

Abstract
Evidence suggests that β-amyloid (Aβ) peptide triggers a pathogenic cascade leading to neuronal loss in Alzheimer’s disease (AD). However, the causal link between Aβ and neuron death in vivo remains unclear since most animal models fail to recapitulate the dramatic cell loss observed in AD. We have recently developed transgenic mice that overexpress human APP and PS1 with five familial AD mutations (5XFAD mice) and exhibit robust neuron death. Here, we demonstrate that genetic deletion of the β-secretase (BACE1) not only abrogates Aβ generation and blocks amyloid deposition but also prevents neuron loss found in the cerebral cortex and subiculum, brain regions manifesting the most severe amyloidosis in 5XFAD mice. Importantly, BACE1 gene deletion also rescues memory deficits in 5XFAD mice. Our findings provide strong evidence that Aβ ultimately is responsible for neuron death in AD and validate the therapeutic potential of BACE1-inhibiting approaches for the treatment of AD.
Keywords: Alzheimer’s disease, β-amyloid (Aβ), β-secretase, BACE1 knockout, Tg6799, gliosis, cognitive impairment, spontaneous alternation, Y-maze
Introduction
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder and the most common cause of dementia among the elderly population, but no treatments addressing the underlying cause of disease have been developed. One of the hallmarks of AD is extensive neuronal death in the brain, approaching 90% loss in certain regions such as entorhinal cortex and nucleus basalis (for review, see Morrison and Hof, 1997). Although the precise mechanisms of AD and related cell loss and memory deficits are not fully determined, data support the hypothesis that amyloid-β (Aβ) peptides trigger a pathologic cascade ultimately leading to neuron death and cognitive impairment in AD (Hardy and Selkoe, 2002; LaFerla and Oddo, 2005; Selkoe and Schenk, 2003; Sisodia and St George-Hyslop, 2002; Turner et al., 2003). Mutations in the genes for amyloid precursor protein (APP) and presenilins 1/2 (PS1/2) cause familial AD (FAD) and increase production of the 42-amino acid form of Aβ (Aβ42). Furthermore, Aβ kills neurons in culture (for review, see Yankner, 1996), and Aβ also appears neurotoxic in vivo. For example, when directly injected into the brains of aged primates, fibrillar Aβ causes neuron death around the injection site (Geula et al., 1998). However, the case for the role of Aβ in neuron death has been challenged because many APP transgenic mice that overproduce Aβ form amyloid plaques and develop memory deficits but do not loose significant numbers of neurons (Irizarry et al., 1997a; Irizarry et al., 1997b; for review, see McGowan et al., 2006). Recently, new APP/PS1 transgenic mouse lines developed by our group and others show considerable neuron loss in the hippocampus (Casas et al., 2004; Schmitz et al., 2004) or the cortex and subiculum (Oakley et al., 2006). These studies suggest that Aβ does kill neurons in vivo, although they cannot formally exclude the possibility that overexpression of APP and PS1 with multiple FAD mutations is the cause of neuron death in these mouse models.
β-Site APP cleaving enzyme 1 (BACE1) has been identified as the β-secretase, the protease that initiates cleavage of APP to generate pathogenic Aβ peptides (Haniu et al., 2000; Hussain et al., 1999; Sinha et al., 1999; Vassar et al., 1999; Yan et al., 1999). As the rate-limiting enzyme for Aβ production, BACE1 is a prime therapeutic target for lowering cerebral Aβ levels in AD. Targeted deletion of the BACE1 gene (BACE1−/−) in mice abrogates the production of Aβ (Cai et al., 2001; Luo et al., 2001; Roberds et al., 2001). Moreover, APP transgenic mice with the BACE1 null genotype do not form amyloid plaques (Laird et al., 2005; Luo et al., 2003) and are prevented from developing Aβ-dependent memory deficits (Laird et al., 2005; Ohno et al., 2006; Ohno et al., 2004; for review, see Ohno, 2006). In the present study, we have used BACE1−/− mice to demonstrate that Aβ, rather than mutant APP or PS1 overexpression, is the cause of neuron loss in our APP/PS1 transgenic mouse line, 5XFAD (Oakley et al., 2006; Ohno et al., 2006). We determined that BACE1-deficient 5XFAD (BACE1−/−·5XFAD) mice do not have the amyloid plaques, astogliosis or memory deficits found in age-matched 5XFAD mice with wild-type BACE1 genes. Most importantly, neuron death in the cerebral cortex and subiculum was prevented in the BACE1−/−·5XFAD mice. To our knowledge, this is the first demonstration of genetic rescue of neuronal loss via ablation of BACE1, and consequently of Aβ, in an Alzheimer’s transgenic mouse model and provides strong evidence that Aβ ultimately kills neurons in vivo.
Materials and methods
Animals
We used APP/PS1 doubly transgenic mice that co-express and co-inherit both human APP and PS1 transgenes with a total of five FAD mutations under transcriptional control of the neuron-specific mouse Thy-1 promoter [5XFAD mice, Tg6799 line; (Oakley et al., 2006; Ohno et al., 2006)]. In 5XFAD mice, an APP transgene carrying triple FAD mutations [the Swedish mutation: K670N, M671L (Mullan et al., 1992); the Florida mutation: I716V (Eckman et al., 1997); the London mutation: V717I (Goate et al., 1991)] and a PS1 transgene carrying double FAD mutations [M146L and L286V (Citron et al., 1998)] were co-injected into the pronucleus of single cell C57BL/6XSJL hybrid mouse embryos. 5XFAD transgenic lines were maintained by crossing heterozygous transgenic mice with B6/SJL F1 breeders. In order to examine the impact of the BACE1 null mutation, hemizygous 5XFAD mice were crossbred to BACE1−/− mice with the BlkSw/129 background (Luo et al., 2001). The resultant F1 progeny were further intercrossed, yielding age-matched littermates with the genotypes of interest in a subset of the F2 progeny. Genotyping was performed by PCR analysis of tail DNA. The following four genotypes were analyzed: BACE1+/+·5XFAD−, BACE1+/+·5XFAD+, BACE1−/−·5XFAD−, and BACE1−/−·5XFAD+. All experiments were done at 15–18 months of age, blind with respect to the genotype of the mice, and were conducted with the approval of the Northwestern University Animal Care and Use Committee.
Spontaneous alternation Y-maze test
Spontaneous alternation performance was tested as described previously (Ohno et al., 2004). Each mouse was placed in the center of the symmetrical Y-maze and was allowed to explore freely through the maze during an 8-min session. The sequence and total number of arms entered were recorded. Arm entry was considered to be complete when the hind paws of the mouse had been completely placed in the arm. Percentage alternation is the number of triads containing entries into all three arms divided by the maximum possible alternations (the total number of arms entered minus 2) X 100.
Histology and immunostaining
After behavioral tests were completed, mice were decapitated under halothane anesthesia. One hemibrain of each mouse was fixed in 4% paraformaldehyde solution in phosphate buffered saline (PBS) and cryoprotected in 30% sucrose solution in PBS containing 0.01% sodium azide. Brains were sectioned sagittally on a freezing microtome at 30 μm and successive sections were stored in azide-PBS solution at 4°C. For estimating neuronal loss, cresyl violet staining was performed to visualize neurons. For immunohistochemical analysis, the sections were stained by the avidin-biotin peroxidase complex method as described previously (Oakley et al., 2006). Briefly, the sections were incubated overnight at room temperature with the following primary antibodies: rabbit anti-Aβ42 (1:5,000; Biosource, Camarillo, CA) and rabbit anti-glial fibrillary acidic protein (GFAP) (1:10,000; G9269, Sigma, St Louis, MO). After washes, the sections were incubated with secondary biotinylated goat anti-rabbit IgG at 1:2,000 for 2 hr at room temperature. The ABC kit (Vector Laboratories, Burlingame, CA) was utilized with 3,3′-diaminobenzidine tetrahydrochloride as a chromogen to visualize the reaction product. The sections were then mounted on charged slides, counterstained with hematoxylin, dehydrated in a series of alcohol, cleared in xylene, and covered with a coverslip. Light microscopy was conducted on a Nikon E800 microscope with a Spot Advanced digital camera for capturing images.
Immunoblotting
After behavioral tests were completed, hemibrain samples were taken from the mice under halothane anesthesia and were snap-frozen for immunoblot assays. Hemibrains were homogenized in 1% Triton X-100, PBS, protease inhibitor cocktail (Calbiochem, La Jolla, CA), and Halt Phosphatase Inhibitor Cocktail (Pierce, Rockford, IL). Following brief sonication, homogenates were diluted 1:1 with sample boiling buffer (60 mM Tris, 10% glycerol, 5% SDS), pH 6.8 and 5% loading dye, and boiled for 5 min. Equal amounts of protein (15 μg) were resolved on 4–12% Bis-Tris NuPAGE gels (Invitrogen, Carlsbad, CA), transferred onto PVDF membranes, and blocked in 5% nonfat milk in Tris-buffered saline containing 0.05% Tween 20. For Aβ immunoblots, as a positive control 100 ng of synthetic Aβ40 (Invitrogen, Carlsbad, CA) dissolved in distilled H2O was electrophoresed along with brain samples. Membranes were probed with primary antibodies and incubated with HRP-conjugated secondary after washing. The following antibodies were used: anti-total Aβ (1:10,000; 6E10, Signet, Dedham, MA), anti-APP (1:5000; 22C11, Chemicon, Temecular, CA), anti-BACE1 (1:1000; BACE1-Cat, J. Zhao, L. Binder, R. Vassar, unpublished), and anti-actin (1:10,000; AC-15, Sigma, St. Louis, MO). Immunoblot signals were detected by enhanced chemiluminescence plus (Amersham Biosciences, Arlington Heights, IL) and quantified using an Eastman Kodak Image Analyzer (Rochester, NY).
Statistical analysis
The significance of differences between the groups was determined by a one-way ANOVA, and post-hoc Fisher’s PLSD tests were performed when appropriate.
Results
APP transgenic mouse models recapitulate several features of AD, such as amyloid pathology, synaptic dysfunction, and behavioral deficits, but there has been little demonstration of extensive neuronal loss in these models (Ashe, 2001; Dodart et al., 2002; German and Eisch, 2004; Irizarry et al., 1997a; Irizarry et al., 1997b; Janus and Westaway, 2001; Kobayashi and Chen, 2005; McGowan et al., 2006). We recently developed a novel APP/PS1 doubly transgenic mouse line [5XFAD mice, Tg6799 line; (Oakley et al., 2006; Ohno et al., 2006)] that harbors APP with three intramolecular FAD mutations (Swedish, Florida, and London mutations) and PS1 with two intramolecular FAD mutations (M146L and L286V). When expressed in combination, these five FAD mutations act together to additively increase Aβ42 production. As a result, 5XFAD mice exhibit dramatically accelerated Aβ42 accumulation, start to develop visible amyloid deposition in the deeper layer of the cerebral cortex (layer 5) and the subiculum at ~2 months of age, and have memory impairments in several behavioral paradigms beginning at ~4 months of age (Oakley et al., 2006; Ohno et al., 2006). In addition, at ~9 months of age, 5XFAD mice show pronounced loss of large pyramidal neurons in cortical layer 5 and subiculum, the same regions that have the greatest amyloid burden.
We hypothesized that neuron loss in 5XFAD brain was the result of Aβ42-induced neurotoxicity. However, we could not exclude the possibility that abnormal interactions among the five FAD mutations in APP and PS1 or overexpression of the mutant transgenes, rather than Aβ42 itself, led to the neuron death in 5XFAD mice. To address this problem, we crossed BACE1−/− mice with 5XFAD transgenic mice to abrogate Aβ generation without blocking APP and PS1 transgene expression. Microscopic examination of cresyl violet-stained brain sections revealed that most of the large pyramidal neurons were lost in cortical layer 5 (Fig. 1C,D) and subiculum (Fig. 2C,D) of 5XFAD mice at 18 months of age, in agreement with our previous study (Oakley et al., 2006). The loss of large pyramidal neurons in 5XFAD cortex and subiculum was clearly obvious by qualitative visual inspection of micrographs, thus making it unnecessary to perform cell counting. Neurons in other cortical layers, hippocampus (including CA1–4 and dentate gyrus), and elsewhere in the brain of 5XFAD mice appeared intact by visual inspection, although we cannot exclude the possibility of less obvious cell loss in other brain regions. We also noted that 5XFAD brain sizes appeared normal.
Fig. 1.
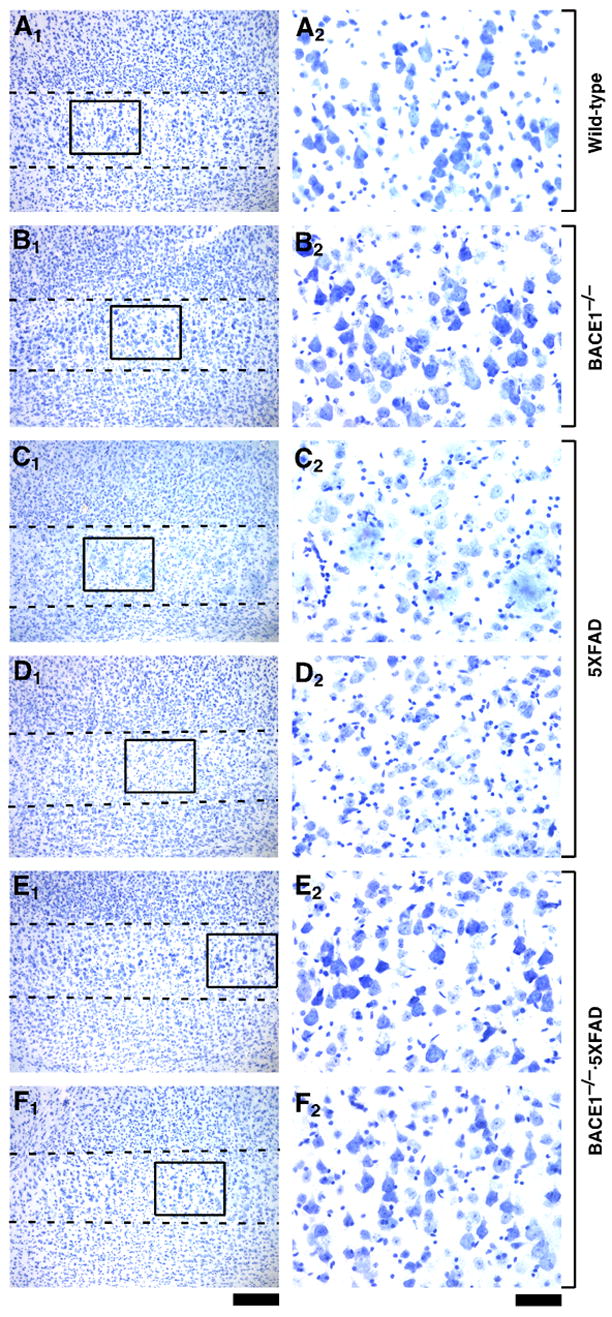
BACE1 null mutation prevents neuronal loss in the cerebral cortex of 5XFAD transgenic mice. Sagittal brain sections from wild-type control (A), BACE1−/− (B), 5XFAD (C,D), and BACE1−/−·5XFAD (E,F) mice at 18 months of age were stained with cresyl violet. Two different mice are presented for each of the 5XFAD and BACE1−/−·5XFAD genotypes. Shown are photomicrographs of cortical layer 5 (A1–F1: areas between the dashed lines) and higher magnification of pyramidal neurons in areas identified by respective rectangles (A2–F2). Note that large pyramidal neurons are nearly completely lost in layer 5 of 5XFAD mice as compared with the other three genotypes. In contrast, large pyramidal neurons are rescued to wild-type levels in BACE1−/−·5XFAD brains. Scale bar = 200 μm in A1–F1; 50 μm in A2–F2.
Fig. 2.
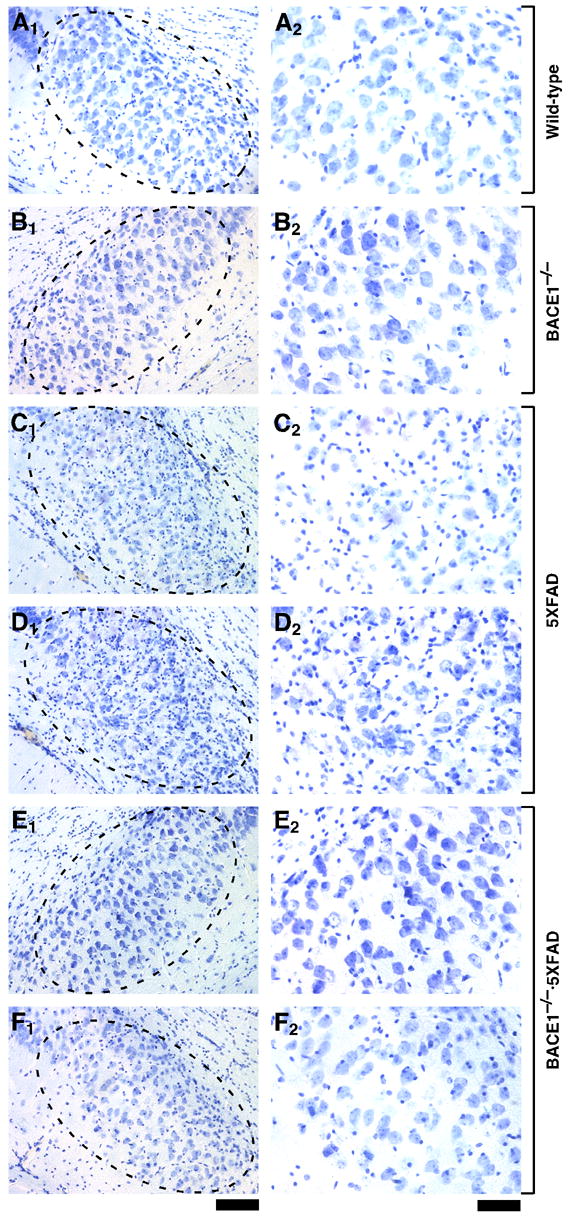
BACE1 null mutation prevents neuronal loss in the subiculum of 5XFAD transgenic mice. Sagittal brain sections from wild-type control (A), BACE1−/− (B), 5XFAD (C,D), and BACE1−/−·5XFAD (E,F) mice at 18 months of age (the same brains shown in Fig. 1) were stained with cresyl violet. Two different mice are presented for each of the 5XFAD and BACE1−/−·5XFAD genotypes. Shown are photomicrographs of the subiculum (A1–F1: areas within dashed ovals) and respective higher magnification images (A2–F2). Note that large pyramidal neurons are significantly reduced in number in subiculum of 5XFAD mice as compared with the other three genotypes; numbers of glia are increased, as indicated by the high density of small cresyl violet-stained nuclei. In contrast, the quantity of large pyramidal neurons in subiculum of BACE1−/−·5XFAD brains appears similar to that of wild-type brains, demonstrating rescue of neuron loss by BACE1 gene deletion. Scale bar = 100 μm in A1–F1; 50 μm in A2–F2.
In contrast to 5XFAD mice, BACE1−/− mice appeared to have normal numbers of large pyramidal neurons both in the cortex (Fig. 1B) and in the subiculum (Fig. 2B), which were indistinguishable from those of wild-type controls (Figs. 1A and 2A), and no overt neural lesions were observed in other BACE1−/− brain regions (Luo et al., 2001). Most importantly, large pyramidal neurons in cortical layer 5 (Fig. 1E,F) and subiculum (Fig. 2E,F) of BACE1-deficient 5XFAD (BACE1−/−·5XFAD) mice were well preserved throughout successive sections and were comparable in number to those of wild-type controls. Furthermore, as noted in our previous study (Oakley et al., 2006), 5XFAD mice had a significantly thinner cortical layer 1 as compared with wild-type controls (data not shown), most likely reflecting the loss of layer 5 neuron dendrites that project to and ramify in layer 1. We also observed that this dendritic loss was prevented in BACE1−/−·5XFAD mice, which showed layer 1 thickness comparable to controls (data not shown). Together, these findings provide compelling evidence that elimination of Aβ via BACE1 deletion prevents neuronal loss found in the 5XFAD mouse model, even though FAD mutant transgene expression remains intact.
It has recently been reported that the expression and activity of β-secretase/BACE1 are significantly elevated in AD brain (Fukumoto et al., 2002; Li et al., 2004; Yang et al., 2003), raising the possibility that increased BACE1 levels may be causally involved in the pathogenesis of AD. Immunoblot analysis demonstrated that BACE1 levels were significantly increased in 5XFAD mouse brains as compared with wild-type controls [F1,8 = 15.34, P < 0.01] (Fig. 3A,B; J. Zhao, L. Binder, and R. Vassar, unpublished observations), validating 5XFAD transgenic mice as useful animal models that recapitulate the BACE1 elevation in AD. A very strong signal in 5XFAD brain homogenate was observed for Aβ (Fig. 3A), which is usually very difficult to detect by immunoblot, illustrating the extremely high cerebral Aβ42 levels found in this mouse model (Oakley et al., 2006; Ohno et al., 2006). We confirmed that BACE1 null mutation completely abolished BACE1 protein and that Aβ peptides were not detectable in BACE1−/−·5XFAD bigenic mouse brains by immunoblot (Fig. 3A). Interestingly, BACE1−/−·5XFAD mice exhibited APP overexpression that was significantly higher than that of 5XFAD [F3,16 = 95.28, P < 0.01] (Fig. 3A,C), indicating that the full-length APP substrate accumulates in the brain if β-secretase cleavage of APP is ablated in 5XFAD mice.
Fig. 3.
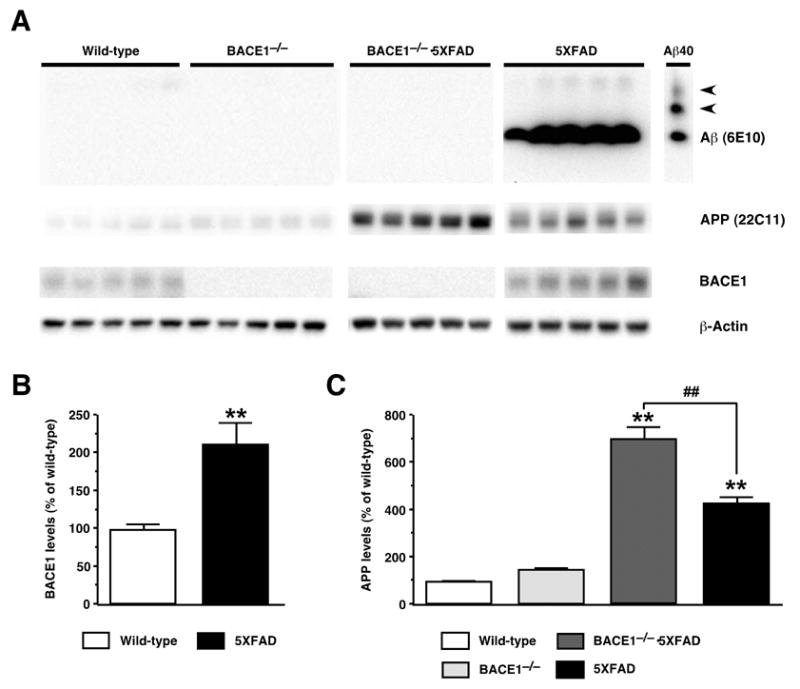
Effects of BACE1 null mutation on cerebral Aβ, BACE1 and full-length APP levels in 5XFAD transgenic mice. (A) Immunoblot analyses of protein extracts (15 μg/lane) from hemibrain homogenates of wild-type control, 5XFAD, BACE1−/−·5XFAD, and BACE1−/− mice at 18 months of age. Shown are immunoblots for Aβ (top panels; anti-total Aβ antibody 6E10), APP (second from top panels; anti-mouse and human APP antibody 22C11), BACE1 (third from top panels; anti-BACE1 antibody BACE1-Cat), and actin (bottom panels; anti-actin antibody AC-15). 5XFAD brain homogenates show a strong Aβ band, while Aβ is below the level of detection by immunoblot analysis in brain extracts from the other three genotypes. The lane labeled “Aβ40” contains 100 ng of synthetic Aβ40 for use as a positive control; the two slower migrating bands (arrowheads) likely represent multimeric Aβ assemblies. (B) Quantification of BACE1 blots in which band intensities were measured by phosphorimaging and expressed as percentage of wild-type control levels. Note that BACE1 levels are significantly elevated in 5XFAD brains. (C) Quantification of APP blots in which band intensities were measured by phosphorimaging and expressed as percentage of wild-type control levels. Note that while 5XFAD mice overexpress human APP approximately fourfold relative to endogenous mouse APP protein, levels of APP are elevated even further in BACE1−/−·5XFAD mice. Data are presented as the mean ± SEM of 5 animals. **P < 0.01 (vs. wild-type controls), ##P < 0.01 (vs. BACE1−/−·5XFAD).
Immunostaining with an Aβ42 end-specific antibody revealed that 5XFAD mice had massive amyloid deposition in the cerebral cortex and subiculum at 18 months of age (Fig. 4C,G). Consistent with our previous study (Oakley et al., 2006), amyloid plaques spread to fill much of the cortex and other hippocampal regions by this age and were also observed in the thalamus, olfactory bulb, and brain stem, although deposition was less severe in these regions (data not shown). Importantly, Aβ deposition was completely abolished in all brain regions, including the cortex and subiculum, of BACE1−/−·5XFAD mice (Fig. 4D,H), which were indistinguishable from wild-type control mice (Fig. 4A,E) and BACE1−/− mice (Fig. 4B,F). Amyloid plaques in APP transgenic mouse brains are often accompanied by glial activation indicative of inflammatory processes reminiscent of AD (Akiyama et al., 2000; Dodart et al., 2002). Immunostaining for the astrocyte marker GFAP revealed that 5XFAD mice had robust astrogliosis in the cerebral cortex (Fig. 5C). In contrast, there was no evidence of astrogliosis in brains of BACE1−/−·5XFAD mice (Fig. 5D) as well as in those of wild-type (Fig. 5A) or BACE1−/− (Fig. 5B) mice, demonstrating that the accumulation of Aβ peptides causes glial activation in APP/PS1 transgenic mice. Taken together, these results show that BACE1 deletion completely prevents AD-related neuropathology, including amyloidosis, gliosis, and neuron death found in the 5XFAD transgenic mouse model.
Fig. 4.
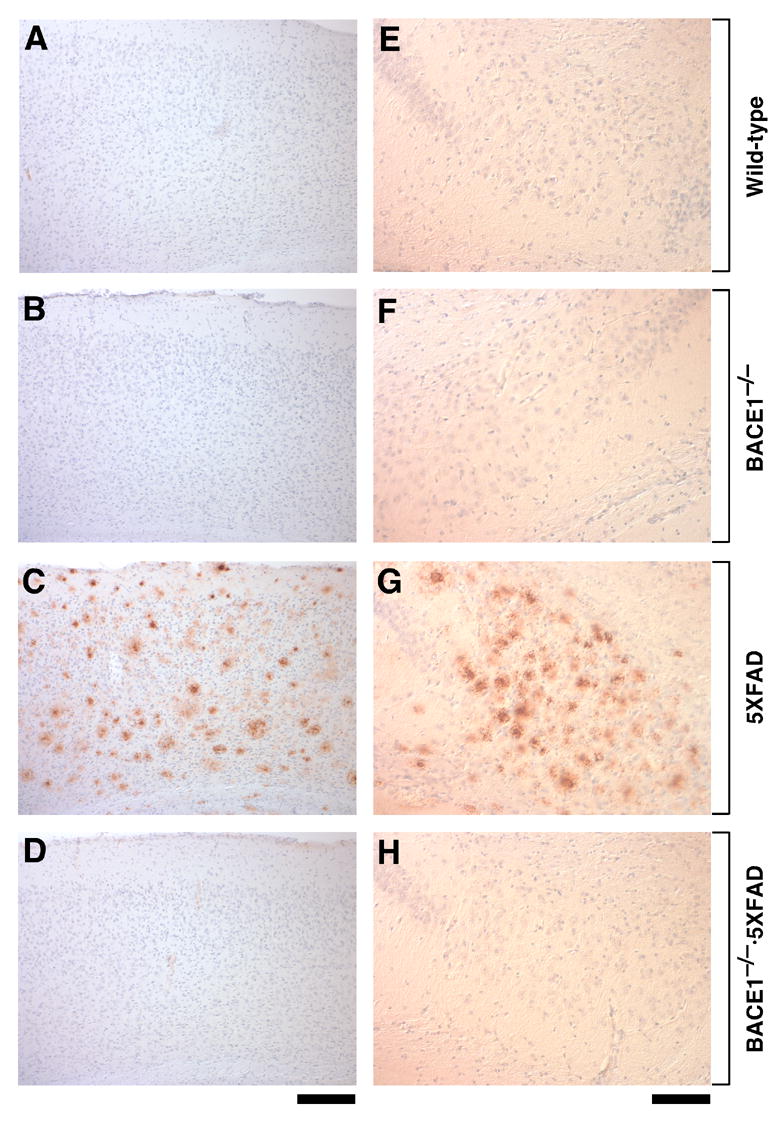
BACE1 null mutation prevents amyloid deposition in 5XFAD transgenic mice. Sagittal brain sections from wild-type control (A,E), BACE1−/− (B,F), 5XFAD (C,G) and BACE1−/−·5XFAD (D,H) mice at 18 months of age were immunostained with antibody specifically recognizing the Aβ42 C-terminus and counterstained with hematoxylin. Shown are photomicrographs of the layers of the cerebral cortex (A–D) and subiculum (E–H). Note that 5XFAD sections have robust Aβ42 staining in both brain regions, while BACE1−/−·5XFAD brain is protected from Aβ42 accumulation. Scale bar = 200 μm in A–D; 100 μm in E–H.
Fig. 5.
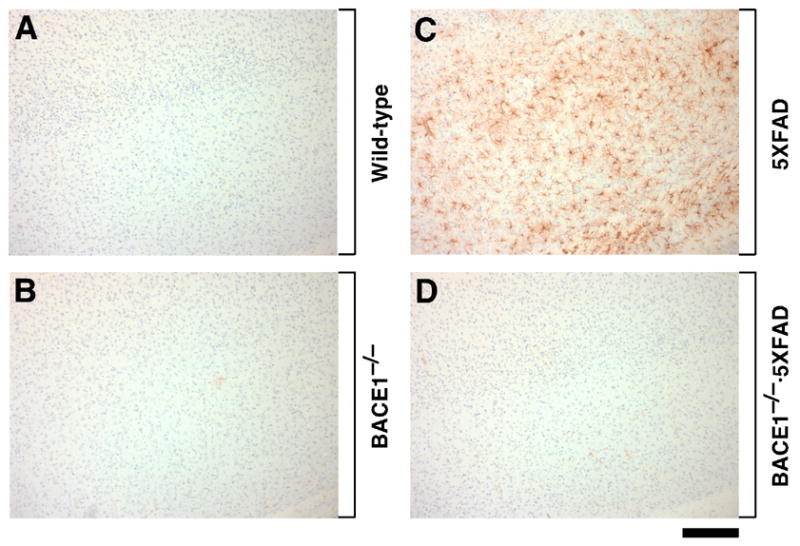
BACE1 null mutation prevents gliosis in 5XFAD transgenic mice. Sagittal sections of cerebral cortex from wild-type control (A), BACE1−/− (B), 5XFAD (C) and BACE1−/−·5XFAD (D) mice at 18 months of age were immunostained for astrocyte marker GFAP and counterstained with hematoxylin. Note that 5XFAD cortex shows extensive astrogliosis, while BACE1−/−·5XFAD brain has control levels of astrocytes. Scale bar = 200 μm.
To investigate the relationship between neuronal loss and memory deficits, we applied the spontaneous alternation Y-maze test at 15–18 months of age to the four genotypic groups of mice obtained by crossbreeding BACE1−/− and 5XFAD mice (Fig. 6). This behavioral assay does not involve any training, reward, or punishment, and allows us to assess spatial working memory. ANOVA revealed significant differences in percent alternation between the groups [F3,38 = 9.17, P < 0.01]. 5XFAD transgenic mice showed dramatically reduced spontaneous alternation performance in the Y-maze as compared with wild-type controls (P < 0.01) (Fig. 6A). Consistent with our previous report (Ohno et al., 2004), BACE1 gene deletion by itself in mice resulted in decreased percent alternation in this task (P < 0.01). Importantly, the spatial working memory deficits were rescued to wild-type control levels in BACE1−/−·5XFAD mice, which showed higher spontaneous alternation than 5XFAD mice (P < 0.01) and BACE1−/− mice (P < 0.05). The total number of arm entries in the alternation Y-maze test was significantly increased in BACE1−/− mice [F3,38 = 5.76, P < 0.01], suggesting that BACE1 null mutants exhibit increased levels of exploratory activity in the Y-maze (Fig. 6B).
Fig. 6.
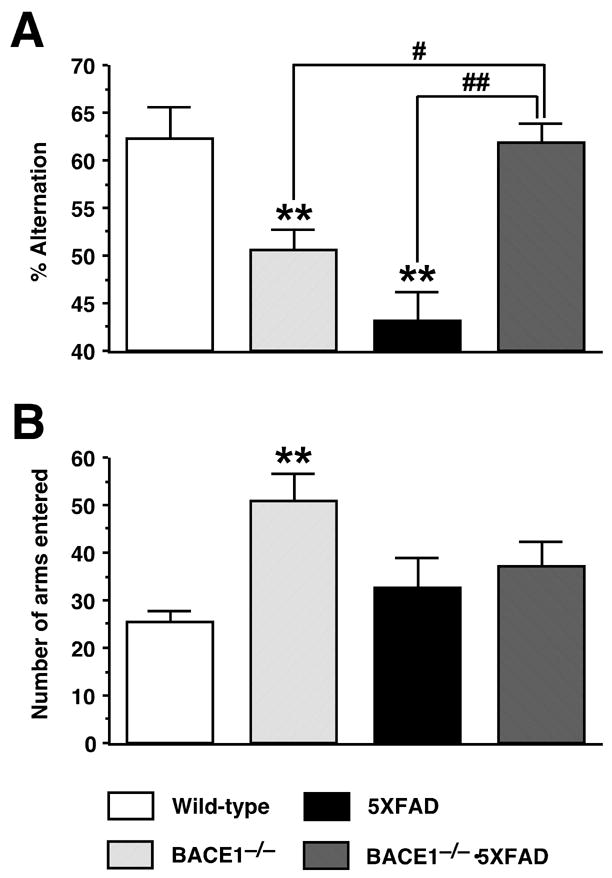
BACE1 null mutation prevents memory deficits in 5XFAD transgenic mice. (A) Spatial working memory of mice at 15–18 months of age was tested by spontaneous alternation performance in the Y-maze. BACE1−/−·5XFAD mice are rescued completely back to wild-type levels of alternation in the Y-maze. Note also that both BACE1−/− mice and 5XFAD mice show significantly lower levels of alternation performance as compared to wild-type control and BACE1−/−·5XFAD mice, although the deficit for 5XFAD tends to be more severe. (B) Total number of arm entries indicates that BACE1−/− mice show significantly higher levels of exploratory activity in the Y-maze as compared to wild-type control mice. Data are presented as the mean ± SEM of 7–14 animals. **P < 0.01 (vs. wild-type controls), #P < 0.05, ##P < 0.01 (vs. BACE1−/−·5XFAD).
Discussion
It is well established that Aβ kills neurons in culture (Pike et al., 1991; Roher et al., 1991; Yankner et al., 1990a; Yankner et al., 1989; Yankner et al., 1990b), but the link between Aβ and neuron loss in vivo has been equivocal. Cerebral injection of Aβ preparations into primates and rodents has produced variable effects, with some experiments demonstrating neurodegenerative changes (Frautschy et al., 1991; Geula et al., 1998; Kowall et al., 1991), while others yielding negative results (Podlisny et al., 1993). Moreover, most transgenic mice that express human APP with FAD mutations and overproduce Aβ lack significant cell loss (for review, see McGowan et al., 2006). For example, no significant neuron loss is reported in the hippocampus or cerebral cortex of Tg2576 and PDAPP mice (Irizarry et al., 1997a; Irizarry et al., 1997b). A small reduction in neuron number occurs in the hippocampal CA1 region of APP23 mice, but no loss is observed in the cortex in this model (Calhoun et al., 1998).
The lack of consistent results among the various earlier reports has cast some doubt upon the role of Aβ in neuron death in AD. However, recent work has begun to strengthen the connection between Aβ and neurodegeneration. For example, Aβ reduction by active or passive Aβ immunization protects against synaptic degeneration as determined by inhibition of progressive loss of synaptophysin in the hippocampus and cortex of PDAPP transgenic mice (Buttini et al., 2005). Moreover, potential toxic effects of intraneuronal Aβ or soluble Aβ oligomers have come to light and may be involved in neurodegeneration (Calhoun et al., 1998; Casas et al., 2004; Dickson, 2004; Schmitz et al., 2004; Urbanc et al., 2002). We have previously shown that 5XFAD transgenic mice have increased levels of the pro-neurodegeneration factor p25, exhibit decreased levels of the synaptic markers synaptophysin, syntaxin and PSD-95, and manifest a unique spatial pattern of large pyramidal neuron loss that occurs in cortical layer 5 and subiculum (Oakley et al., 2006). It is important to note that these same brain regions of 5XFAD have the highest levels of intraneuronal Aβ accumulation and extracellular amyloid plaques, both of which label with thioflavin S and therefore consist of β-pleated sheet aggregates. Our results are similar to those with another APP/PS1 transgenic mouse line having intraneuronal Aβ and marked death in the hippocampal CA1/2 pyramidal cell layer (Casas et al., 2004; Schmitz et al., 2004). 5XFAD brain also shows elevated levels of soluble Aβ oligomers (Ohno et al., 2006). Most importantly, the present study demonstrates that neuron loss as well as intraneuronal and extracellular Aβ deposition are prevented in brains of BACE1−/−·5XFAD mice. Notably, APP or PS1 transgene expression was not decreased by BACE1−/− genotype in this study as well as in our previous investigation (Ohno et al., 2006), suggesting that mere overexpression of FAD mutant APP and PS1 does not contribute to cell loss in 5XFAD mice. BACE1 deficiency also abolishes soluble oligomeric Aβ assemblies in 5XFAD mice (Ohno et al., 2006). Although it is difficult to unequivocally determine the form of Aβ that is responsible for neurodegeneration, our investigation provides direct evidence that Aβ, or a BACE1 cleavage fragment of APP, is ultimately responsible for neuronal loss in 5XFAD mice.
Since 5XFAD mice generate Aβ42 almost exclusively (Oakley et al., 2006; Ohno et al., 2006), it is very likely that Aβ42 was the trigger of the neuronal loss that we observed. However, we cannot completely exclude the possibility that neurotoxicity in 5XFAD mice may be caused by build-up of β-secretase-cleaved C-terminal fragment C99, which is also eliminated by BACE1−/− genotype in APP and APP/PS1 transgenic mouse brains (Laird et al., 2005; Luo et al., 2001). Transgenic overexpression of the potentially amyloidogenic C99 results in progressive neurodegeneration in the cerebral cortex and hippocampus and causes memory deficits (Berger-Sweeney et al., 1999; Lee et al., 2006; Nalbantoglu et al., 1997). Furthermore, C99 has been shown to impair learning and memory when it is centrally administered (Choi et al., 2001; Song et al., 1998). Therefore, it is reasonable to speculate that both amyloidogenic proteins, Aβ and C99, may contribute to cell death and memory deficits in 5XFAD mice.
We have previously reported that BACE1 gene deletion and consequent reductions in Aβ prevent deficits in olfactory, temporal, and spatial (working and reference) memories in young plaque-free Tg2576 transgenic mice (Ohno et al., 2004) or plaque-bearing 5XFAD mice (Ohno et al., 2006). Consistent with these results, other groups have shown that lowering BACE1 levels in vivo by null mutation or lentiviral delivery of small interfering RNA ameliorates amyloid pathology and spatial memory impairments in APP or APP/PS1 transgenic mice (Laird et al., 2005; Singer et al., 2005; for review, see Ohno, 2006). Here, we show that BACE1 deletion also rescues memory deficits in 5XFAD mice at advanced age, when animals with BACE1+/+ genotype have developed dramatic loss of large pyramidal neurons in cortical layer 5 and subiculum. In this regard, it is interesting to note that neuron loss is also observed in both of these regions in AD brain (Bobinski et al., 1997; Giannakopoulos et al., 1996). Importantly, it is plausible that ablation of these neurons contributes to memory decline, since layer 5 pyramidal neurons receive the highest density of cholinergic innervation (Houser et al., 1985) and greater atrophy in the subiculum (which is part of the hippocampus) is associated with increased risk for conversion from mild cognitive impairment to AD (Apostolova et al., 2006). Our finding that BACE1 deletion prevents neuronal loss, amyloidosis, astrogliosis, and spatial working memory deficits in the Y-maze in 5XFAD mice of advanced age suggests a beneficial effect of BACE1 inhibition and Aβ reduction on later phases of cognitive impairments and AD-related pathologies. Inducible genetic and pharmacologic strategies to block BACE1 will be important for testing the reversibility of Aβ-dependent cognitive deficits by suppressing BACE1 at different stages of amyloid pathology and determining the extent and duration of ameliorative effects of BACE1 inhibition following treatment.
While cognitive rescue by BACE1 ablation is consistent across different transgenic mouse models of AD and various behavioral assays, BACE1−/− genotype by itself affects memory performance in some learning paradigms. The present study as well as others (Laird et al., 2005; Ohno et al., 2004) demonstrates that BACE1−/− mice are impaired in spatial working memory, as assessed by the spontaneous alternation Y-maze test. Spatial reference memory in a Morris water maze is also significantly impaired in BACE1−/− mice (Laird et al., 2005; Ohno et al., 2006). In contrast, BACE1 deletion does not significantly impair olfactory memory assessed by the social recognition test (Ohno et al., 2004) or temporal associative memory assayed by trace fear conditioning (Ohno et al., 2006). It is therefore interesting to speculate that a BACE1-dependent mechanism may play a role in certain types of cognition, especially spatial components of learning and memory (for review, see Ohno, 2006). Since a variety of possible BACE1 physiological substrates have been reported in addition to APP [e.g., α-2,6-sialyltransferase (Kitazume et al., 2001), P-selectin glycoprotein ligand-1 (Lichtenthaler et al., 2003), β-subunits of voltage-gated sodium channels (Wong et al., 2005), low density lipoprotein receptor-related protein (von Arnim et al., 2005), and neuregulin-1 (Willem et al., 2006)], we cannot exclude the possibility that changes in the proteolytic products of these substrates may contribute to cognitive deficits in BACE1−/− mice.
In the present study, we show that both 5XFAD and BACE1−/− mice have spatial memory impairments, while BACE1−/−·5XFAD bigenic mice are rescued from these cognitive deficits. Similar results were observed in our previous study analyzing Tg2576, BACE1−/−, and BACE1−/−·Tg2576 bigenic mice (Ohno et al., 2004) and in another investigation (Laird et al., 2005) (for review, see Ohno, 2006). Numerous studies suggest that the memory impairments of APP transgenic mice are caused by excessive levels of cerebral Aβ (for review, see McGowan et al., 2006), but the deficits of BACE1−/− mice and the rescue of BACE1−/−·APP bigenic mice are more difficult to explain. Genetic ablation of BACE1 in Tg2576 lowered cerebral Aβ level to only a few percent of that observed in BACE1+/+·Tg2576 mice (Ohno et al., 2004), clearly demonstrating that BACE1 is the predominant β-secretase enzyme responsible for Aβ generation in the brain. However, a residual amount of brain Aβ was measured in BACE1−/−·Tg2576 mice that was approximately equivalent to the level of endogenous Aβ observed in non-transgenic wild-type brain. Endogenous levels of Aβ are normally quite low and difficult to detect in non-transgenic mouse brain, so only sight leakiness of Aβ generation in BACE1−/−·Tg2576 brain would be required to produce the residual levels of Aβ that we measured. Importantly, Aβ levels in both BACE1−/−·Tg2576 and wild-type brains were significantly higher than the background levels of Aβ found in BACE1−/− monogenic brains. From this evidence, we hypothesized that an optimal level of Aβ was necessary to maintain normal memory function (Ohno et al., 2004). This notion is also consistent with the observation that spatial memory is impaired in APP-deficient mice (Dawson et al., 1999). Our results imply that both excessive (e.g., AD and APP transgenic) and deficient (e.g., BACE1−/− and APP−/−) Aβ levels may impair cognition. Although we have not quantified Aβ levels in our current study, we speculate that residual levels of Aβ in the brains of BACE1−/−·5XFAD mice may be responsible for the behavioral rescue of these bigenic animals.
The enzyme responsible for generating residual Aβ in BACE1−/−·APP bigenic mice is unknown. In our previous study (Ohno et al., 2004), we measured cerebral Aβ levels with an Aβ ELISA that detected endogenous mouse and transgenic human Aβ with equal sensitivity and that did not discriminate among Aβ peptides with heterogeneous N-termini. We have shown that BACE1 is responsible for generating the major Aβ peptides starting at Asp1 and Glu11 (Vassar et al., 1999). Work from Selkoe and colleagues identified minor Aβ forms beginning at Val-3 and Ile-6, demonstrated that these peptides are not made by β-secretase, and showed that inhibition of β-secretase activity in cells leads to elevated production of these minor Aβ species (Haass et al., 1995; Citron et al., 1996). Unlike Asp1 and Glu11 Aβ, the Val-3 and Ile-6 Aβ are not increased upon BACE1 overexpression (Vassar et al., 1999), indicating that they are indeed generated by a different enzyme(s). We speculate that the residual levels of Aβ measured in BACE1−/−·Tg2576 brain (Ohno et al., 2004) represented Aβ peptides beginning at Val-3 and Ile-6. Genetic ablation of BACE1 would increase the generation of Val-3 and Ile-6 Aβ, similar to the effects of β-secretase inhibition (Haass et al., 1995; Citron et al., 1996). Moreover, transgenic overexpression of APP would provide greater levels of substrate for cleavage at Val-3 and Ile-6. Given these arguments, it appears likely that a small amount of residual Aβ beginning at Val-3 and Ile-6 would be generated in BACE1−/−·5XFAD brains, thus providing an explanation for the rescue of memory deficits in these mice. Alternatively, BACE2, which also cleaves at Asp1 (but with lower efficiency than BACE1) and is expressed in the brain (but at much lower levels than BACE1), may play a role in producing residual Aβ in BACE1−/−·5XFAD brain. Consistent with recent evidence suggesting potential physiological roles for Aβ peptides (Esteban, 2004; Kamenetz et al., 2003; Pearson and Peers, 2006; Plant et al., 2006; Yu et al., 2001), our findings favor the idea that normal levels of Aβ may be necessary for neuronal functions involved in learning and memory, although further investigation is required to elucidate potential underlying mechanisms.
In summary, the results presented here strongly support the hypothesis that Aβ is neurotoxic and initiates the pathogenic cascade leading to neuronal loss in AD. We conclude that inhibition of the β-secretase, BACE1, should prove beneficial as a disease-modifying therapeutic strategy to counter the cell death and cognitive impairments of this intractable neurodegenerative disorder.
Acknowledgments
This work was supported by National Institutes of Health grants R01 MH067251 (M.O.), R01 AG022560 (R.V.), P01 AG021184 (R.V. and R.B.) and R37 AG08796 (J.F.D).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, O’Banion MK, Pachter J, Pasinetti G, Plata-Salaman C, Rogers J, Rydel R, Shen Y, Streit W, Strohmeyer R, Tooyoma I, Van Muiswinkel FL, Veerhuis R, Walker D, Webster S, Wegrzyniak B, Wenk G, Wyss-Coray T. Inflammation and Alzheimer’s disease. Neurobiol Aging. 2000;21:383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apostolova LG, Dutton RA, Dinov ID, Hayashi KM, Toga AW, Cummings JL, Thompson PM. Conversion of mild cognitive impairment to Alzheimer disease predicted by hippocampal atrophy maps. Arch Neurol. 2006;63:693–699. doi: 10.1001/archneur.63.5.693. [DOI] [PubMed] [Google Scholar]
- Ashe KH. Learning and memory in transgenic mice modeling Alzheimer’s disease. Learn Mem. 2001;8:301–308. doi: 10.1101/lm.43701. [DOI] [PubMed] [Google Scholar]
- Berger-Sweeney J, McPhie DL, Arters JA, Greenan J, Oster-Granite ML, Neve RL. Impairments in learning and memory accompanied by neurodegeneration in mice transgenic for the carboxyl-terminus of the amyloid precursor protein. Brain Res Mol Brain Res. 1999;66:150–162. doi: 10.1016/s0169-328x(99)00014-5. [DOI] [PubMed] [Google Scholar]
- Bobinski M, Wegiel J, Tarnawski M, Bobinski M, Reisberg B, de Leon MJ, Miller DC, Wisniewski HM. Relationships between regional neuronal loss and neurofibrillary changes in the hippocampal formation and duration and severity of Alzheimer disease. J Neuropathol Exp Neurol. 1997;56:414–420. doi: 10.1097/00005072-199704000-00010. [DOI] [PubMed] [Google Scholar]
- Buttini M, Masliah E, Barbour R, Grajeda H, Motter R, Johnson-Wood K, Khan K, Seubert P, Freedman S, Schenk D, Games D. β-Amyloid immunotherapy prevents synaptic degeneration in a mouse model of Alzheimer’s disease. J Neurosci. 2005;25:9096–9101. doi: 10.1523/JNEUROSCI.1697-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai H, Wang Y, McCarthy D, Wen H, Borchelt DR, Price DL, Wong PC. BACE1 is the major β-secretase for generation of Aβ peptides by neurons. Nat Neurosci. 2001;4:233–234. doi: 10.1038/85064. [DOI] [PubMed] [Google Scholar]
- Calhoun ME, Wiederhold KH, Abramowski D, Phinney AL, Probst A, Sturchler-Pierrat C, Staufenbiel M, Sommer B, Jucker M. Neuron loss in APP transgenic mice. Nature. 1998;395:755–756. doi: 10.1038/27351. [DOI] [PubMed] [Google Scholar]
- Casas C, Sergeant N, Itier JM, Blanchard V, Wirths O, van der Kolk N, Vingtdeux V, van de Steeg E, Ret G, Canton T, Drobecq H, Clark A, Bonici B, Delacourte A, Benavides J, Schmitz C, Tremp G, Bayer TA, Benoit P, Pradier L. Massive CA1/2 neuronal loss with intraneuronal and N-terminal truncated Aβ42 accumulation in a novel Alzheimer transgenic model. Am J Pathol. 2004;165:1289–1300. doi: 10.1016/s0002-9440(10)63388-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi SH, Park CH, Koo JW, Seo JH, Kim HS, Jeong SJ, Lee JH, Kim SS, Suh YH. Memory impairment and cholinergic dysfunction by centrally administered Aβ and carboxyl-terminal fragment of Alzheimer’s APP in mice. FASEB J. 2001;15:1816–1818. doi: 10.1096/fj.00-0859fje. [DOI] [PubMed] [Google Scholar]
- Citron M, Diehl TS, Capell A, Haass C, Teplow DB, Selkoe DJ. Inhibition of amyloid β-protein production in neural cells by the serine protease inhibitor AEBSF. Neuron. 1996;17:171–179. doi: 10.1016/s0896-6273(00)80290-1. [DOI] [PubMed] [Google Scholar]
- Citron M, Eckman CB, Diehl TS, Corcoran C, Ostaszewski BL, Xia W, Levesque G, St George Hyslop P, Younkin SG, Selkoe DJ. Additive effects of PS1 and APP mutations on secretion of the 42-residue amyloid β-protein. Neurobiol Dis. 1998;5:107–116. doi: 10.1006/nbdi.1998.0183. [DOI] [PubMed] [Google Scholar]
- Dawson GR, Seabrook GR, Zheng H, Smith DW, Graham S, O’Dowd G, Bowery BJ, Boyce S, Trumbauer ME, Chen HY, Van der Ploeg LH, Sirinathsinghji DJ. Age-related cognitive deficits, impaired long-term potentiation and reduction in synaptic marker density in mice lacking the β-amyloid precursor protein. Neuroscience. 1999;90:1–13. doi: 10.1016/s0306-4522(98)00410-2. [DOI] [PubMed] [Google Scholar]
- Dickson DW. Building a more perfect beast: APP transgenic mice with neuronal loss. Am J Pathol. 2004;164:1143–1146. doi: 10.1016/S0002-9440(10)63202-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodart JC, Mathis C, Bales KR, Paul SM. Does my mouse have Alzheimer’s disease? Genes Brain Behav. 2002;1:142–155. doi: 10.1034/j.1601-183x.2002.10302.x. [DOI] [PubMed] [Google Scholar]
- Eckman CB, Mehta ND, Crook R, Perez-tur J, Prihar G, Pfeiffer E, Graff-Radford N, Hinder P, Yager D, Zenk B, Refolo LM, Prada CM, Younkin SG, Hutton M, Hardy J. A new pathogenic mutation in the APP gene (I716V) increases the relative proportion of Aβ42(43) Hum Mol Genet. 1997;6:2087–2089. doi: 10.1093/hmg/6.12.2087. [DOI] [PubMed] [Google Scholar]
- Esteban JA. Living with the enemy: a physiological role for the β-amyloid peptide. Trends Neurosci. 2004;27:1–3. doi: 10.1016/j.tins.2003.10.008. [DOI] [PubMed] [Google Scholar]
- Frautschy SA, Baird A, Cole GM. Effects of injected Alzheimer β-amyloid cores in rat brain. Proc Natl Acad Sci USA. 1991;88:8362–8366. doi: 10.1073/pnas.88.19.8362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukumoto H, Cheung BS, Hyman BT, Irizarry MC. β-Secretase protein and activity are increased in the neocortex in Alzheimer disease. Arch Neurol. 2002;59:1381–1389. doi: 10.1001/archneur.59.9.1381. [DOI] [PubMed] [Google Scholar]
- German DC, Eisch AJ. Mouse models of Alzheimer’s disease: insight into treatment. Rev Neurosci. 2004;15:353–369. doi: 10.1515/revneuro.2004.15.5.353. [DOI] [PubMed] [Google Scholar]
- Geula C, Wu CK, Saroff D, Lorenzo A, Yuan M, Yankner BA. Aging renders the brain vulnerable to amyloid β-protein neurotoxicity. Nat Med. 1998;4:827–831. doi: 10.1038/nm0798-827. [DOI] [PubMed] [Google Scholar]
- Giannakopoulos P, Hof PR, Kovari E, Vallet PG, Herrmann FR, Bouras C. Distinct patterns of neuronal loss and Alzheimer’s disease lesion distribution in elderly individuals older than 90 years. J Neuropathol Exp Neurol. 1996;55:1210–1220. doi: 10.1097/00005072-199612000-00004. [DOI] [PubMed] [Google Scholar]
- Goate A, Chartier-Harlin MC, Mullan M, Brown J, Crawford F, Fidani L, Giuffra L, Haynes A, Irving N, James L, Mant R, Newton P, Rooke K, Roques P, Talbot C, Pericak-Vance M, Roses A, Williamson R, Rossor M, Owen M, Hardy J. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature. 1991;349:704–706. doi: 10.1038/349704a0. [DOI] [PubMed] [Google Scholar]
- Haass C, Capell A, Citron M, Teplow DB, Selkoe DJ. The vacuolar H+-ATPase inhibitor bafilomycin A1 differentially affects proteolytic processing of mutant and wild-type β-amyloid precursor protein. J Biol Chem. 1995;270:6186–6192. doi: 10.1074/jbc.270.11.6186. [DOI] [PubMed] [Google Scholar]
- Haniu M, Denis P, Young Y, Mendiaz EA, Fuller J, Hui JO, Bennett BD, Kahn S, Ross S, Burgess T, Katta V, Rogers G, Vassar R, Citron M. Characterization of Alzheimer’s β-secretase protein BACE. A pepsin family member with unusual properties. J Biol Chem. 2000;275:21099–21106. doi: 10.1074/jbc.M002095200. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Houser CR, Crawford GD, Salvaterra PM, Vaughn JE. Immunocytochemical localization of choline acetyltransferase in rat cerebral cortex: a study of cholinergic neurons and synapses. J Comp Neurol. 1985;234:17–34. doi: 10.1002/cne.902340103. [DOI] [PubMed] [Google Scholar]
- Hussain I, Powell D, Howlett DR, Tew DG, Meek TD, Chapman C, Gloger IS, Murphy KE, Southan CD, Ryan DM, Smith TS, Simmons DL, Walsh FS, Dingwall C, Christie G. Identification of a novel aspartic protease (Asp 2) as β-secretase. Mol Cell Neurosci. 1999;14:419–427. doi: 10.1006/mcne.1999.0811. [DOI] [PubMed] [Google Scholar]
- Irizarry MC, McNamara M, Fedorchak K, Hsiao K, Hyman BT. APPSw transgenic mice develop age-related Aβ deposits and neuropil abnormalities, but no neuronal loss in CA1. J Neuropathol Exp Neurol. 1997a;56:965–973. doi: 10.1097/00005072-199709000-00002. [DOI] [PubMed] [Google Scholar]
- Irizarry MC, Soriano F, McNamara M, Page KJ, Schenk D, Games D, Hyman BT. Aβ deposition is associated with neuropil changes, but not with overt neuronal loss in the human amyloid precursor protein V717F (PDAPP) transgenic mouse. J Neurosci. 1997b;17:7053–7059. doi: 10.1523/JNEUROSCI.17-18-07053.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janus C, Westaway D. Transgenic mouse models of Alzheimer’s disease. Physiol Behav. 2001;73:873–886. doi: 10.1016/s0031-9384(01)00524-8. [DOI] [PubMed] [Google Scholar]
- Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, Sisodia S, Malinow R. APP processing and synaptic function. Neuron. 2003;37:925–937. doi: 10.1016/s0896-6273(03)00124-7. [DOI] [PubMed] [Google Scholar]
- Kitazume S, Tachida Y, Oka R, Shirotani K, Saido TC, Hashimoto Y. Alzheimer’s β-secretase, β-site amyloid precursor protein-cleaving enzyme, is responsible for cleavage secretion of a Golgi-resident sialyltransferase. Proc Natl Acad Sci USA. 2001;98:13554–13559. doi: 10.1073/pnas.241509198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi DT, Chen KS. Behavioral phenotypes of amyloid-based genetically modified mouse models of Alzheimer’s disease. Genes Brain Behav. 2005;4:173–196. doi: 10.1111/j.1601-183X.2005.00124.x. [DOI] [PubMed] [Google Scholar]
- Kowall NW, Beal MF, Busciglio J, Duffy LK, Yankner BA. An in vivo model for the neurodegenerative effects of β amyloid and protection by substance P. Proc Natl Acad Sci USA. 1991;88:7247–7251. doi: 10.1073/pnas.88.16.7247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaFerla FM, Oddo S. Alzheimer’s disease: Aβ, tau and synaptic dysfunction. Trends Mol Med. 2005;11:170–176. doi: 10.1016/j.molmed.2005.02.009. [DOI] [PubMed] [Google Scholar]
- Laird FM, Cai H, Savonenko AV, Farah MH, He K, Melnikova T, Wen H, Chiang HC, Xu G, Koliatsos VE, Borchelt DR, Price DL, Lee HK, Wong PC. BACE1, a major determinant of selective vulnerability of the brain to amyloid-β amyloidogenesis, is essential for cognitive, emotional, and synaptic functions. J Neurosci. 2005;25:11693–11709. doi: 10.1523/JNEUROSCI.2766-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KW, Im JY, Song JS, Lee SH, Lee HJ, Ha HY, Koh JY, Gwag BJ, Yang SD, Paik SG, Han PL. Progressive neuronal loss and behavioral impairments of transgenic C57BL/6 inbred mice expressing the carboxy terminus of amyloid precursor protein. Neurobiol Dis. 2006;22:10–24. doi: 10.1016/j.nbd.2005.09.011. [DOI] [PubMed] [Google Scholar]
- Li R, Lindholm K, Yang LB, Yue X, Citron M, Yan R, Beach T, Sue L, Sabbagh M, Cai H, Wong P, Price D, Shen Y. Amyloid β peptide load is correlated with increased β-secretase activity in sporadic Alzheimer’s disease patients. Proc Natl Acad Sci USA. 2004;101:3632–3637. doi: 10.1073/pnas.0205689101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtenthaler SF, Dominguez DI, Westmeyer GG, Reiss K, Haass C, Saftig P, De Strooper B, Seed B. The cell adhesion protein P-selectin glycoprotein ligand-1 is a substrate for the aspartyl protease BACE1. J Biol Chem. 2003;278:48713–48719. doi: 10.1074/jbc.M303861200. [DOI] [PubMed] [Google Scholar]
- Luo Y, Bolon B, Damore MA, Fitzpatrick D, Liu H, Zhang J, Yan Q, Vassar R, Citron M. BACE1 (β-secretase) knockout mice do not acquire compensatory gene expression changes or develop neural lesions over time. Neurobiol Dis. 2003;14:81–88. doi: 10.1016/s0969-9961(03)00104-9. [DOI] [PubMed] [Google Scholar]
- Luo Y, Bolon B, Kahn S, Bennett BD, Babu-Khan S, Denis P, Fan W, Kha H, Zhang J, Gong Y, Martin L, Louis JC, Yan Q, Richards WG, Citron M, Vassar R. Mice deficient in BACE1, the Alzheimer’s β-secretase, have normal phenotype and abolished β-amyloid generation. Nat Neurosci. 2001;4:231–232. doi: 10.1038/85059. [DOI] [PubMed] [Google Scholar]
- McGowan E, Eriksen J, Hutton M. A decade of modeling Alzheimer’s disease in transgenic mice. Trends Genet. 2006;22:281–289. doi: 10.1016/j.tig.2006.03.007. [DOI] [PubMed] [Google Scholar]
- Morrison JH, Hof PR. Life and death of neurons in the aging brain. Science. 1997;278:412–419. doi: 10.1126/science.278.5337.412. [DOI] [PubMed] [Google Scholar]
- Mullan M, Crawford F, Axelman K, Houlden H, Lilius L, Winblad B, Lannfelt L. A pathogenic mutation for probable Alzheimer’s disease in the APP gene at the N-terminus of β-amyloid. Nat Genet. 1992;1:345–347. doi: 10.1038/ng0892-345. [DOI] [PubMed] [Google Scholar]
- Nalbantoglu J, Tirado-Santiago G, Lahsaini A, Poirier J, Goncalves O, Verge G, Momoli F, Welner SA, Massicotte G, Julien JP, Shapiro ML. Impaired learning and LTP in mice expressing the carboxy terminus of the Alzheimer amyloid precursor protein. Nature. 1997;387:500–505. doi: 10.1038/387500a0. [DOI] [PubMed] [Google Scholar]
- Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, Guillozet-Bongaarts A, Ohno M, Disterhoft J, Van Eldik L, Berry R, Vassar R. Intraneuronal β-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J Neurosci. 2006;26:10129–10140. doi: 10.1523/JNEUROSCI.1202-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno M. Genetic and pharmacological basis for therapeutic inhibition of β- and γ-secretases in mouse models of Alzheimer’s memory deficits. Rev Neurosci. 2006;17:429–454. doi: 10.1515/revneuro.2006.17.4.429. [DOI] [PubMed] [Google Scholar]
- Ohno M, Chang L, Tseng W, Oakley H, Citron M, Klein WL, Vassar R, Disterhoft JF. Temporal memory deficits in Alzheimer’s mouse models: rescue by genetic deletion of BACE1. Eur J Neurosci. 2006;23:251–260. doi: 10.1111/j.1460-9568.2005.04551.x. [DOI] [PubMed] [Google Scholar]
- Ohno M, Sametsky EA, Younkin LH, Oakley H, Younkin SG, Citron M, Vassar R, Disterhoft JF. BACE1 deficiency rescues memory deficits and cholinergic dysfunction in a mouse model of Alzheimer’s disease. Neuron. 2004;41:27–33. doi: 10.1016/s0896-6273(03)00810-9. [DOI] [PubMed] [Google Scholar]
- Pearson HA, Peers C. Physiological roles for amyloid β peptides. J Physiol. 2006;575:5–10. doi: 10.1113/jphysiol.2006.111203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pike CJ, Walencewicz AJ, Glabe CG, Cotman CW. In vitro aging of β-amyloid protein causes peptide aggregation and neurotoxicity. Brain Res. 1991;563:311–314. doi: 10.1016/0006-8993(91)91553-d. [DOI] [PubMed] [Google Scholar]
- Plant LD, Webster NJ, Boyle JP, Ramsden M, Freir DB, Peers C, Pearson HA. Amyloid β peptide as a physiological modulator of neuronal ‘A’-type K+ current. Neurobiol Aging. 2006;27:1673–1683. doi: 10.1016/j.neurobiolaging.2005.09.038. [DOI] [PubMed] [Google Scholar]
- Podlisny MB, Stephenson DT, Frosch MP, Tolan DR, Lieberburg I, Clemens JA, Selkoe DJ. Microinjection of synthetic amyloid β-protein in monkey cerebral cortex fails to produce acute neurotoxicity. Am J Pathol. 1993;142:17–24. [PMC free article] [PubMed] [Google Scholar]
- Roberds SL, Anderson J, Basi G, Bienkowski MJ, Branstetter DG, Chen KS, Freedman SB, Frigon NL, Games D, Hu K, Johnson-Wood K, Kappenman KE, Kawabe TT, Kola I, Kuehn R, Lee M, Liu W, Motter R, Nichols NF, Power M, Robertson DW, Schenk D, Schoor M, Shopp GM, Shuck ME, Sinha S, Svensson KA, Tatsuno G. BACE knockout mice are healthy despite lacking the primary β-secretase activity in brain: implications for Alzheimer’s disease therapeutics. Hum Mol Genet. 2001;10:1317–1324. doi: 10.1093/hmg/10.12.1317. [DOI] [PubMed] [Google Scholar]
- Roher AE, Ball MJ, Bhave SV, Wakade AR. β-Amyloid from Alzheimer disease brains inhibits sprouting and survival of sympathetic neurons. Biochem Biophys Res Commun. 1991;174:572–579. doi: 10.1016/0006-291x(91)91455-l. [DOI] [PubMed] [Google Scholar]
- Schmitz C, Rutten BP, Pielen A, Schafer S, Wirths O, Tremp G, Czech C, Blanchard V, Multhaup G, Rezaie P, Korr H, Steinbusch HW, Pradier L, Bayer TA. Hippocampal neuron loss exceeds amyloid plaque load in a transgenic mouse model of Alzheimer’s disease. Am J Pathol. 2004;164:1495–1502. doi: 10.1016/S0002-9440(10)63235-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ, Schenk D. Alzheimer’s disease: molecular understanding predicts amyloid-based therapeutics. Annu Rev Pharmacol Toxicol. 2003;43:545–584. doi: 10.1146/annurev.pharmtox.43.100901.140248. [DOI] [PubMed] [Google Scholar]
- Singer O, Marr RA, Rockenstein E, Crews L, Coufal NG, Gage FH, Verma IM, Masliah E. Targeting BACE1 with siRNAs ameliorates Alzheimer disease neuropathology in a transgenic model. Nat Neurosci. 2005;8:1343–1349. doi: 10.1038/nn1531. [DOI] [PubMed] [Google Scholar]
- Sinha S, Anderson JP, Barbour R, Basi GS, Caccavello R, Davis D, Doan M, Dovey HF, Frigon N, Hong J, Jacobson-Croak K, Jewett N, Keim P, Knops J, Lieberburg I, Power M, Tan H, Tatsuno G, Tung J, Schenk D, Seubert P, Suomensaari SM, Wang S, Walker D, Zhao J, McConlogue L, John V. Purification and cloning of amyloid precursor protein β-secretase from human brain. Nature. 1999;402:537–540. doi: 10.1038/990114. [DOI] [PubMed] [Google Scholar]
- Sisodia SS, St George-Hyslop PH. γ-Secretase, Notch, Aβ and Alzheimer’s disease: where do the presenilins fit in? Nat. Rev Neurosci. 2002;3:281–290. doi: 10.1038/nrn785. [DOI] [PubMed] [Google Scholar]
- Song DK, Won MH, Jung JS, Lee JC, Kang TC, Suh HW, Huh SO, Paek SH, Kim YH, Kim SH, Suh YH. Behavioral and neuropathologic changes induced by central injection of carboxyl-terminal fragment of β-amyloid precursor protein in mice. J Neurochem. 1998;71:875–878. doi: 10.1046/j.1471-4159.1998.71020875.x. [DOI] [PubMed] [Google Scholar]
- Turner PR, O’Connor K, Tate WP, Abraham WC. Roles of amyloid precursor protein and its fragments in regulating neural activity, plasticity and memory. Prog Neurobiol. 2003;70:1–32. doi: 10.1016/s0301-0082(03)00089-3. [DOI] [PubMed] [Google Scholar]
- Urbanc B, Cruz L, Le R, Sanders J, Ashe KH, Duff K, Stanley HE, Irizarry MC, Hyman BT. Neurotoxic effects of thioflavin S-positive amyloid deposits in transgenic mice and Alzheimer’s disease. Proc Natl Acad Sci USA. 2002;99:13990–13995. doi: 10.1073/pnas.222433299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, Teplow DB, Ross S, Amarante P, Loeloff R, Luo Y, Fisher S, Fuller J, Edenson S, Lile J, Jarosinski MA, Biere AL, Curran E, Burgess T, Louis JC, Collins F, Treanor J, Rogers G, Citron M. β-Secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999;286:735–741. doi: 10.1126/science.286.5440.735. [DOI] [PubMed] [Google Scholar]
- von Arnim CA, Kinoshita A, Peltan ID, Tangredi MM, Herl L, Lee BM, Spoelgen R, Hshieh TT, Ranganathan S, Battey FD, Liu CX, Bacskai BJ, Sever S, Irizarry MC, Strickland DK, Hyman BT. The low density lipoprotein receptor-related protein (LRP) is a novel β-secretase (BACE1) substrate. J Biol Chem. 2005;280:17777–17785. doi: 10.1074/jbc.M414248200. [DOI] [PubMed] [Google Scholar]
- Willem M, Garratt AN, Novak B, Citron M, Kaufmann S, Rittger A, Destrooper B, Saftig P, Birchmeier C, Haass C. Control of peripheral nerve myelination by the β-secretase BACE1. Science. 2006;314:664–666. doi: 10.1126/science.1132341. [DOI] [PubMed] [Google Scholar]
- Wong HK, Sakurai T, Oyama F, Kaneko K, Wada K, Miyazaki H, Kurosawa M, De Strooper B, Saftig P, Nukina N. β Subunits of voltage-gated sodium channels are novel substrates of β-site amyloid precursor protein-cleaving enzyme (BACE1) and γ-secretase. J Biol Chem. 2005;280:23009–23017. doi: 10.1074/jbc.M414648200. [DOI] [PubMed] [Google Scholar]
- Yan R, Bienkowski MJ, Shuck ME, Miao H, Tory MC, Pauley AM, Brashier JR, Stratman NC, Mathews WR, Buhl AE, Carter DB, Tomasselli AG, Parodi LA, Heinrikson RL, Gurney ME. Membrane-anchored aspartyl protease with Alzheimer’s disease β-secretase activity. Nature. 1999;402:533–537. doi: 10.1038/990107. [DOI] [PubMed] [Google Scholar]
- Yang LB, Lindholm K, Yan R, Citron M, Xia W, Yang XL, Beach T, Sue L, Wong P, Price D, Li R, Shen Y. Elevated β-secretase expression and enzymatic activity detected in sporadic Alzheimer disease. Nat Med. 2003;9:3–4. doi: 10.1038/nm0103-3. [DOI] [PubMed] [Google Scholar]
- Yankner BA. Mechanisms of neuronal degeneration in Alzheimer’s disease. Neuron. 1996;16:921–932. doi: 10.1016/s0896-6273(00)80115-4. [DOI] [PubMed] [Google Scholar]
- Yankner BA, Caceres A, Duffy LK. Nerve growth factor potentiates the neurotoxicity of β amyloid. Proc Natl Acad Sci USA. 1990a;87:9020–9023. doi: 10.1073/pnas.87.22.9020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yankner BA, Dawes LR, Fisher S, Villa-Komaroff L, Oster-Granite ML, Neve RL. Neurotoxicity of a fragment of the amyloid precursor associated with Alzheimer’s disease. Science. 1989;245:417–420. doi: 10.1126/science.2474201. [DOI] [PubMed] [Google Scholar]
- Yankner BA, Duffy LK, Kirschner DA. Neurotrophic and neurotoxic effects of amyloid β protein: reversal by tachykinin neuropeptides. Science. 1990b;250:279–282. doi: 10.1126/science.2218531. [DOI] [PubMed] [Google Scholar]
- Yu H, Saura CA, Choi SY, Sun LD, Yang X, Handler M, Kawarabayashi T, Younkin L, Fedeles B, Wilson MA, Younkin S, Kandel ER, Kirkwood A, Shen J. APP processing and synaptic plasticity in presenilin-1 conditional knockout mice. Neuron. 2001;31:713–726. doi: 10.1016/s0896-6273(01)00417-2. [DOI] [PubMed] [Google Scholar]