. Pellets were resuspended in 50 μL of HBSS and aliquots were stored at −80°C for use as retroviral stocks.
The titer of the retroviral stocks of LEGFP, FGFRdn-FLAG, PMXs, and PMXs-FGFRdn were determined as described (Nielsen et al., 2004). The control LEGFP plasmid typically yielded higher titer supernatants (1.0 × 106 colony forming unit (CFU)/mL; CFU/mL), which were then diluted with supernatant from a mock-transfected dish to achieve the same colony CFU/mL in the control LEGFP, FGFRdn-FLAG, PMXs, and PMXs-FGFRdn viral stocks. Polybrene was added to the supernatant to a final concentration of 6 μg/mL. For infection of OPCs, PDGF and FGF2 were added to a 10 ng/mL final concentration in the infection medium.
Immunocytochemistry
After fixation with 4% paraformaldehyde, cells were permeabilized with 0.05% Triton-X 100 for 5 min Anti-FLAG (M2 mouse IgG; Sigma; St. Louis, MO) was added at a 1:2,000 dilution and incubated overnight at 4°C. Following blocking with 10% normal donkey serum, the primary anti-FLAG antibody was detected with donkey anti-mouse IgG Cy3. Double immunolabeling with FLAG and O1 was preformed as previously described (Nielsen et al., 2004). After immunostaining with anti-FLAG, cultures were incubated with O1 antibody (1:10 dilution of mouse IgM hybridoma supernatant), which was detected with donkey anti-mouse IgM-FITC or Cy3. To identify astrocytes, cultures were immunostained with anti-glial fibrillary acidic protein (GFAP; rabbit polyclonal antibody diluted 1:100; DAKO; Carpenteria, CA), which was detected with donkey anti-rabbit Cy3. FGF receptor substrate 2 alpha (FRS2) phosphorylated on tyrosine 436 was detected by immunustaining with a rabbit polyclonal antibody (1:100; Cell Signaling Technology, Danvers, MA) followed by donkey anti-rabbit Cy3. All secondary antibodies were purchased from Jackson Immunoresearch (West Grove, PA).
PLP mRNA In Situ Hybridization
In situ hybridization for PLP mRNA was performed as previously described (Nielsen et al., 2004). Briefly, cells were fixed with 4% paraformaldehyde, acetylated, and prehybridized with RNA hybridization buffer (DAKO; Carpenteria, CA). A 980 bp cDNA corresponding to the entire coding region of the mouse PLP gene (Hudson et al., 1987), served as a template to incorporate digoxigenin-11-UTP (Roche Applied Science, Indianapolis, IN) using in vitro transcription (Ambion; Austin, Texas). The riboprobe hybridization was detected using an anti-digoxigenin antibody conjugated with alkaline phosphatase (Roche Applied Science, Indianapolis, IN) followed by a 2 h NBT/BCIP colorimetric substrate reaction (DAKO; Carpenteria, CA), which was previously established to be within the linear range for accumulation of NBT/BCIP signal in this reaction (Nielsen et al., 2004). The colorimetric reaction was stopped by washing in water, and the cells were then processed for immunolabeling with anti-GFP (1:100 dilution of mouse IgG; Clontech). Semi-quantitative analysis of relative PLP mRNA expression was performed with Metamorph software. The average signal intensity per pixel was measured within the area of the cell body for infected cells and multiple adjacent noninfected cells in cultures infected with control PMXs and PMXs-FGFRdn retrovirus.
Cell Proliferation Assays
OPCs, generated as described above, were seeded in 24-well plates at density of 60,000 cells/well for FGFRdn analysis or 35,000 cells/well for siRNA assays. After growing for 48 h in medium containing mitogens, the cultures were infected with retrovirus (see above) and grown for an additional 48 h in defined medium with mitogens as noted for specific experiments. Bromodeoxyuridine (BrdU; 10 μM; Sigma) was added during the final 20 h before fixation. Cultures were fixed overnight in 1% paraformaldehyde in PBS containing 0.01% Tween-20. For cell permeabilization, cultures were treated for 10 min with 0.1% Triton X-100 in PBS followed by incubation in DNAse I (50 U/mL) at 37°C for 30 min. Cultures were then incubated for 1 h with anti-BrdU antibody (Roche Applied Science, Indianapolis, IN) followed by detection with donkey anti-mouse IgG Cy3 (Jackson Immunoresearch, West Grove, PA). The BrdU data was collected from three independent experiments with at least three wells/condition and greater than 10 fields/well included in the quantitation.
Clonal analysis was also used to estimate OPC proliferation, as in Nielsen et al. (2004). Infection at a relatively low retroviral titer was used such that there were approximately 10 clones per well of a 24-well plate. A clone was defined as a group of GFP labeled cells surrounded by a border of at least 600 μm in width that had no labeled cells. The number of cells per clone was used to estimate proliferation during the infection period.
Retrovirus Injection and Immunohistochemistry
As described in Murtie et al., 2005a, 7-day-old male C57Bl/6 mice were placed on cooling blocks and anesthetized with isoflurane using the A.D.S. 1000 anesthesia delivery system (Engler Engineering Corporation; Hialeah, FL) with a mouse anesthesia mask (Stoelting Physiology Research Instruments; Wood Dale, IL). Partial laminectomy was performed to expose the L5 spinal cord through the intervertebral space. Retrovirus (100 CFU in 1 μL) was injected into the dorsal columns through a pulled glass pipette (outer diameter ~50 μm) mounted on a Hamilton syringe at a rate of 0.2 μL per min (Levison et al., 1999). After surgery, the wounds were closed with a single wound clip and pups were returned to their mother. At 28 days of age, mice injected with retrovirus at P7 were transcardially perfused with 3% paraformaldehyde in 0.1 M phosphate buffer followed by fixation overnight at 4°C in 3% paraformaldehyde solution. Tissues were cryoprotected in 30% sucrose overnight at 4°C, embedded in OCT compound, and stored at −80°C. Cryoprotected tissues were sectioned and immunostained with CC1 antibody (Oncogene Research Products, Cambridge, MA). CC1 identifies cell bodies of mature oligodendrocyte without labeling myelin (Fuss et al., 2000; Murtie et al., 2005a,b). CC1 antibody was detected with donkey anti-mouse lgG F(ab)2 fragment conjugated with Cy3 (Jackson Immunoresearch).
RNA Interference Assays
For RNA interference from retroviral vectors, short hairpin RNA constructs were designed to target FGFR1, FGFR2, and FGFR3. Each shRNA contains a fragment of a 19 nt sense sequence and a fragment of a 19 nt antisense sequence joined by a 9 nt hairpin loop. Separate shRNAs were designed for specific knockdown of each FGFR isoform. A scrambled 19 nt siRNA sequence was used to generate a control fragment. The resulting siRNA target sequences have at least 3 nt mismatches with other genes, including other FGFR types, but match exactly between mouse and rat FGFR genes. FGFR1 was targeted with a sequence of 5′-GUGGCC-GUGAAGAUGUUGA-3′; FGFR2 was targeted with a sequence of 5′-GGAGCACCGUACUGGACCA-3′; and FGFR3 with a sequence of 5′-GCUAUUGGCAUCGA-CAAGG-3′. For constructing each siRNA expression vector, complementary 67 bp oligonucleotides (including both the sense and antisense sequences of the corresponding 19 bp siRNA, the hairpin-forming sequence, and the adapter sequences for cloning) were annealed to form a double-stranded DNA fragment. Each fragment was ligated into pSIREN-RetroQ-ZsGreen vector (BD Biosciences), a replication-incompetent retroviral plasmid containing a ZsGreen fluorescent reporter sequence (Matz et al., 1999). Retroviral stocks were generated in GP2-293 packaging cells, as described above.
The efficacy of siRNA knockdown of gene expression was verified using quantitative RT-PCR of FGFR1, FGFR2, and FGFR3 mRNA abundance after transfection of each retroviral plasmid into mouse C3H10T1/2 cells (Zhou et al., 2000). RNA was isolated, using RNA-queous-4PCR kit (Ambion, Austin, TX), 96 h after transfection. Two micrograms of total RNA was reverse transcribed using random hexamer primers. Murine FGFR1, FGFR2, and FGFR3 primer and probe sets were obtained from PE Applied Biosystems (Foster City, CA) for kinetic quantitative PCR using the ABI 7500 Real-Time PCR System (PE Applied Biosystems). The input RNA values and RT reaction efficiency were normalized by measuring endogenous18S rRNA for each sample in parallel. The results were quantified as transfected cells relative to mock transfected control cells.
Image Acquisition and Statistical Analysis
Images for Figs. 1–3, 5, and 6 were acquired with an Olympus IX70 epifluorescence microscope equipped with a Spot2 digital camera. Signal intensities and area measurements were calculated using Metamorph software (Universal Imaging Corporation; West Chester, PA). Higher resolution images (see Fig. 4) were acquired on a Zeiss Pascal confocal microscope. Figure panels were prepared in Adobe Photoshop (Adobe; San Jose, California). All quantification was based on data combined from at least 3 independent preparations of cells from separate litters of animals. The Chi-Square test with the Mantel-Haenzel subgroup analysis was used to compare each percentage of cells with a specific phenotype (i.e. immunostained for O1, GFAP, CC1, or BrdU) among the population of retrovirally labeled cells. The Student’s t test was used to compare clone size and absolute values of PLP mRNA intensities between conditions.
Fig. 1.

Retroviral expression of dominant negative FGFR blocks FGF2 inhibition of OPC differentiation in vitro. A: Replication-incompetent retroviral constructs used to monitor OPC differentiation into mature oligodendrocytes, with or without expression of a dominant negative form of FGFR (FGFRdn). Truncated murine FGFR2 (nucleotides 607–2016) was used to generate FGFRdn, which lacks the cytoplasmic domain required for signaling after ligand binding and homo- or hetero- dimerization with endogenous FGFRs. Each retroviral vector contains 5′ and 3′ long terminal repeat (LTR) regions. The LEGFP control and FGFRdn-FLAG retroviral vectors are generated from a matched plasmid backbone, with either a FLAG epitope tag or enhanced GFP as the reporter. The FGFRdn-FLAG retrovirus generates a fusion protein of FGFRdn and FLAG to facilitate detection of FGFRdn expression and localization. PMXs-FGFRdn was created by inserting the FGFRdn fragment into the PMXs retroviral vector, which contains the cytomegalovirus promoter (CMV) to drive constitutive expression of inserted sequences followed by IRES and then GFP. With the PMXs-FGFRdn retrovirus, the FGFRdn fragment and GFP are generated as a single RNA transcript that is translated as two separate proteins to avoid interference from the GFP reporter. B–D: OPCs infected with each retrovirus expressed the appropriate reporter and differentiation was assessed by immunostaining with O1, an oligodendrocyte marker. Scale bars = 20 μm. Bar for C and D shown in D. B: Immunolabeling of the FLAG epitope tag (red) and O1 antigen (green). Nuclei are labeled with DAPI (blue). Functional block of FGF2 inhibition of OPC differentiation is indicated by acquisition of O1 and elaboration of branched processes. C, D: Immunolabeling of O1 antigen (red) with GFP (green) detection of PMXs (C) or PMXs-FGFRdn (D). In the presence of FGF2, PMXs infected cells remain immature as small, bipolar cells (C) that are not labeled with O1 while cells infected with PMXs-FGFRdn elaborate extensive branched processes and membrane sheets immunolabeled with O1 (D). E: Quantitation of O1 immunolabeling among GFP expressing cells in cultures infected with PMXs or with PMXs-FGFRdn retrovirus. Among cells infected with the PMXs control retrovirus, FGF2 treatment (+) prevents OPC differentiation and acquisition of O1 immunolabeling, as compared with defined medium without FGF2 (−). In contrast, cells infected with PMXs-FGFRdn retrovirus differentiate irregardless of FGF2 treatment, indicating FGFRdn expression blocks endogenous FGFR responsiveness to FGF2. Values shown are the mean of three independent experiments, with at least 300 cells counted for each retrovirus condition, and error bars denote standard error of the proportion. The asterisk (*) indicates a significance difference of P < 0.001 for PMXs versus PMXs-FGFRdn in the presence of FGF2.
Fig. 3.

FGFRdn expression blocks OPC proliferation in response to FGF2, but not PDGF. OPC cultures were infected with retrovirus and grown in defined medium containing FGF2 (A, B) or PDGF with BrdU added during the final 20 h. A, B: Cells infected with either PMXs (A, green) or PMXs-FGFRdn (B, green) exhibited BrdU incorporation (red in A, B) and active mitosis (A, left side). Scale bar = 20 μm. C: Quantitation of the percentage of BrdU labeled cells among retrovirally labeled cells expressing GFP. In medium containing FGF2, BrdU incorporation was significantly decreased among cells infected with PMXs-FGFRdn as compared with the control PMXs infections (*P < 0.05, n = 4 independent experiments with at least 400 cells counted for each retrovirus condition). In contrast, BrdU incorporation is not altered by infection with PMXs versus PMXs-FGFRdn among OPCs with PDGF as the mitogen (P ≫ 0.05, n = 4 independent experiments with at least 400 cells counted for each retrovirus condition).
Fig. 5.

Retroviral siRNA targeting FGFR1 reduces FGF2 inhibition of OPC differentiation in vitro. A–C: OPC cultures were infected with retroviruses expressing siRNA targeted to FGFR1 (A), FGFR2 (B) and FGFR3 (C), which was detected with ZsGreen reporter (green). OPCs that differentiated into mature oligodendrocytes exhibited O1 immunostaining of elaborately branched processes and membrane sheets (red in A, C). Scale bars = 20 μm. D: Quantitation of O1 immunoreactivity among infected cells expressing ZsGreen. FGF2 treatment inhibits OPC differentiation in the cultures infected with a control scrambled siRNA sequence. OPC cultures infected with siRNA to knock down FGFR1 expression exhibited a significant increase in the percentage of cells that differentiated and acquired O1 immunoreactivity in presence of FGF2 (*P < 0.001; n = 3 independent experiments with at least 300 cells counted for each retrovirus condition).
Fig. 6.

Retroviral siRNA targeting FGFR1 reduces immunoreactivity for phosphorylated FGF receptor substrate 2 alpha (FRS2). OPC cultures were infected with retroviruses expressing siRNA targeted to FGFR1 with ZsGreen reporter and then immunostained for phosphorylated FRS2, indicative of the activated form of FRS2. In the presence of FGF2, cells expressing FGFR1 siRNA (A, and green in C) had branched processes indicative of differentiation. Adjacent progenitor cells showed cytoplasmic immunoreactivity for phosphorylated FRS2 (B, and red in C) while this immunoreactivity was attenuated in the cells expressing FGFR1 siRNA. Scale bar shown in merged image (C) = 20 μm.
Fig. 4.

Retroviral expression of FGFRdn increases the frequency of OPC differentiation into mature oligodendrocytes during myelination in vivo. PMXs control or PMXs-FGFRdn retrovirus was injected into the lumbar dorsal column to monitor cumulative differentiation spanning the peak period of myelination (P7-P28). A, B: Confocal microscopy of tissue sections was used to image GFP retroviral expression (green) along with immunostaining for CC1 (red), to identify mature oligodendrocytes. A: Cells infected with PMXs more frequently maintained an immature progenitor phenotype and were not immunolabeled by CC1. B: Cells infected with PMXs-FGFRdn typically exhibited CC1 immunolabeling and elaborated processes that bifurcated (example indicated by arrow) along the plane of axons, indicative of a mature oligodendrocyte phenotype, and often extended along the axons (examples at arrowheads). Scale bar = 20 μm. C: Quantitation of CC1 immunolabeling among infected cells expressing GFP indicates a significant increase in OPC differentiation into mature oligodendrocytes with PMXs-FGFRdn retrovirus as compared with the PMXs control retrovirus (*P < 0.001; n = 4 mice injected for each retrovirus and over 300 cells counted for each retrovirus condition).
RESULTS
Retroviral Expression of FGFRdn Blocks FGF2 Inhibition of OPC Differentiation Along the Oligodendrocyte Pathway
We previously used retroviral lineage tracing to monitor in vivo differentiation of endogenous cycling cells into mature oligodendrocytes and demonstrated an increased frequency of OPC differentiation into oligodendrocytes in FGF2 null versus wild type mice (Murtie et al., 2005a). To examine the mechanism underlying this effect of FGF2 expression on OPC differentiation, we first needed to identify whether direct FGFR activation in OPCs could produce this effect, or whether indirect signaling through neighboring cells may need to be considered. Retroviral constructs (see Fig. 1) were designed to express a reporter with, or without, the FGFRdn fragment. This truncated form of FGFR2 lacks the cytoplasmic domain that is required to initiate intracellular signaling. Characterization of similar FGFR constructs has shown that this design takes advantage of the ligand-dependent homo- and heterodimerization among the different high-affinity FGFRs (FGFR1, 2, and 3) to block signaling through multiple endogenous FGFR types (Li et al., 2001; Ueno et al., 1992). With this approach, the differentiation of a limited number of retrovirally labeled cells can be analyzed in the context of an otherwise normal CNS environment. Since the retroviruses are replication-incompetent, only retrovirally labeled cells expressing FGFRdn will have impaired signaling of endogenous FGFRs.
Retrovirus expression was detected using either of two complementary reporter proteins for analysis of FGFRdn function. Initial studies tested retroviral expression of FGFRdn directly fused with a ‘‘FLAG’’ epitope tag as a direct reporter of FGFRdn expression and localization (Figs. 1A,B). Expression of FGFRdn-FLAG was detected by FLAG immunostaining and compared with infections from LEGFP, a control retrovirus expressing GFP from a similar vector construction. The same FGFRdn construct was also inserted into the PMXs retroviral vector to generate PMXs-FGFRdn (Figs. 1A,C,D) to facilitate subsequent in vivo studies. PMXs-FGFRdn expresses FGFRdn followed by IRES and GFP sequences. The IRES results in a single transcript for FGFRdn and GFP that is translated as two separate proteins. Therefore, cells that express GFP will also express the FGFRdn, without potential interference from a reporter as a direct protein fusion.
Infection of cultured OPCs was used to demonstrate retroviral expression and assay function of the FGFRdn construct in response to FGF2 treatment (see Fig. 1). OPC cultures were infected with retrovirus in the presence of mitogens to maximize proliferation, since retroviral integration into chromatin occurs after mitosis. Expression of the retroviral reporter was typically detectable within 24 h of infection (data not shown). At 48 h postinfection, the cultures were transferred to defined medium containing T3 and high insulin to promote differentiation along the oligodendrocytic pathway. Cultures were grown with or without FGF2 in this differentiation medium. At 96 h postinfection, differentiation was assessed using O1 immunostaining to identify mature oligodendrocytes.
Quantitation of O1 immunostaining among neonatal OPCs expressing FGFRdn demonstrated functional attentuation of the response to FGF2 (Fig. 1E). In control PMXs infected cultures, FGF2 treatment inhibited OPC differentiation and acquisition of O1 immunostaining. Importantly, in cultures infected with PMXs-FGFRdn, FGF2 did not effect OPC differentiation as measured by O1 immunostaining. A similar block of FGF2 inhibition of OPC differentiation was obtained in separate experiments with expression of FGFRdn-FLAG. FGF2 reduced O1 immunostaining by 78% among LEGFP infected cells but this response was not observed among cells infected with FGFRdn-FLAG (37.7% ± 1.9% without FGF2; 37.4% ± 1.0% with FGF2).
We also generated a PMXs-FGFRdn retrovirus expressing truncated FGFR1 lacking the cytoplasmic domain, which also blocked FGF2 inhibition of OPC differentiation in similarly prepared cultures (data not shown). Therefore, FGFRdn was effective when the construct was generated from FGFR1 or FGFR2, as expected for heterodimerization of the truncated FGFR isoforms with endogenously expressed FGFR1–3 (Li et al., 2001; Ueno et al., 1992). Our siRNA analysis (see below) also supports the interpretation that the FGFRdn construct generated from FGFR2 acts through heterodimerization with FGFR1, and possibly FGFR3 as well.
To confirm that these results using O1 immunostaining reflected differences in OPC terminal differentiation, cultures were analyzed for expression of PLP, a myelin-specific gene. PLP mRNA levels increase dramatically as OPCs differentiate into mature oligodendrocytes (Macklin et al., 1991; Nielsen et al., 2004; Shiota et al., 1991). Neonatal OPCs were infected with retrovirus, as in the above experiments, but the culture period in mitogens was reduced to 24 h, while the period for growth in differentiation medium with FGF2 was extended to 72 h to allow accumulation of PLP mRNA transcripts. The average intensity of PLP mRNA in situ hybridization signal (pixel intensity/area of cell body) of cells infected with PMXs-FGFRdn was increased 2.27-fold relative to cells infected with the control PMXs retrovirus (2.33 ± 0.65 for PMXs, 5.28 ± 0.10 for PMXs-FGFRdn, P < 0.05. Values are means and standard errors of three independent sets of cultures with at least 30 cells analyzed for each retrovirus). Therefore, both the analysis of O1 immunostaining and PLP in situ hybridization indicates that FGFRdn abrogates FGF2 inhibition of OPCs differentiation along the oligodendrocyte pathway.
FGFRdn Expression Does Not Alter OPC Differentiation Along the Astrocytic Pathway
OPCs can differentiate into either oligodendrocytes, as in the above culture conditions, or Type 2 astrocytes in medium containing high levels of FBS (Raff et al., 1983). Therefore, we examined retroviral infections of OPC under conditions that promote astrocytic differentiation (see Fig. 2). Cultures of neonatal OPCs were infected with PMXs-FGFRdn or control PMXs retrovirus, maintained in mitogens to allow retroviral integration, and then grown for 48 h in medium with 10% FBS to induce astrocytic differentiation. The cultures were immunostained for GFAP, an intermediate filament protein that distinguishes astrocytes (Raff et al., 1983) (Figs. 2A,B). Among the GFP cells expressing the GFP reporter, a similar proportion of astrocytes immunostained for GFAP was observed with infection from either PMXs-FGFRdn or the control PMXs retrovirus (Fig. 2C). Normal OPC differentiation into Type 2 astrocytes with FGFRdn expression is consistent with reports that the astrocytic pathway is induced by several factors present in FBS (Lillien and Raff, 1990; Raff and Lillien, 1988), and indicates specificity of FGFRdn in FGF2 inhibition of OPC differentiation into oligodendrocytes.
Fig. 2.

FGFRdn expression does not alter OPC differentiation into type 2 astrocytes. OPC cultures were infected with retrovirus and then grown in Dulbecco modified eagle medium (DMEM) with 10% FBS to promote OPC differentiation along the astrocytic pathway. A, B: Cells infected with either PMXs (A, green) or PMXs-FGFRdn (B, green) exhibited immunoreactivity for GFAP (red in A, B), characteristic of an astrocytic phenotype. The typical processed morphology of type 2 astrocytes (B, left side) contrasts with the flattened, fibroblastic morphology of type 1 astrocytes (B, right side). Scale bar = 20 μm. C: Quantitation of GFAP immunoreactivity shows a similar frequency of astrocytic differentiation among GFP expressing cells infected with PMXs compared with PMXs-FGFRdn (P ≫ 0.05; values shown are the mean of three independent experiments with at least 300 cells counted for each retrovirus condition).
FGFRdn Inhibits FGF2 Induced OPC Proliferation
FGF2 is a well characterized mitogen for cultured neonatal and adult OPCs (Frost et al., 2003; McKinnon et al., 1990; Wolswijk and Noble, 1992). The OPC proliferative response was examined to demonstrate that FGFRdn could block this second FGF2 stimulated response (see Fig. 3). Actively proliferating OPCs were infected with the PMXs-FGFRdn or control PMXs retrovirus and at 48 h postinfection the cells were transferred into differentiation medium with FGF2 for an additional 48 h. With a low frequency of infection in a culture, proliferation of initially infected cells generates distinct clones of adjacent cohort cells labeled with GFP. The number of cells generated per clone was markedly reduced by expression of FGFRdn as compared with the control PMXs retrovirus (6.8 ± 0.3 cells/clone for PMXs; 3.2 ± 0.2 cells/clone for PMXs-FGFRdn; P < 0.05; mean of three independent sets of cultures with at least 300 cells counted for each retrovirus). To more specifically assess proliferation, BrdU was added to the cultures as a 20-hour terminal pulse and detected by immunostaining for BrdU with GFP imaging of infected cells (Figs. 3A,B). BrdU incorporation was decreased 57% among cells infected with the PMXs-FGFRdn retrovirus as compared with cells infected with the control PMXs retrovirus (Fig. 3C).
FGFRdn Does Not Block PDGF-Induced OPC Proliferation
PDGF is also a potent mitogen for cultured neonatal OPCs (Noble et al., 1988; Richardson et al., 1988). The PDGF receptor alpha, which is expressed by OPCs, is a tyrosine kinase receptor that is closely related to the FGFR. Therefore, OPC proliferation in response to PDGF was examined to demonstrate ligand and receptor signaling specificity of FGFRdn expression (Fig. 3C). OPC cultures were infected and grown in defined medium, as described above for Fig. 3, with PDGF used in place of FGF2 during the final 48 h of the culture period. BrdU incorporation was similar among cells infected with either PMXs-FGFRdn or the PMXs control retrovirus. These BrdU labeling results provide additional evidence of specificity of FGFRdn attenuation of FGF2 stimulated FGFR responses in OPCs.
FGFRdn Attenuates Inhibition of OPC Differentiation in Developing White Matter
In our previous studies of retroviral lineage analysis in FGF2 null mice, we demonstrated that oligodendrocyte lineage cell differentiation could be monitored in vivo during oligodendrogenesis (Murtie et al., 2005a). We now performed similar studies to test the in vivo effect of expression of PMXs-FGFRdn (see Fig. 4). We predicted that a direct effect of FGFR signaling on OPC differentiation would be revealed by an increased frequency of differentiation into mature oligodendrocytes among infected cells expressing PMXs-FGFRdn, as compared with the control PMXs retrovirus.
PMXs-FGFRdn or control PMXs retrovirus was injected into the dorsal column white matter of postnatal day 7 (P7) C57Bl/6 mice to label endogenous cycling cells prior to the peak period of oligodendrocyte generation, which occurs around P15. Mice were perfused at P28 and spinal cord sections were examined using GFP detection combined with immunostaining for CC1 to identify mature oligodendrocytes (Fuss et al., 2000; Murtie et al., 2005a,b). The specificity of CC1 immunostaining for mature oligodendrocytes was supported by the morphology of the GFP labeled cells (see Fig. 4). Mature oligodendrocytes have multiple processes that bifurcate forming T-shaped intersections with extensions that run parallel to axons, while OPC have numerous fine processes do not form such intersections or extensions along axons (Levison and Goldman, 1993). Quantitation of the proportion of GFP labeled cells that were identified as oligodendrocytes by CC1 immunostaining revealed that mature oligodendrocytes were generated at a significantly higher frequency among cells infected with PMXs-FGFRdn as compared with those infected with the control PMXs retrovirus (Fig. 4C). This effect of FGFRdn expression is very similar to the effect of the FGF2 null genotype in our previous retroviral lineage analysis (Murtie et al., 2005a) and supports a model of FGF2 inhibiting OPC differentiation in vivo by acting directly through OPC FGFRs.
siRNA Knockdown of FGFR1 Blocks FGF2 Inhibition of OPC Differentiation
To determine which FGFR type(s) can mediate FGF2 inhibition of OPC differentiation, we integrated in vitro retroviral monitoring of differentiation with stable siRNA knockdown of individual FGFR genes. Retroviruses were constructed to express the ZsGreen reporter and a shRNA sequence that would produce small interfering RNA (siRNA) sequences targeted to either FGFR1, FGFR2, or FGFR3. The efficacy of siRNA knockdown of expression of each targeted FGFR type was confirmed by quantitative RT-PCR analysis of transient transfections in C3H10T1/2 cells, a cell line that endogenously expresses FGFR1–3 (Zhou et al., 2000). Three days after transfection of shRNA plasmids, RNA abundance of the corresponding FGFR type was reduced approximately 90% relative to mock transfected C3H10T1/2 cells (89% ± 2.6% for FGFR1; 93% ± 5.9% for FGFR2; 94% ± 4.5% for FGFR3; each value is the mean ± SE derived from three independent experiments). The siRNA targeted to FGFR2 and FGFR3 reduced only the targeted FGFR type, and not the other FGFR types. The siRNA targeted to FGFR1 partially reduced expression of FGFR3 54% ± 7.5%. The FGFR1 target region is not present in the FGFR3 gene, indicating that FGFR1 signaling may modulate expression of FGFR3.
OPC cultures were infected with each retrovirus to determine the effect of siRNA knockdown of each FGFR type on differentiation (see Fig. 5). OPCs were infected, grown in mitogens for 48 h to enable retrovirus integration and expression, transferred to differentiation medium for 48 h, and then immunostained with O1 to identify oligodendrocytes among the infected cells expressing the ZsGreen reporter. In the presence of FGF2, OPC differentiation was inhibited in cells infected with a control retrovirus expressing a scrambled siRNA sequence. A significantly greater proportion of infected cells differentiated into O1 immunolabeled oligodendrocytes using retrovirus expressing siRNA targeted to knockdown FGFR1 (Fig. 5D). OPC differentiation was not altered by retroviral expression of siRNA sequences targeted to knock down FGFR2 or FGFR3 (Fig. 5D).
This effect of FGFR1 shRNA expression is consistent with our results from expression of FGFRdn (see Fig. 1). To further correlate expression of FGFR1 shRNA with FGFR signaling, cultures were treated with FGF2 and immunostained for phophorylated FGF receptor substrate 2 alpha (FRS2) as an indicator of activated FGFR signaling (Ridyard and Robbins, 2003; Hoch and Soriano, 2006). Cytoplasmic immunoreactivity for FRS2 was observed in noninfected cells in the presence of FGF2 but was typically very weak in cells expressing siRNA targeted to FGFR1 (see Fig. 6). A similar reduction of immunoreactivity for FRS2 was observed in cells expressing FGFRdn (data not shown), confirming appropriate FRS2 association with FGFR signaling in this immunostaining assay. These RNA interference studies indicate that FGFR1 is the predominant isoform by which FGF2 regulates OPC differentiation. FGFR1 modulation of FGFR3 expression may contribute to this effect.
siRNA Knockdown of FGFR1 Decreases OPC Proliferation Stimulated by FGF2, But Not PDGF
As noted above, FGF2 and PDGF are both effective mitogens and closely related tyrosine kinase receptors. We performed proliferation analysis of OPCs infected with siRNA retroviruses to determine which FGFR type(s) mediate this response to FGF2 and to demonstrate the specificity of the siRNA effect relative to PDGF signaling (see Fig. 7). OPCs were infected, grown in FGF2 and PDGF for 48 hours to enable retrovirus integration and expression, and then transferred to differentiation medium containing only FGF2 or PDGF for 48 h with BrdU added during the terminal 20 h. In medium containing FGF2 alone, immunodetection of BrdU incorporation demonstrated that proliferation was significantly reduced with retroviral expression of siRNA targeted to knockdown FGFR1, as compared with expression of a control retrovirus expressing a scrambled siRNA sequence (Fig. 7B). Proliferation stimulated by FGF2 was not altered by infection with retrovirus expressing siRNA targeted to FGFR2 or FGFR3 (Fig. 7B). In medium containing PDGF as the mitogen, OPC proliferation was not altered by infection with retrovirus expressing siRNA targeted to any of the FGFR types. The proportion of BrdU labeled cells among those expressing the ZsGreen retroviral reporter was 37.7% ± 0.6% for the control, 35.1% ± 0.4% for FGFR1, 36.6% ± 0.5% for FGFR2, and 36.5% ± 1.5% for FGFR3. Values are the mean, and SE, from four independent experiments of PDGF treatment with at least 400 cells expressing ZsGreen counted for each retrovirus condition. This proliferation analysis demonstrates the specificity of the siRNA inactivation and indicates a significant role for FGFR1 in the OPC proliferation response to FGF2.
Fig. 7.
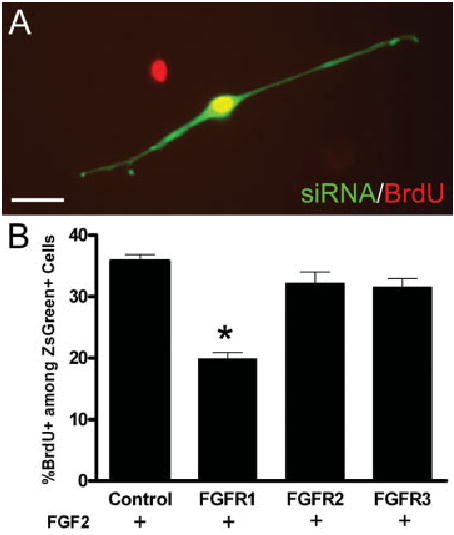
Retroviral siRNA targeting FGFR1 reduces FGF2 stimulated OPC proliferation in vitro. OPC cultures were infected with retroviruses expressing siRNA targeted to FGFR1, FGFR2, or FGFR3. Retrovirus expression was detected with ZsGreen reporter and proliferation was estimated from BrdU incorporation during a 20 h terminal pulse. A: Example of a bipolar OPC expressing ZsGreen reporter (green; FGFR2 siRNA) with nuclear BrdU immunoreactivity (red; double labeling appears yellow). Scale bar = 20 μm. B: Quantitation of BrdU labeling among infected cells expressing siRNA and ZsGreen. In medium containing FGF2, as compared with control infections, BrdU incorporation was significantly decreased using siRNA that targeted FGFR1 expression (*P < 0.05) but was not altered using siRNA that targeted FGFR2 (P ≫ 0.05) or FGFR3 (P ≫ 0.05) (n = 4 independent experiments with at least 400 ZsGreen labeled cells counted for each retrovirus condition).
DISCUSSION
Environmental signals strongly impact the differentiation program of immature cells. In the postnatal CNS, myelination progresses with a spatiotemporal pattern that corresponds to the functional maturation of axons in a given fiber tract (Miller et al., 1985; Small et al., 1987). The environmental interactions that regulate the process of OPC differentiation into myelinating oligodendrocytes are important to elucidate to improve our understanding of CNS development. Signals regulating OPC differentiation are also important to understand in the context of demyelinated lesions since OPCs and premyelinating oligodendrocytes may be present in MS lesions yet often fail to remyelinate effectively (Chang et al., 2000, 2002).
Our previous work using FGF2 null mice indicated that endogenous FGF2 inhibits differentiation of OPCs into myelinating oligodendrocytes in both developmental and remyelination contexts (Armstrong et al., 2002; Murtie et al., 2005a,b). Following chronic demyelination, FGF2 limits the effective remyelination of lesion areas (Armstrong et al., 2006). The present study demonstrates that FGF2 inhibition of OPC differentiation in vivo occurs through direct activation of FGFR signaling. Further, among the three high-affinity FGFR isoforms expressed in the oligodendrocyte lineage, FGFR1 expression has a predominant role in regulating FGF2 inhibition of OPC differentiation. FGFR1 may also maintain FGFR3 expression, which may contribute to this OPC response to FGF2.
The in vivo effect of FGF2 signaling on OPC differentiation was determined using retroviral lineage tracing to directly monitor the progression from endogenous cycling cell to mature oligodendrocyte. Retroviral expression of an FGFRdn fragment resulted in a higher frequency of progenitor differentiation into mature oligodendrocytes (see Fig. 4). Similarly, using a control retrovirus expressing only the GFP reporter, we found that a greater proportion of retrovirally labeled cells differentiated into mature oligodendrocytes in FGF2 null mice than in wild type mice (Murtie et al., 2005a). Interestingly, in FGF2 null mice, fewer oligodendrocytes overall were generated during the peak of oligodendrogenesis and the retrovirally labeled cells produced smaller clones. Therefore, FGF2 inhibition of oligodendrocyte progenitor differentiation may prevent precocious progenitor differentiation to a postmitotic state that would inappropriately limit the generation of oligodendrocytes (Murtie et al., 2005a).
In our previous retroviral lineage analysis during myelination (Murtie et al., 2005a) and the current study, cells in dorsal column white matter that were infected during ongoing myelination (P7) gave rise to a cohort of GFP-labeled OPCs and oligodendrocytes, with only rare retroviral labeling of astrocytes (Murtie et al., 2005a). Therefore, retroviral expression of FGFRdn, a dominant negative form of FGFR, was targeted for expression in developing oligodendrocyte lineage cells. Importantly, this FGFRdn expression in oligodendrocyte lineage cells resulted in a greater proportion of retrovirally labeled cells differentiating into mature oligodendrocytes than with infection by the control retrovirus that expressed only the GFP reporter. Although many cell types in the CNS can express diverse FGFR types, our findings indicate a direct effect of endogenous FGF2 on differentiation of oligodendrocyte lineage cells. This interpretation is consistent with the finding of increased myelination in transgenic mice that express a dominant negative form of FGFR driven from the promoter of the myelin basic protein gene (Harari et al., 1997).
The current study tested the potential contribution of each of the three FGFR types (FGFR1, 2, and 3) that are expressed in oligodendrocyte lineage cells and could mediate FGF2 inhibition of OPC differentiation. FGFR1–3 expression is differentially regulated with progressive maturation in the oligodendrocyte lineage during development and in the adult rodent CNS (Bansal et al., 1996,2003; Messersmith et al., 2000). Antibody blocking studies reported that inactivation of both FGFR1 and FGFR3 is necessary to significantly block FGF2 inhibition of oligodendrocyte development in vitro (Fortin et al., 2005). Our retroviral expression of siRNA demonstrated that inactivation of FGFR1, but not FGFR2 or FGFR3, was sufficient to specifically block FGF2 inhibition of OPC differentiation. Interestingly, although the siRNA target for FGFR1 is not shared with FGFR3, a partial reduction of FGFR3 mRNA levels occurred with expression of the FGFR1 siRNA. Taken together, these findings indicate that direct activation of FGFR1 signaling in OPCs inhibits differentiation, and may modulate FGFR3 levels in regulating this response. Further, our results with specific siRNA knockdown of only FGFR3 do not support a previously hypothesized role for FGFR3 in promoting oligodendrocyte differentiation, at least not as direct effect on OPCs (Oh et al., 2003).
FGF2 administration can also stimulate OPC proliferation, and it is not yet clear which FGFRs mediate this effect or the extent to which FGF2 acts as a mitogen in vivo during development and remyelination. In vitro, FGF2 alone or in combination with PDGF can induce proliferation of OPCs isolated from neonatal CNS, normal adult CNS, and experimentally demyelinated CNS (Bogler et al., 1990; Frost et al., 2003; McKinnon et al., 1990; Wolswijk and Noble, 1992). Our current siRNA experiments implicate FGFR1 in regulating FGF2 mediated OPC proliferation. Adenoviral overexpression of FGF2 increased the numbers of OPCs and oligodendrocytes in mice with experimental allergic encephalomyelitis (Ruffini et al., 2001). Injections to elevate FGF2 increased proliferation of neural stem cells in the subventricular zone, which may have contributed to repopulation of corpus callosum lesions (Decker et al., 2002). In contrast, our in vivo studies of development and remyelination used FGF2 null mice to eliminate endogenous FGF2 and did not detect any significant differences in OPC proliferation relative to wild type mice (Armstrong et al., 2002; Murtie et al., 2005a,b). Corresponding studies of development and remyelination indicate that endogenous PDGF signaling regulates OPC proliferation (Calver et al., 1998; Fruttiger et al., 1999; Murtie et al., 2005b). Therefore, while experimentally elevated FGF2 may induce OPC proliferation in vitro and in vivo, endogenous FGF2 may not contribute significantly as an OPC mitogen in vivo.
FGF2 may interact with other signaling pathways in regulating oligodendrocyte lineage differentiation during development and remyelination. Notch1 inhibits oligodendrocyte differentiation and myelination during CNS development (Wang et al., 1998). Conditional inactivation of Notch1 in the oligodendrocyte lineage resulted in ectopic and precocious OPC differentiation (Genoud et al., 2002). Pharmacological inactivation of notch signaling promoted remyelination and clinical recovery in a model of experimental demyelination in mice (Jurynczyk et al., 2005). In MS lesions, nonremyelinating areas exhibited Notch1 expression on immature oligodendrocytes and corresponding expression of the Notch1 ligand, Jagged1, by hypertrophic astrocytes (John et al., 2002). Interestingly, studies of inactivation of Notch1 signaling and those of inactivation of FGF2 signaling indicate that removing signals that inhibit remyelination can be neuroprotective for denuded or regenerating axons (Jungnickle et al., 2004; Jurynczyk et al., 2005). Additional studies will be needed to determine the interactions of these molecular signals in inhibiting OPC differentiation, as well as to further understand the dynamic balance of inhibitory signals relative to those that promote OPC differentiation.
Our findings also demonstrate the advantage of in vivo application of retroviral lineage tracing with expression of specific gene constructs within developing oligodendrocyte lineage cells. Retroviral expression of a GFP reporter has been useful for lineage tracing in embryonic, postnatal, and adult CNS (Kakita et al., 2003; Murtie et al., 2005a,b; Noctor et al., 2001; van Praag et al., 2002). Further, the FGFRdn construct is expressed sufficiently in vivo to produce a significant effect on cell fate that is similar to the effect of the FGF2 null genotype (Murtie et al., 2005a). Since the retrovirus infects only a relatively small population of endogenous cycling cells, this approach enables monitoring of a subset of labeled cells within an otherwise normal CNS environment. This approach should be useful for analysis of gene mutations that might otherwise be lethal at early developmental stages. Moreover, in vivo transfer of siRNA expressed from retroviral vectors into selected populations of CNS progenitor cells should be an attractive method for studying gene function.
FGF2 activation of specific FGFR isoforms in oligodendrocyte lineage cells may have potential implications for demyelinating diseases as well as other forms of CNS injury. Expression of FGF2, FGFR1, and FGFR3 is increased within and near lesions induced in multiple models of experimental demyelination (Liu et al., 1998; Messersmith et al., 2000). Reactive astrocytes of acute and chronic MS plaques can express FGF2 (Holley et al., 2003). Our in vitro rodent findings may also extend to human tissues since FGF2 inhibited the differentiation of preoligodendrocytes into oligodendrocytes in cultures from human temporal lobe white matter (Armstrong et al., 1992). Both FGF2 and FGFR1 are also increased in CNS injuries that are not specifically related to demyelination, such as trauma (Logan et al., 1992), ischemia (Masumura et al., 1996), and neurotoxicity (Belluardo et al., 1997). FGF2 and FGFR1 levels may be important in the relative degree of astroglial scarring that develops in a CNS lesion (Smith et al., 2001) and thus can impact the environmental conditions for axonal survival and regeneration (Silver and Miller, 2004). However, FGF2 is among the most effective neuroprotective growth factors in diverse models of CNS injury, including stroke, trauma, excitotoxicity, and axotomy (Alzheimer and Werner, 2002; Teng et al., 1999). Presumably, other FGF family members can compensate in FGF2 null mice for the loss of FGF2 neuroprotection (reviewed in Reuss and von Bohlen und Halbach, 2003) but cannot fully compensate for the role of FGF2 on differentiation of the oligodendrocyte lineage. This interpretation would be consistent with our similar results from retroviral lineage tracing of oligodendrocyte progenitor differentiation in development in FGF2 null mice or with expression of FGFRdn, and in remyelination in FGF2 null mice. Teasing out the mechanism and context effects of FGF2 actions in the normal and pathological CNS will be necessary for the design of therapeutic molecules to take advantage of this potent signaling pathway.
Acknowledgments
We thank Dr.Toshio Kitamura for the PMXs retroviral vector and Dr. Alka Mansukhani for the plasmid encoding FGFR2. We also thank Dr. Nicole Dobson, Dr. Diane Borst, Dr. Joe Nielsen, Dr. Tong Lin, and Adam Vana for critical reading of the manuscript.
Footnotes
Grant sponsor: National Institutes of Health; Grant number: NS39293; Grant sponsor: National Multiple Sclerosis; Grant number: RG3515; Grant sponsor: Uniformed Services University of the Health Sciences; Grant number: HO70QS.
Joshua C. Murtie is currently at The Children’s Hospital Boston, Enders 270, 300 Longwood Ave, Boston, MA 02115.
This article is a US government work and, as such, is in the public domain of the United States of America.
References
- Alzheimer C, Werner S. Fibroblast growth factors and neuroprotection. Adv Exp Med Biol. 2002;513:335–351. doi: 10.1007/978-1-4615-0123-7_12. [DOI] [PubMed] [Google Scholar]
- Armstrong RC. Isolation and characterization of immature oligodendrocyte lineage cells. Methods. 1998;16:282–292. doi: 10.1006/meth.1998.0685. [DOI] [PubMed] [Google Scholar]
- Armstrong RC, Dorn HH, Kufta CV, Friedman E, Dubois-Dalcq ME. Pre-oligodendrocytes from adult human CNS. J Neurosci. 1992;12:1538–1547. doi: 10.1523/JNEUROSCI.12-04-01538.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong RC, Kim JG, Hudson LD. Expression of myelin transcription factor I (MyTI), a “zinc-finger” DNA-binding protein, in developing oligodendrocytes. Glia. 1995;14:303–321. doi: 10.1002/glia.440140407. [DOI] [PubMed] [Google Scholar]
- Armstrong RC, Le TQ, Frost EE, Borke RC, Vana AC. Absence of fibroblast growth factor 2 promotes oligodendroglial repopulation of demyelinated white matter. J Neurosci. 2002;22:8574–8585. doi: 10.1523/JNEUROSCI.22-19-08574.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong RC, Le TQ, Flint NC, Vana AC, Zhou YX. Endogenous cell repair of chronic demyelination. J Neuropathol Exp Neurol. 2006;65(3):245–256. doi: 10.1097/01.jnen.0000205142.08716.7e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asai T, Wanaka A, Kato H, Masana Y, Seo M, Tohyama M. Differential expression of two members of FGF receptor gene family, FGFR-1 and FGFR-2 mRNA, in the adult rat central nervous system. Brain Res Mol Brain Res. 1993;17:174–178. doi: 10.1016/0169-328x(93)90088-7. [DOI] [PubMed] [Google Scholar]
- Ballabriga J, Pozas E, Planas AM, Ferrer I. bFGF and FGFR-3 immunoreactivity in the rat brain following systemic kainic acid administration at convulsant doses: Localization of bFGF and FGFR-3 in reactive astrocytes, and FGFR-3 in reactive microglia. Brain Res. 1997;752:315–318. doi: 10.1016/s0006-8993(96)01308-x. [DOI] [PubMed] [Google Scholar]
- Bansal R, Kumar M, Murray K, Morrison RS, Pfeiffer SE. Regulation of FGF receptors in the oligodendrocyte lineage. Mol Cell Neurosci. 1996;7:263–275. doi: 10.1006/mcne.1996.0020. [DOI] [PubMed] [Google Scholar]
- Bansal R, Lakhina V, Remedios R, Tole S. Expression of FGF receptors 1, 2, 3 in the embryonic and postnatal mouse brain compared with Pdgfralpha, Olig2 and Plp/dm20: Implications for oligodendrocyte development. Dev Neurosci. 2003;25:83–95. doi: 10.1159/000072258. [DOI] [PubMed] [Google Scholar]
- Belluardo N, Wu G, Mudo G, Hansson AC, Pettersson R, Fuxe K. Comparative localization of fibroblast growth factor receptor-1, -2, and -3 mRNAs in the rat brain: In situ hybridization analysis. J Comp Neurol. 1997;379:226–246. [PubMed] [Google Scholar]
- Bogler O, Wren D, Barnett SC, Land H, Noble M. Cooperation between two growth factors promotes extended self-renewal and inhibits differentiation of oligodendrocyte-type-2 astrocyte (O-2A) progenitor cells. Proc Natl Acad Sci USA. 1990;87:6368–6372. doi: 10.1073/pnas.87.16.6368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calver AR, Hall AC, Yu WP, Walsh FS, Heath JK, Betsholtz C, Richardson WD. Oligodendrocyte population dynamics and the role of PDGF in vivo. Neuron. 1998;20:869–882. doi: 10.1016/s0896-6273(00)80469-9. [DOI] [PubMed] [Google Scholar]
- Chang A, Nishiyama A, Peterson J, Prineas J, Trapp BD. NG2-positive oligodendrocyte progenitor cells in adult human brain and multiple sclerosis lesions. J Neurosci. 2000;20:6404–6412. doi: 10.1523/JNEUROSCI.20-17-06404.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang A, Tourtellotte WW, Rudick R, Trapp BD. Premyelinating oligodendrocytes in chronic lesions of multiple sclerosis. N Engl J Med. 2002;346:165–173. doi: 10.1056/NEJMoa010994. [DOI] [PubMed] [Google Scholar]
- Decker L, Picard-Riera N, Lachapelle F, Baron-Van Evercooren A. Growth factor treatment promotes mobilization of young but not aged adult subventricular zone precursors in response to demyelination. J Neurosci Res. 2002;69:763–771. doi: 10.1002/jnr.10411. [DOI] [PubMed] [Google Scholar]
- Eccleston PA, Silberberg DH. The differentiation of oligodendrocytes in a serum-free hormone-supplemented medium. Brain Res. 1984;318:1–9. doi: 10.1016/0165-3806(84)90056-7. [DOI] [PubMed] [Google Scholar]
- Fortin D, Rom E, Sun H, Yayon A, Bansal R. Distinct fibroblast growth factor (FGF)/FGF receptor signaling pairs initiate diverse cellular responses in the oligodendrocyte lineage. J Neurosci. 2005;25:7470–7479. doi: 10.1523/JNEUROSCI.2120-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost EE, Nielsen JA, Le TQ, Armstrong RC. PDGF and FGF2 regulate oligodendrocyte progenitor responses to demyelination. J Neurobiol. 2003;54:457–472. doi: 10.1002/neu.10158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fruttiger M, Karlsson L, Hall AC, Abramsson A, Calver AR, Bostrom H, Willetts K, Bertold CH, Heath JK, Betsholtz C, Richardson WD. Defective oligodendrocyte development and severe hypomyelination in PDGF-A knockout mice. Development. 1999;126:457–467. doi: 10.1242/dev.126.3.457. [DOI] [PubMed] [Google Scholar]
- Fuss B, Mallon B, Phan T, Ohlemeyer C, Kirchhoff F, Nishiyama A, Macklin WB. Purification and analysis of in vivo-differentiated oligodendrocytes expressing the green fluorescent protein. Dev Biol. 2000;218:259–274. doi: 10.1006/dbio.1999.9574. [DOI] [PubMed] [Google Scholar]
- Genoud S, Lappe-Siefke C, Goebbels S, Radtke F, Aguet M, Scherer SS, Suter U, Nave KA, Mantei N. Notch1 control of oligodendrocyte differentiation in the spinal cord. J Cell Biol. 2002;158:709–718. doi: 10.1083/jcb.200202002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harari D, Finkelstein D, Bernard O. FGF plays a subtle role in oligodendrocyte maintenance in vivo. J Neurosci Res. 1997;49:404–415. [PubMed] [Google Scholar]
- Hoch RV, Soriano P. Context-specific requirements for Fgfr1 signaling through Frs2 and Frs3 during mouse development. Development. 2006;133:663–673. doi: 10.1242/dev.02242. [DOI] [PubMed] [Google Scholar]
- Holley JE, Gveric D, Newcombe J, Cuzner ML, Gutowski NJ. Astrocyte characterization in the multiple sclerosis glial scar. Neuropathol Appl Neurobiol. 2003;29:434–444. doi: 10.1046/j.1365-2990.2003.00491.x. [DOI] [PubMed] [Google Scholar]
- Hudson LD, Berndt JA, Puckett C, Kozak CA, Lazzarini RA. Aberrant splicing of proteolipid protein mRNA in the dysmyelinating jimpy mutant mouse. Proc Natl Acad Sci USA. 1987;84:1454–1458. doi: 10.1073/pnas.84.5.1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John GR, Shankar SL, Shafit-Zagardo B, Massimi A, Lee SC, Raine CS, Brosnan CF. Multiple sclerosis: Re-expression of a developmental pathway that restricts oligodendrocyte maturation. Nat Med. 2002;8:1115–1121. doi: 10.1038/nm781. [DOI] [PubMed] [Google Scholar]
- Jungnickel J, Claus P, Gransalke K, Timmer M, Grothe C. Targeted disruption of the FGF-2 gene affects the response to peripheral nerve injury. Mol Cell Neurosci. 2004;25:444–452. doi: 10.1016/j.mcn.2003.11.007. [DOI] [PubMed] [Google Scholar]
- Jurynczyk M, Jurewicz A, Bielecki B, Raine CS, Selmaj K. Inhibition of Notch signaling enhances tissue repair in an animal model of multiple sclerosis. J Neuroimmunol. 2005;170:3–10. doi: 10.1016/j.jneuroim.2005.10.013. [DOI] [PubMed] [Google Scholar]
- Kakita A, Zerlin M, Takahashi H, Goldman JE. Some glial progenitors in the neonatal subventricular zone migrate through the corpus callosum to the contralateral cerebral hemisphere. J Comp Neurol. 2003;458:381–388. doi: 10.1002/cne.10597. [DOI] [PubMed] [Google Scholar]
- Levison SW, Goldman JE. Both oligodendrocytes and astrocytes develop from progenitors in the subventricular zone of postnatal rat forebrain. Neuron. 1993;10(2):201–212. doi: 10.1016/0896-6273(93)90311-e. [DOI] [PubMed] [Google Scholar]
- Levison SW, Young GM, Goldman JE. Cycling cells in the adult rat neocortex preferentially generate oligodendroglia. J Neurosci Res. 1999;57:435–446. [PubMed] [Google Scholar]
- Li X, Chen Y, Scheele S, Arman E, Haffner-Krausz R, Ekblom P, Lonai P. Fibroblast growth factor signaling and basement membrane assembly are connected during epithelial morphogenesis of the embryoid body. J Cell Biol. 2001;153:811–822. doi: 10.1083/jcb.153.4.811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lillien LE, Raff MC. Differentiation signals in the CNS: Type-2 astrocyte development in vitro as a model system. Neuron. 1990;5:111–119. doi: 10.1016/0896-6273(90)90301-u. [DOI] [PubMed] [Google Scholar]
- Liu X, Mashour GA, Webster HF, Kurtz A. Basic FGF, FGF receptor 1 are expressed in microglia during experimental autoimmune encephalomyelitis: Temporally distinct expression of midkine and pleiotrophin. Glia. 1998;24:390–397. doi: 10.1002/(sici)1098-1136(199812)24:4<390::aid-glia4>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- Logan A, Frautschy SA, Gonzalez AM, Baird A. A time course for the focal elevation of synthesis of basic fibroblast growth factor and one of its high-affinity receptors (flg) following a localized cortical brain injury. J Neurosci. 1992;12:3828–3837. doi: 10.1523/JNEUROSCI.12-10-03828.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macklin WB, Gardinier MV, Obeso ZO, King KD, Wight PA. Mutations in the myelin proteolipid protein gene alter oligodendrocyte gene expression in jimpy and jimpymsd mice. J Neurochem. 1991;56:163–171. doi: 10.1111/j.1471-4159.1991.tb02576.x. [DOI] [PubMed] [Google Scholar]
- Mansukhani A, Dell’Era P, Moscatelli D, Kornbluth S, Hanafusa H, Basilico C. Characterization of the murine BEK fibroblast growth factor (FGF) receptor: Activation by three members of the FGF family and requirement for heparin. Proc Natl Acad Sci USA. 1992;89:3305–3309. doi: 10.1073/pnas.89.8.3305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masumura M, Murayama N, Inoue T, Ohno T. Selective induction of fibroblast growth factor receptor-1 mRNA after transient focal ischemia in the cerebral cortex of rats. Neurosci Lett. 1996;213:119–122. doi: 10.1016/0304-3940(96)12841-x. [DOI] [PubMed] [Google Scholar]
- Matz MV, Fradkov AF, Labas YA, Savitsky AP, Zaraisky AG, Markelov ML, Lukyanov SA. Fluorescent proteins from nonbioluminescent Anthozoa species. Nat Biotechnol. 1999;17:969–973. doi: 10.1038/13657. [DOI] [PubMed] [Google Scholar]
- McKinnon RD, Matsui T, Dubois-Dalcq M, Aaronson SA. FGF modulates the PDGF-driven pathway of oligodendrocyte development. Neuron. 1990;5:603–614. doi: 10.1016/0896-6273(90)90215-2. [DOI] [PubMed] [Google Scholar]
- Messersmith DJ, Murtie JC, Le TQ, Frost EE, Armstrong RC. Fibroblast growth factor 2 (FGF2) and FGF receptor expression in an experimental demyelinating disease with extensive remyelination. J Neurosci Res. 2000;62:241–256. doi: 10.1002/1097-4547(20001015)62:2<241::AID-JNR9>3.0.CO;2-D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller RH, David S, Patel R, Abney ER, Raff MC. A quantitative immunohistochemical study of macroglial cell development in the rat optic nerve: In vivo evidence for two distinct astrocyte lineages. Dev Biol. 1985;111:35–41. doi: 10.1016/0012-1606(85)90432-4. [DOI] [PubMed] [Google Scholar]
- Miyake A, Hattori Y, Ohta M, Itoh N. Rat oligodendrocytes and astrocytes preferentially express fibroblast growth factor receptor-2 and -3 mRNAs. J Neurosci Res. 1996;45:534–441. doi: 10.1002/(SICI)1097-4547(19960901)45:5<534::AID-JNR3>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Murtie JC, Zhou YX, Le TQ, Armstrong RC. In vivo analysis of oligodendrocyte lineage development in postnatal FGF2 null mice. Glia. 2005a;49:542–554. doi: 10.1002/glia.20142. [DOI] [PubMed] [Google Scholar]
- Murtie JC, Zhou YX, Le TQ, Vana AC, Armstrong RC. PDGF and FGF2 pathways regulate distinct oligodendrocyte lineage responses in experimental demyelination with spontaneous remyelination. Neurobiol Dis. 2005b;19:171–182. doi: 10.1016/j.nbd.2004.12.006. [DOI] [PubMed] [Google Scholar]
- Nielsen JA, Berndt JA, Hudson LD, Armstrong RC. Myelin transcription factor 1 (Myt1) modulates the proliferation and differentiation of oligodendrocyte lineage cells. Mol Cell Neurosci. 2004;25:111–123. doi: 10.1016/j.mcn.2003.10.001. [DOI] [PubMed] [Google Scholar]
- Noble M, Murray K, Stroobant P, Waterfield MD, Riddle P. Platelet-derived growth factor promotes division and motility and inhibits premature differentiation of the oligodendrocyte/type-2 astrocyte progenitor cell. Nature. 1988;333:560–562. doi: 10.1038/333560a0. [DOI] [PubMed] [Google Scholar]
- Noctor SC, Flint AC, Weissman TA, Dammerman RS, Kriegstein AR. Neurons derived from radial glial cells establish radial units in neocortex. Nature. 2001;409:714–720. doi: 10.1038/35055553. [DOI] [PubMed] [Google Scholar]
- Oh LY, Denninger A, Colvin JS, Vyas A, Tole S, Ornitz DM, Bansal R. Fibroblast growth factor receptor 3 signaling regulates the onset of oligodendrocyte terminal differentiation. J Neurosci. 2003;23:883–894. doi: 10.1523/JNEUROSCI.23-03-00883.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raff MC, Lillien LE. Differentiation of a bipotential glial progenitor cell: What controls the timing and the choice of developmental pathway? J Cell Sci Suppl. 1988;10:77–83. doi: 10.1242/jcs.1988.supplement_10.6. [DOI] [PubMed] [Google Scholar]
- Raff MC, Miller RH, Noble M. A glial progenitor cell that develops in vitro into an astrocyte or an oligodendrocyte depending on culture medium. Nature. 1983;303:390–396. doi: 10.1038/303390a0. [DOI] [PubMed] [Google Scholar]
- Reuss B, von Bohlen und Halbach O. Fibroblast growth factors and their receptors in the central nervous system. Cell Tissue Res. 2003;313:139–157. doi: 10.1007/s00441-003-0756-7. [DOI] [PubMed] [Google Scholar]
- Richardson WD, Pringle N, Mosley MJ, Westermark B, Dubois-Dalcq M. A role for platelet-derived growth factor in normal gliogenesis in the central nervous system. Cell. 1988;53:309–319. doi: 10.1016/0092-8674(88)90392-3. [DOI] [PubMed] [Google Scholar]
- Ridyard MS, Robbins SM. Fibroblast growth factor-2-induced signaling through lipid raft-associated fibroblast growth factor receptor substrate 2 (FRS2) J Biol Chem. 2003;278:13803–13809. doi: 10.1074/jbc.M210245200. [DOI] [PubMed] [Google Scholar]
- Ruffini F, Furlan R, Poliani PL, Brambilla E, Marconi PC, Bergami A, Desina G, Glorioso JC, Comi G, Martino G. Fibroblast growth factor-II gene therapy reverts the clinical course and the pathological signs of chronic experimental autoimmune encephalomyelitis in C57BL/6 mice. Gene Ther. 2001;8:1207–1213. doi: 10.1038/sj.gt.3301523. [DOI] [PubMed] [Google Scholar]
- Shiota C, Ikenaka K, Mikoshiba K. Developmental expression of myelin protein genes in dysmyelinating mutant mice: Analysis by nuclear run-off transcription assay, in situ hybridization, and immunohistochemistry. J Neurochem. 1991;56:818–826. doi: 10.1111/j.1471-4159.1991.tb01997.x. [DOI] [PubMed] [Google Scholar]
- Silver J, Miller JH. Regeneration beyond the glial scar. Nat Rev Neurosci. 2004;5:146–156. doi: 10.1038/nrn1326. [DOI] [PubMed] [Google Scholar]
- Small RK, Riddle P, Noble M. Evidence for migration of oligodendrocyte–type-2 astrocyte progenitor cells into the developing rat optic nerve. Nature. 1987;328:155–157. doi: 10.1038/328155a0. [DOI] [PubMed] [Google Scholar]
- Smith C, Berry M, Clarke WE, Logan A. Differential expression of fibroblast growth factor-2 and fibroblast growth factor receptor 1 in a scarring and nonscarring model of CNS injury in the rat. Eur J Neurosci. 2001;13:443–456. doi: 10.1046/j.1460-9568.2001.01400.x. [DOI] [PubMed] [Google Scholar]
- Teng YD, Mocchetti I, Taveira-DaSilva AM, Gillis RA, Wrathall JR. Basic fibroblast growth factor increases long-term survival of spinal motor neurons and improves respiratory function after experimental spinal cord injury. J Neurosci. 1999;19:7037–7047. doi: 10.1523/JNEUROSCI.19-16-07037.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno H, Gunn M, Dell K, Tseng A, Jr, Williams L. A truncated form of fibroblast growth factor receptor 1 inhibits signal transduction by multiple types of fibroblast growth factor receptor. J Biol Chem. 1992;267:1470–1476. [PubMed] [Google Scholar]
- van Praag H, Schinder AF, Christie BR, Toni N, Palmer TD, Gage FH. Functional neurogenesis in the adult hippocampus. Nature. 2002;415:1030–1034. doi: 10.1038/4151030a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Sdrulla AD, diSibio G, Bush G, Nofziger D, Hicks C, Weinmaster G, Barres BA. Notch receptor activation inhibits oligodendrocyte differentiation. Neuron. 1998;21:63–75. doi: 10.1016/s0896-6273(00)80515-2. [DOI] [PubMed] [Google Scholar]
- Wolswijk G. Chronic stage multiple sclerosis lesions contain a relatively quiescent population of oligodendrocyte precursor cells. J Neurosci. 1998;18:601–609. doi: 10.1523/JNEUROSCI.18-02-00601.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolswijk G. Oligodendrocyte precursor cells in the demyelinated multiple sclerosis spinal cord. Brain. 2002;125:338–349. doi: 10.1093/brain/awf031. [DOI] [PubMed] [Google Scholar]
- Wolswijk G, Noble M. Cooperation between PDGF and FGF converts slowly dividing O-2Aadult progenitor cells to rapidly dividing cells with characteristics of O-2Aperinatal progenitor cells. J Cell Biol. 1992;118:889–900. [Google Scholar]
- Zhou YX, Xu X, Chen L, Li C, Brodie SG, Deng CX. A Pro250Arg substitution in mouse Fgfr1 causes increased expression of Cbfa1 and premature fusion of calvarial sutures. Hum Mol Genet. 2000;9:2001–2008. doi: 10.1093/hmg/9.13.2001. [DOI] [PubMed] [Google Scholar]