

Abstract
In addition to inhibiting the mitochondrial respiratory chain, toxins known to cause Parkinson's disease (PD), such as 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine and rotenone, also strongly depolymerize microtubules and increase tubulin degradation. Microtubules are polymers of tubulin α/β heterodimers, whose correct folding requires coordinated actions of cellular chaperonins and cofactors. Misfolded tubulin monomers are highly toxic and quickly degraded through a hitherto unknown mechanism. Here we report that parkin, a protein–ubiquitin E3 ligase linked to PD, was tightly bound to microtubules in taxol-mediated microtubule coassembly assays. In lysates from the rat brain or transfected human embryonic kidney (HEK) 293 cells, α-tubulin and β-tubulin were strongly coimmunoprecipitated with parkin at 4°C in the presence of colchicine, a condition in which tubulin exits as α/β heterodimers. At the subcellular level, parkin exhibited punctate immunostaining along microtubules in rat brain sections, cultured primary neurons, glial cells, and cell lines. This pattern of subcellular localization was abolished in cells treated with the microtubule-depolymerizing drug colchicine. The binding between parkin and tubulin apparently led to increased ubiquitination and accelerated degradation of α- and β-tubulins in HEK293 cells. Similarly ubiquitinated tubulins were also observed in rat brain lysates. Furthermore, parkin mutants found in PD patients did not ubiquitinate or degrade either tubulin. Taken together, our results show that parkin is a novel tubulin-binding protein, as well as a microtubule-associated protein. Its ability to enhance the ubiquitination and degradation of misfolded tubulins may play a significant role in protecting neurons from toxins that cause PD.
Keywords: parkin, Parkinson's disease, tubulin, ubiquitination, misfolding, microtubule, dopamine
Introduction
Microtubules play critical roles in diverse cellular functions. The polymerization of tubulin α/β heterodimers into microtubules is a dynamically regulated process that is influenced by many factors, among which the concentration of free tubulin heterodimers is the driving force. The synthesis of α- and β-tubulins is coordinated such that excess tubulin monomers are never produced in any significant amount, because the overexpression of either tubulin gene alone is toxic to the cell (Burke et al., 1989;Weinstein and Solomon, 1990). After translation, the correct folding of tubulin monomers and the formation of functional α/β heterodimers require a series of coordinated actions of cellular chaperonins and cofactors (for review, see Lewis et al., 1997).
The complex and reversible nature of the folding process for α/β tubulin heterodimers, in addition to the mechanisms that regulate the synthesis of tubulin polypeptides, make it unavoidable for the cell to produce misfolded tubulin monomers, which are quickly degraded through an unknown mechanism to prevent cytotoxicity. Several lines of evidence indicate that misfolded tubulin may be involved in Parkinson's disease (PD). First, both ubiquitin (Lowe et al., 1988; Mayer et al., 1989) and tubulin (Galloway et al., 1992) are major components of the Lewy body, suggesting that ubiquitinated tubulin may be present in this histological hallmark of Parkinson's disease. Second, 1-methyl-4-phenylpyridinium (MPP+), a neurotoxin that kills dopamine (DA) neurons and induces PD-like symptoms (Langston et al., 1983; Przedborski and Jackson-Lewis, 1998), depolymerizes microtubules in PC12 cells (Cappelletti et al., 1999) as well as in vitro (Cappelletti et al., 2001). Third, the long-term systemic administration of rotenone leads to selective degeneration of nigral DA neurons and locomotor problems resembling PD (Betarbet et al., 2000). In addition to its widely recognized ability to inhibit mitochondrial complex I (Higgins and Greenamyre, 1996), rotenone also potently depolymerizes microtubules in vivoand in vitro (Brinkley et al., 1974), by binding to the colchicine site on tubulin heterodimers (Marshall and Himes, 1978). Because the depolymerization of microtubules leads to increased tubulin degradation (Cleveland, 1989), the cellular mechanism for the degradation of tubulin in response to these neurotoxins may play a role in PD.
Parkin, a gene linked to autosomal recessive juvenile PD (AR-JP) (Kitada et al., 1998) has been found to be a protein–ubiquitin E3 ligase (Shimura et al., 2000). A variety of mutations exist in the parkin gene of patients with autosomal recessive (Lucking et al., 2000) or sporadic PD (Scott et al., 2001). Many of the mutations appear to be clustered in the ubiquitin-like domain and the RING (Really Interesting New Gene) finger domains (Giasson and Lee, 2001), suggesting that the E3 ligase activity of parkin is crucial for PD. Although previous studies have identified several substrates of parkin in the cell (Zhang et al., 2000; Chung et al., 2001; Imai et al., 2001;Shimura et al., 2001), it is clear that parkin may have additional substrates that contribute to the degeneration of nigral DA neurons. Here we report the discovery of parkin as a novel tubulin-binding protein, as well as an E3 ligase for α- and β-tubulins.
Materials and Methods
Antibodies and cDNAs. Polyclonal antibodies against parkin (P304 and P124) were generated by immunizing rabbits with KLH-conjugated peptides derived from mouse parkin sequence (amino acids 304–322 and 124–141, respectively). Antisera were purified by affinity chromatography using the same peptide immobilized on SulfoLink gel matrix (Pierce, Rockford, IL) according to the manufacturer's protocol. Monoclonal antibodies against synaptophysin (SVP-38), α-tubulin (DM1A), β-tubulin (TUB2.1), FLAG (M2), anti-FLAG-conjugated (M2) agarose, and rhodamine-conjugated phalloidin were purchased from Sigma (St. Louis, MO). Anti-ubiquitin, anti-microtubule-associated protein 1A (MAP1A), and rhodamine- or FITC-conjugated secondary antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Monoclonal anti-hemagglutinin was purchased from Roche (Indianapolis, IN). Monoclonal anti-postsynaptic density 95 (PSD-95) was purchased from Affinity BioReagents (Golden, CO). TO-PRO-3 (a DNA-binding dye) was purchased from Molecular Probes (Eugene, OR). Monoclonal anti-MAP2 was purchased from Upstate Biotechnology (Lake Placid, NY). Mouse parkin cDNA was amplified by reverse transcription PCR from brain total RNA. It was completely sequenced to ensure that no mutation was introduced by PCR. The FLAG epitope tag was added to the 5′ end by PCR, and the tagged cDNA was subcloned into pcDNA3.1. The HA-tagged ubiquitin construct was described previously (Wang et al., 2000).
Taxol-mediated microtubule coassembly assay. The experiments were performed according to the protocol of Vallee (1986). Briefly, 3-week-old male Sprague Dawley rats were decapitated after being anesthetized with halothane (Sigma). A whole brain was homogenized in PEM buffer (0.1 m PIPES, 1 mm EGTA, and 1 mmMgSO4) on ice, centrifuged at 4°C first at 30,000 × g for 15 min then at 180,000 ×g for 90 min. The supernatant fraction contained the cytosol (C). Taxol (Sigma) and GTP were added to C to a final concentration of 20 μm and 1 mm, respectively. The solution was warmed up to 37°C and centrifuged through a layer of sucrose. The supernatant fraction was designated S1, and the pellet was washed with PEM buffer and resuspended in PEM buffer contain taxol and GTP at 37°C (P1), which was centrifuged again. The supernatant fraction was designated S2, and the pellet was washed with PEM buffer and resuspended in PEM buffer containing taxol and GTP at 37°C (P2), which was separated into three equal portions. Each portion was mixed with one-tenth, one-third, or one-half volume of MAP dissociation buffer (PEM + taxol + GTP + 4 m NaCl) at 37°C to elute MAPs at 0.36, 1, or 2 m of NaCl, respectively. After the mixtures were centrifuged, the supernatant fractions were designated S3, S4, and S5, respectively, and pellet fractions were rinsed in PEM buffer at 37°C and resuspended in PEM buffer at 4°C to depolymerize microtubules (P3, P4, and P5, respectively). Equal amounts of total proteins (10 μg) from each fraction were boiled and separated on 7.5% SDS–polyacrylamide gel and analyzed by Western blots with antibodies against MAP1A, MAP2, parkin and α-tubulin, respectively.
Transfection, immunoprecipitation, and Western blot. Human embryonic kidney (HEK) 293, SH-SY5Y, and BE(2)C cells were purchased from American Type Culture Collection (Manassas, VA). They were maintained in DMEM with 10% FCS and antibiotics. Transfection of various constructs was performed using Fugene 6 (Roche, Indianapolis, IN) according to the manufacturer's protocol. Sixty hours after transfection, cells cultured in 10 cm dishes were lysed on ice in cold lysis buffer (containing the following: 1% Triton X-100, 10 mm Tris pH 7.6, 50 mmNaCl, 30 mm sodium pyrophosphate, 50 mm NaF, 5 mm EDTA, and 0.1 mmNa3VO4) for 20 min. Lysates were centrifuged at 4°C at 16,000 × g and supernatant fractions were incubated with antibody against α- or β-tubulin for 1 hr at 4°C, followed by incubation with protein A/G plus agarose (Santa Cruz Biotechnology) at the same condition. Immunoprecipitates were washed three times with the lysis buffer, then boiled in 2× SDS loading buffer for 5 min and separated on 7.5% SDS-polyacrylamide gel. Half of the immunoprecipitates were for Western blot with anti-HA, and the other half for Western blot with anti-α-tubulin or anti-β-tubulin (loading control). Western blots were carried out using the ECL method according to the manufacturer's protocol (Amersham Biosciences, Piscataway, NJ). In some experiments, cell lysates containing equal amounts of total protein (100 μg) were boiled in sample buffer, separated on 7.5% SDS-polyacrylamide gel, and analyzed by Western blot with anti-HA or anti-FLAG. For experiments using rat brain homogenates, one whole brain was homogenized in 15 ml of ice-cold lysis buffer on ice in a tissue grinder (Fisher Scientific, Pittsburgh, PA). The homogenate was centrifuged at 16,000 × g for 20 min and ultracentrifuged at 338,000 × g for 30 min. The supernatant fraction was used in immunoprecipitation or Western blot as described above.
Immunohistochemistry. Adult male Sprague Dawley rats were anesthetized and transcardially perfused with PBS, followed by 4% paraformaldehyde (PFA; Sigma) in PBS. Fixed brain chucks were cut on a cryostat to obtain sagittal sections (14 μm thick), which were floated on PBS and picked up with slides precoated with 0.2% polyethylenimine (Sigma). After the sections were air-dried, they were permeablized in 0.1% Triton X-100 in PBS for 15 min, then blocked with 3% BSA for 1 hr. This was followed by incubation in primary antibodies (anti-parkin and anti-β-tubulin) for 1 d at 4°C and secondary antibodies (rhodamine-conjugated goat anti-rabbit IgG and FITC-conjugated goat anti-mouse IgG) for another day at 4°C. Slides were counterstained with the DNA-binding dye TO-PRO-3 (1:1000 in PBS, emitting fluorescence in the Cy5 range) for 5 min, and mounted with VectaShield (Vector Laboratories, Burlingame, CA).
Embryonic neuronal culture and immunocytochemistry. Primary neuronal cultures were performed with brains from rat embryos at embryonic day 18. The cortex, hippocampus, or midbrain (containing substantia nigra) was dissected from each embryo for separate cultures. Minced brain tissue was digested in CGBD buffer (0.2 mg/ml dl-cysteine hydrochloride monohydrate, 5 mg/ml glucose, 0.2 mg/ml BSA, and 0.01 mg/ml DNaseI in PBS) containing 9 U/ml of papain (Worthington Biochemical, Lakewood, NJ). Dissociated neurons were plated on coverslips in 12-well plates at a density of 1 × 105cells/well in Neurobasal media supplemented with B27 (Invitrogen, Carlsbad, CA). Cytosine arabinoside (1 mm) was added to culture media from the fourth day to inhibit glia growth and the medium was changed every 4–5 d. Coverslips were precoated with 0.2% polyethylenimine in 0.15m of sodium borate, pH 8.4. Neurons cultured for 3 weeks were fixed in 4% PFA for 15 min at room temperature or cold methanol for 20 min at −20°C. PFA-fixed neurons were permeablized in 0.1% Triton X-100 in PBS for 15 min. Immunostaining was the same as the procedures described for immunohistochemistry.
Confocal microscopy and image analysis. Immunofluorescence images were acquired on a confocal microscope from Bio-Rad (Hercules, CA). Monochrome images (512 × 512 pixels) were pseudocolored and merged with the software Confocal Assistant (freeware by Todd Clark Brelje). Colocalization of signals was assessed by the colocalization analysis function in the LaserSharp software (Bio-Rad). The degree of colocalization is expressed in Pearson's correlation coefficient, which calculates, for example, the proportion of all red intensities that have green components among all red intensities (Manders et al., 1993).
Results
Parkin coassembles with microtubules
To identify substrates of parkin, we performed yeast two-hybrid screening with a mouse brain cDNA library. MAP1A came up as a positive clone in several independent experiments. However, the binding between parkin and MAP1A in coimmunoprecipitation assays was quite weak and apparently dependent on the temperature during immunoprecipitation. The amount of MAP1A coimmunoprecipitated with parkin at 37°C was significantly more than that at 4°C (Feng and Lin, 2001). This result hints that parkin may bind to MAP1A indirectly, through microtubules, because the polymerization of microtubules is temperature-dependent and optimal at 37°C.
To explore this possibility, we carried out taxol-mediated microtubule coassembly experiments using ultracentrifuged rat brain homogenate (Vallee, 1986). As shown in Figure1, α-tubulin and two known MAPs (MAP1A and MAP2) were highly enriched in the pellet fractions containing microtubules (P1 and P2), compared with the soluble fractions (S1 and S2). Parkin was found exclusively in the pellet fractions, indicating that it was bound to microtubules rather than freely floating in the cytosol. When the pellets in P2 were incubated at 37°C in MAP dissociation buffer with varying concentrations of NaCl, MAP1A and MAP2 were eluted into the supernatant fractions (S3, S4, and S5) at 0.36m of NaCl or above. In contrast, parkin could not be dissociated from the pellet fractions even with 2 m of NaCl (P5). These results show that parkin binds to microtubules much more tightly than MAP1A or MAP2 does, perhaps through hydrophobic rather than electrostatic interactions.
Fig. 1.

Coassembly of parkin and microtubules. The cytosolic fraction (C) from ultracentrifuged rat brain homogenate was subjected to two cycles of taxol-mediated assembly. Supernatant and pellet fractions from each cycle were designated as S1, P1, S2, and P2, respectively. The P2 pellet was resuspended and separated into three equal parts, each incubated at 37°C in MAP dissociation buffer containing 0.36, 1, or 2m of NaCl, respectively, to release MAPs into the supernatant fractions (S3, S4, or S5, respectively), whereas the pellet fractions (P3, P4, or P5, respectively) contained mostly microtubules. Ten micrograms of total proteins from each fraction were analyzed by Western blots with antibodies against MAP1A, MAP2, α-tubulin, or parkin, respectively. Note that parkin was always in the pellet fractions, in which microtubules were enriched. The large space in lane S5 on α-tubulin and parkin blots was attributable to distortion caused by high salt concentration of the samples as they migrate to the bottom. The faint signal of MAP1A in C was attributable to its relatively low abundance in the 10 μg of total cytosolic proteins loaded. The experiment was repeated three times, each with similar results.
To ensure the specificity of the parkin antibody used in our studies, we performed Western blots on lysates from the rat brain and HEK293 cells transfected with or without FLAG-tagged parkin. Of the five antibodies that we generated against peptides derived from different regions of parkin, the P304 antibody recognized parkin specifically. As shown in Figure 2, this antibody (PK) detected the same 52 kDa band in the samples, without recognizing anything else. The identity of this band was confirmed by the transfected FLAG-tagged mouse parkin, which migrated at a slightly higher position because of the epitope tag. Preincubation of the antibody with its antigenic peptide completely eliminated the signals, further demonstrating the specificity of this antibody.
Fig. 2.

Generation of a monospecific antibody against parkin. Rat brain homogenates (B) and lysates from HEK293 cells transfected without (C) or with (P) FLAG-tagged mouse parkin were Western blotted (WB) with the antibody against parkin, or the same antibody preincubated with the antigenic peptide, or anti-FLAG. m, Prestained molecular weight marker. The amount of total proteins loaded in lanes B and C was 100 μg, whereas that in lane P was 5 μg, because FLAG-parkin was overexpressed. The experiment was repeated three times, each with similar results.
Parkin binds to α/β tubulin heterodimers
The strong affinity between parkin and microtubules and the fact that very little traditional MAP can remain bound to microtubules in the presence of 2 m NaCl made us suspect that parkin may bind to tubulin directly. To test this hypothesis, we performed coimmunoprecipitation experiments in rat brain lysate. Rat brains were homogenized in a lysis buffer containing 1% Triton X-100, without GTP or taxol. After ultracentrifugation, the supernatant factions were incubated at 4°C in the absence or presence of 25 μm of colchicine for 15 min; then they were immunoprecipitated with the parkin antibody preincubated with or without the antigenic peptide. The immunoprecipitates were analyzed by Western blots with monoclonal antibodies against α- or β-tubulin, respectively. As shown in Figure 3A, both α- and β-tubulins were strongly coimmunoprecipitated with parkin. The signals were completely eliminated when the parkin antibody was preincubated with its antigenic peptide, confirming that the coimmunoprecipitation was indeed caused by parkin. The binding between parkin and α- or β-tubulin was not affected by colchicine treatment at 4°C, indicating that parkin binds to tubulin α/β heterodimers, which are the predominant form of tubulin at this condition.
Fig. 3.
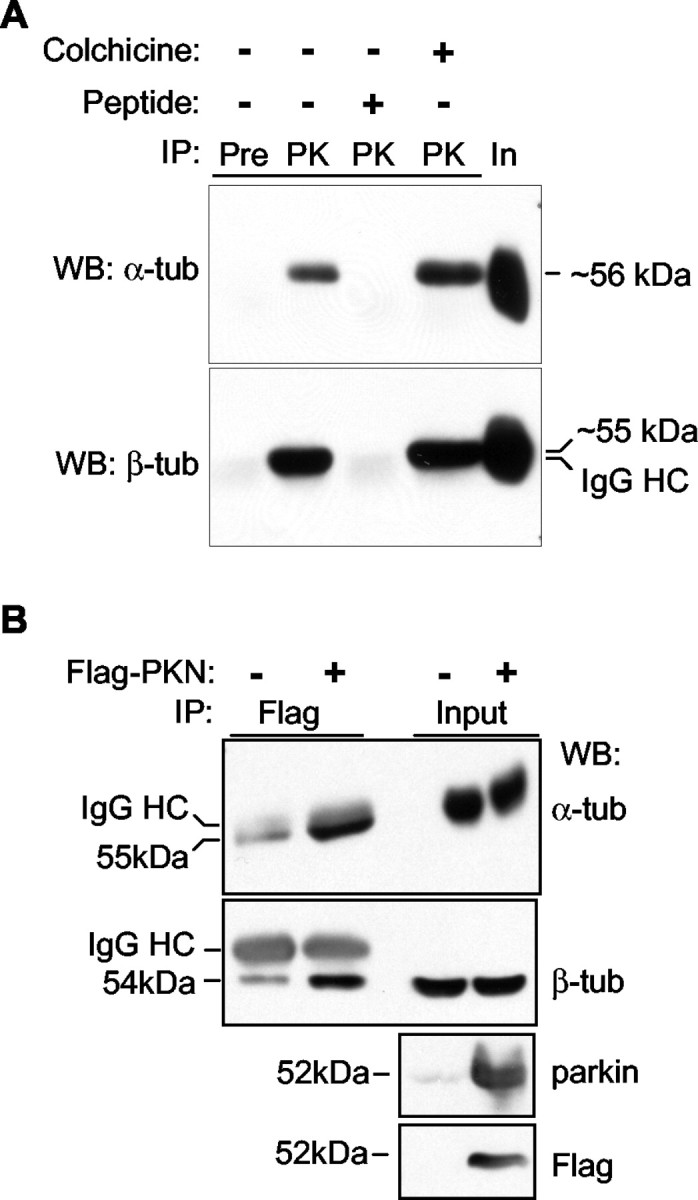
The binding between parkin and α/β tubulins.A, Ultracentrifuged rat brain homogenates were incubated in the absence or presence of 25 μm of colchicine at 4°C to ensure the depolymerization of microtubules. They were then immunoprecipitated (IP) with preimmune serum (Pre) or the antibody against parkin (PK) in the absence or presence of the antigenic peptide. The immunoprecipitates and 1% of input (In) were separated by SDS-PAGE, and blotted with antibodies against α- or β-tubulin, respectively. Both tubulins were coimmunoprecipitated with parkin. HC, Heavy chain. B, Cleared lysates from HEK293 cells transfected with or without FLAG-tagged parkin were immunoprecipitated with anti-FLAG. Precipitated proteins and 1% of input were analyzed by Western blotting with antibodies against α- or β-tubulin, respectively. The lysates were also blotted with anti-parkin and anti-FLAG to show the expression level of transfected parkin. All experiments were repeated at least three times.
Similar results were obtained when we transfected FLAG-tagged parkin into HEK293 cells and examined its interaction with endogenous α- and β-tubulins. As shown in Figure 3B, the expression of FLAG-tagged parkin led to a significant increase in its coimmunoprecipitation with α- or β-tubulin, respectively. The background level of coimmunoprecipitated tubulins in untransfected cells may be attributable to nonspecific binding between tubulins and protein A/G-plus agarose used in immunoprecipitation.
Parkin is localized along microtubules in a punctate manner
The strong binding between parkin and microtubules suggests that they may be colocalized. To test this, we costained rat embryonic cortical neurons cultured for 21 d in vitro with anti-parkin (Fig. 4A), anti-α-tubulin (Fig. 4B), and the DNA-binding dye TO-PRO-3 (merged in Fig. 4C). The punctate subcellular localization of parkin largely coincided with microtubules, as shown by the strong yellow signals, especially in the processes (Fig.4C and inset). Colocalization analysis indicated that 98% of the parkin signals coincided with α-tubulin signals, whereas 70% of α-tubulin signals coincided with those of parkin (Fig.4D). On the other hand, neither parkin nor α-tubulin signals were in the nucleus (colocalization coefficient, 0.04) (Fig. 4E,F), which is consistent with their colocalization and the well-established absence of microtubules in the nucleus. To ensure the specificity of the parkin antibody, we stained cultured neurons with the primary antibodies preincubated with the parkin antigenic peptide and obtained no significant signal from the rhodamine channel (Fig. 4G), whereas signals from other channels were intact (data not shown). The cultures that we used also contained a small number of glial cells. The subcellular localization of parkin in these cells was the same as that in neurons: punctate dots along microtubules (Fig. 4Hand inset). In addition, we have cultured neurons from different brain regions, such as the substantia nigra and hippocampus. There was no difference in the subcellular localization of parkin in neurons from different regions, nor was there any difference between dopaminergic neurons and nondopaminergic neurons (data not shown). To ascertain whether what we saw in cultured neurons represented the situation in the brain, we costained adult rat brain sections with anti-parkin (red), anti-β-tubulin (green), and TO-PRO-3 (blue). As shown in the merged image in Figure 4I, parkin immunostaining was punctate, decorating along β-tubulin signals, which were most prominent in the processes. Thus, the subcellular localization of parkin appears very similar in culture and in vivo: punctate dots along microtubules.
Fig. 4.
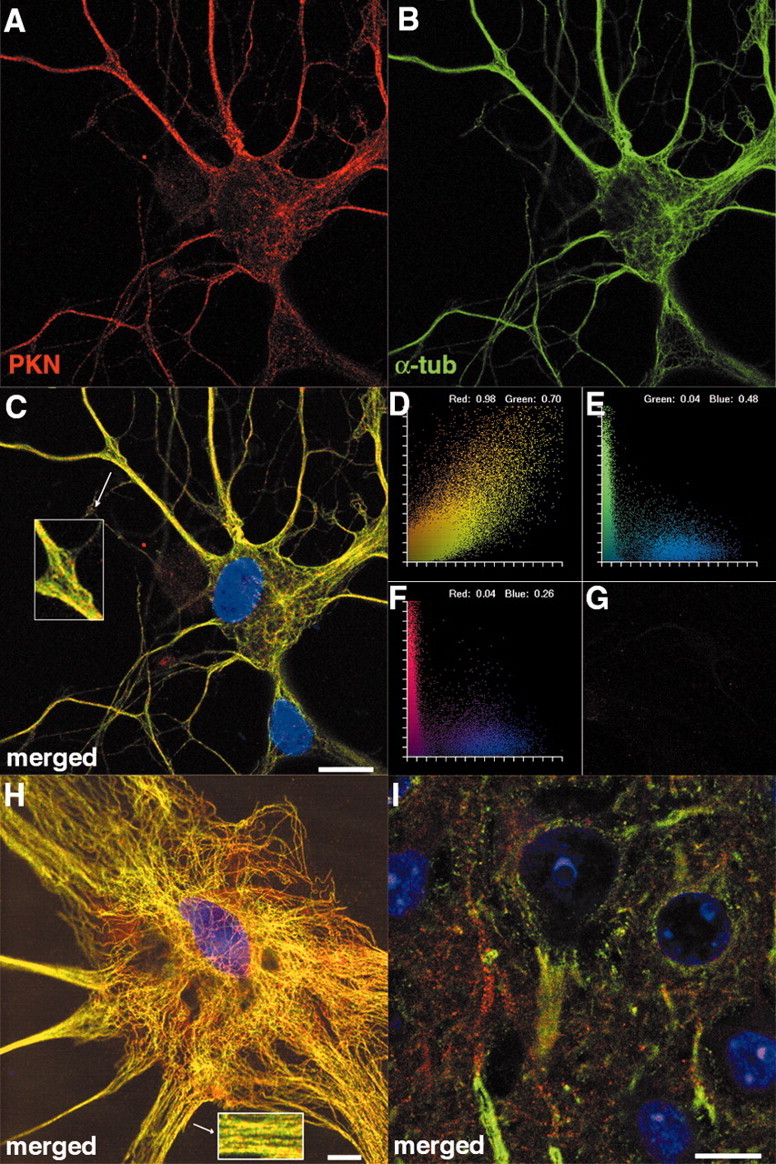
Punctate subcellular localization of parkin along microtubules in rat neurons and glial cells. Cultured rat embryonic cortical neurons were costained with anti-parkin (A), anti-α-tubulin (B), and the DNA-binding dye TO-PRO-3. Confocal images from rhodamine channel (parkin, red), FITC channel (α-tubulin, green), and Cy5 channel (DNA, blue) were merged (C). Inset, Enlarged portion of the composite showing punctate staining of parkin along microtubules. D, Fluorogram showing the degree of colocalization between red signals (parkin) and green signals (microtubule). Colocalization coefficients indicate that 98% of the red signals have green components, whereas 70% of the green signals have red components. E, Fluorogram showing the degree of colocalization between green signals (α-tubulin) and blue signals (DNA). The lack of α-tubulin in the nucleus is indicated by the small coefficient (0.04). F, Fluorogram showing the degree of colocalization between red signals (parkin) and blue signals (DNA). The lack of parkin in the nucleus is indicated by the small coefficient (0.04). G, No significant signal was observed in the rhodamine channel when the parkin antigenic peptide was mixed with the primary antibodies, demonstrating the specificity of parkin staining. Signals from other channels were unaffected (data not shown).H, Merged images of a glial cell in the same culture showing punctate staining of parkin (red) along microtubules (α-tubulin, green). Inset, Enlarged portion of the composite.I, Merged images of an adult rat brain section showing punctate staining of parkin (red) along microtubules (β-tubulin, green). Scale bars, 10 μm. At least 10 sets of images were taken from 5 coverslips, all with similar results.
To confirm that parkin puncta are indeed localized on microtubules, we treated rat neuronal cultures (Fig.5A,B), human neuroblastoma cell line SH-SY5Y (Fig.5C,D), and mouse fibroblast cell line NIH 3T3 (Fig. 5E,F) with or without the microtubule-depolymerizing drug colchicine (25 μm for 12 hr). The treatment changed the subcellular localization of parkin from puncta along microtubules (Fig.5A,C,E) to diffusely cytosolic (Fig.5B,D,F). Taken together with other lines of evidence described above, it demonstrates that parkin puncta are indeed associated with microtubules. The data also show that the microtubule-associated, punctate subcellular localization of parkin is a general phenomenon in a variety of cell types, including neurons, glial cells (Fig. 5A), and drastically different cell lines (Fig. 5C,E), despite the varying level of its expression.
Fig. 5.

Disruption of the punctate subcellular localization of parkin by colchicine. Primary rat neuronal cultures (A, B), human neuroblastoma cell line SH-SY5Y (C, D) and mouse fibroblast cell line NIH 3T3 were treated with (+Col, B, D, F) or without (−Col, A, C, E) colchicine, and costained with anti-parkin (red), anti-α-tubulin (green) and TO-PRO-3 (blue). Colchicine depolymerized microtubules and changed the localization pattern of parkin from punctate to diffusely cytosolic. Insets, Enlarged portion of the main image showing punctate localization of parkin along microtubules. Scale bar, 10 μm.
A previous report (Fallon et al., 2002) showed that parkin was colocalized with calmodulin-sensitive kinase in a punctate manner in postsynaptic densities (PSDs). To ascertain whether the punctate staining pattern of parkin that we saw was PSD or not, we costained rat cortical neurons with antibodies against parkin and PSD-95 (a marker for PSD). As shown in Figure6A, the subcellular localization of parkin and PSD95 appeared to be separated, with almost no yellow spots. Furthermore, using our parkin antibody (P304) in Western blots, we saw that the majority of parkin was in the Triton X-100–soluble fraction, rather than the pellet fraction, which contains PSD. To provide more evidence, we costained culture neurons with rhodamine-conjugated phalloidin and anti-parkin. In mature neurons, the actin-binding compound phalloidin recognizes dendritic spines, where most PSDs are localized. The lack of overlapping between phalloidin puncta and parkin staining in Figure 6Bshowed that parkin was not colocalized with PSD. These results, combined with the same pattern of the subcellular localization of parkin in non-neuronal cells which do not have PSD (Fig.5C,E), demonstrate that parkin puncta are not in the PSD. Costaining of parkin and synaptophysin showed that parkin was not colocalized with presynaptic terminals either (Fig. 6C), which is consistent with previous reports (Fallon et al., 2002).
Fig. 6.

Parkin is not colocalized with PSDs or presynaptic terminals. A, Cultured rat cortical neurons were stained with antibodies against parkin (PKN) and PSD95. Confocal images from rhodamine channel (parkin, red) and FITC channel (PSD95, green) were pseudocolored and merged. B, Cultured rat cortical neurons were stained with rhodamine-conjugated phalloidin (Pha) and anti-parkin (PKN). Confocal images from the rhodamine channel (staining actin in spines, red) and FITC channel (parkin, green) were pseudocolored and merged. C, Cultured rat cortical neurons were stained with antibodies against parkin and synaptophysin (SYN). Confocal images from the rhodamine channel (parkin, red) and FITC channel (synaptophysin, green) were pseudocolored and merged. Insets, Enlarged portions of the main pictures showing lack of colocalization of signals.
In addition to the monospecific antibody against parkin (P304), we also used another antibody, P124, which recognized parkin as a major band and a few minor bands as well on Western blots. Nevertheless, the P124 antibody gave similar results with regard to the subcellular localization of parkin, coassembly with microtubules and binding to tubulin heterodimers, albeit with higher background noise (data not shown).
Parkin enhances ubiquitination of α- and β-tubulins
The above results show that parkin binds to microtubules and tubulin and is localized in a punctate manner along microtubules. Because parkin is an E3 protein–ubiquitin ligase, it is possible that parkin may enhance the ubiquitination of tubulin. To investigate this possibility, we cotransfected HA-tagged ubiquitin without or with FLAG-tagged parkin into HEK293 cells. Cell lysates were immunoprecipitated with antibodies against α-tubulin (Fig.7A) or β-tubulin (Fig.7C). Precipitated proteins were Western-blotted with anti-HA to examine the ubiquitination of endogenous α- or β-tubulin. As shown in Figure 7A, the overexpression of parkin led to an increased amount of polyubiquitinated α-tubulin. When the cells were treated with lactacystin (1 μm for 12 hr), which specifically inhibits proteases in the 26S proteasome (Fenteany and Schreiber, 1998), an additional increase in the amount of polyubiquitinated α-tubulin was observed. Lactacystin treatment alone (lane 5) did not cause any significant increase over the basal level of polyubiquitinated α-tubulin (lane 2), which may be produced by either endogenous parkin or other E3 ligases in HEK293 cells. Similar effects were observed for β-tubulin in Figure 7C. The same set of α- or β-tubulin immunoprecipitates were also separated side by side with cell lysate and blotted with antibodies against α-tubulin (Fig.7B) or β-tubulin (Fig. 7D). The amount of α- or β-tubulins immunoprecipitated were the same across the lanes, respectively. These results show that parkin enhances the ubiquitination of α- and β-tubulins in HEK293 cells. To understand the full extent of the E3 ligase activity of parkin, Western blots using anti-HA were performed on total cell lysates from the same set of samples. As shown in Figure 7E, parkin enhanced the ubiquitination of many proteins in HEK293 cells (lane 3 vs lane 2). An equal expression level of transfected parkin was shown in the anti-FLAG blot in Figure 7F.
Fig. 7.

Parkin enhances the ubiquitination of α- and β-tubulins in HEK293 cells. HEK293 cells were transfected without or with HA-tagged ubiquitin (HA-Ub) and Flag-tagged parkin (F-PKN), and were treated without or with 1 μm lactacystin (Lac) for 12 hr. Cleared cell lysates were immunoprecipitated with antibodies against α-tubulin (A, B) or β-tubulin (C, D). Half of the precipitates were blotted with anti-HA (A, C), whereas the other half were blotted with anti-α-tubulin (B) or anti-β-tubulin (D). Cell lysates (10 μg total proteins, lane 6 in B and D) were loaded next to the lanes containing tubulin immunoprecipitates to indicate the position of α- or β-tubulin (arrow). Ubiquitinated tubulin proteins are marked with a bracket. *IgG heavy chain. The same set of cell lysates (100 μg total proteins in each lane) were also blotted with anti-HA (E) to show ubiquitinated proteins, or with anti-FLAG (F) to show the expression level of transfected parkin; #, nonspecific bands recognized by anti-HA in total cell lysates.
To ascertain whether the ubiquitination of α- and β-tubulin observed in HEK293 cells occurs under normal physiological conditions in the brain, we immunoprecipitated α- or β-tubulin from ultracentrifuged rat brain lysates and analyzed the precipitated proteins by Western blotting with anti-ubiquitin. As shown in the left panel of Figure 8, the α-tubulin antibody immunoprecipitated two proteins (bands a and c) that can be recognized by both anti-ubiquitin and anti-α-tubulin. The size of the two bands suggests that a is probably penta-ubiquitinated α-tubulin and c is probably partially degraded ubiquitinated α-tubulin. The ability of the α-tubulin antibody to immunoprecipitate α-tubulin itself was demonstrated by the presence of band b in the α-tubulin blot, which migrated at the same size as the band recognized by the Western blot of the rat brain lysate. Similar results were obtained for β-tubulin in Figure 8(right). Distinct bands of polyubiquitinated β-tubulin were recognized by both anti-ubiquitin and anti-β-tubulin (bands d), whereas bands f probably represented partially degraded ubiquitinated β-tubulin. The ability of the β-tubulin antibody to immunoprecipitate the protein itself was confirmed by the presence of band e, which was a little smaller in molecular weight than α-tubulin (band b). The tight binding between α- and β-tubulins means that either antibody would pull down α/β tubulin heterodimers. The specificity of these well established antibodies and their recognition of the same bands seen in ubiquitin blots indicate that both tubulins are ubiquitinated in vivo. The sizes of the largest ubiquitinated α- and β-tubulin species in Figure 8 are very similar to those ∼100 kDa bands in Figure 7, A and B, which suggests that the situation in transfected HEK293 cells largely mimics the conditions in the rat brain. The high-molecular-weight species of ubiquitinated tubulins (Fig.7A,B, 200–300 kDa bands) were not observed in rat brain lysate. They are perhaps additionally ubiquitinated products of the ∼100 kDa species by parkin overexpressed in HEK293 cells.
Fig. 8.

Ubiquitinated α- and β-tubulin in rat brain lysate. Ultracentrifuged rat brain lysates were immunoprecipitated with an irrelevant polyclonal antibody (anti-neurabin, n), anti-α-tubulin (α) or anti-β-tubulin (β). Immunoprecipitates were analyzed by Western blotting with anti-ubiquitin (Ub), anti-α-tubulin or anti-β-tubulin. Rat brain lysates (5 μg total proteins, −) were loaded next to the lanes containing tubulin immunoprecipitates to indicate the position of α- or β-tubulin. Bands: a, d, ubiquitinated α- or β-tubulin; b, e, α- or β-tubulin; c, f, partially degraded ubiquitinated α- or β-tubulin; HC, IgG heavy chain; LC, IgG light chain.
Parkin mutants found in PD patients do not ubiquitinate α- or β-tubulin
To provide more evidence that parkin is an E3 ligase for tubulin and to test whether such enzymatic activity is affected by mutations found in PD patients, we transfected HEK293 cells with FLAG-tagged human wild-type parkin or three point mutants (K161N, T240R, and C431F) (Fig. 9A) that are linked to AR-JP (Giasson and Lee, 2001) along with HA-tagged ubiquitin. After the cell lysates were immunoprecipitated with antibodies against α- or β-tubulin, respectively, precipitated proteins were analyzed by Western blotting with anti-HA. As shown in Figure 9B,only wild-type parkin caused a significant increase in the ubiquitination of α- and β-tubulins, and none of the mutants were capable of ubiquitinating either tubulin. When we analyzed the cell lysates by HA Western blot, it was clear that these mutants did not have any significant E3 ligase activity over the background level caused by other E3 ligases in the cell (Fig. 9C). This is consistent with previous studies using these mutants (Giasson and Lee, 2001). The expression levels of wild-type and mutant parkin constructs were comparable in these experiments (Fig. 9D).
Fig. 9.

Parkin mutants found in PD patients cannot ubiquitinate α- and β-tubulin. A, Diagram showing the location of three point mutations found in PD patients. U, Ubiquitin-like domain; R, RING finger domain; I, in-between RING finger domain. B, HEK293 cells were transfected with FLAG-tagged human wild-type (w) or mutant (a, b, or c as shown inA) parkin- and HA-tagged ubiquitin. Cell lysates were immunoprecipitated with antibodies against α-tubulin (left) or β-tubulin (right). Precipitated proteins were blotted with anti-HA to show the ubiquitination of either tubulin (indicated by the bracket). *IgG heavy chain. The same set of cell lysates was also blotted with anti-HA to show the E3 ligase activity of wild-type and mutant parkin (C) or anti-FLAG to show the expression level of these parkin constructs (D). #, Nonspecific bands recognized by anti-HA in HEK293 cell lysates. All experiments were repeated at least three times, each with similar results.
Parkin, but not its mutants linked to PD, accelerates degradation of α- and β-tubulins
Because parkin enhanced the ubiquitination of α- and β-tubulins, it should also facilitate their degradation by the 26S proteasome. To test this, we transfected HEK293 cells with or without parkin and treated the cells with the protein synthesis inhibitor puromycin (100 μm) for various durations. We also transfected cells with the three point mutants that did not ubiquitinate α- or β-tubulin (Fig. 9) to see if these PD-linked mutations affect the ability of parkin to degrade tubulins. Equal fractions (1%) of total cell lysates were separated on SDS-PAGE and analyzed by Western blots with antibodies against α-tubulin, β-tubulin, or parkin, respectively. Without synthesis of new proteins, the level of α- or β-tubulin in these cells should reflect their rates of degradation. As shown in Figure10A, the amount of α-tubulin decreased much faster in HEK293 cells transfected with wild-type parkin, compared with cells untransfected or transfected with any one of the three point mutants. Similar effects were observed for β-tubulin degradation. The amount of endogenous parkin in untransfected cells was significantly reduced at 12 and 24 hr of puromycin treatment. However, the level of transfected wild-type or mutant parkin was not significantly affected by the puromycin treatment, perhaps because of the overexpression of the constructs.
Fig. 10.

Parkin, but not its mutant-linked PD, accelerates the degradation of α- and β-tubulin. HEK293 cells were transfected without or with various parkin constructs [wild-type (WT), K161N, T240R, or C431F mutant] and treated with puromycin (100 μm) for different duration. Equal fractions (1%) of total cell lysates were Western-blotted with anti-α-tubulin, anti-β-tubulin, or anti-parkin (A). Only wild-type parkin significantly accelerated the degradation of α- and β-tubulin at 12 and 24 hr of treatment, compared with cells untransfected or transfected with any one of the point mutants. Expression levels of transfected parkin constructs were not significantly affected by puromycin treatment. Results from four experiments were quantified and normalized against the amount of α-tubulin (B) or β-tubulin (C) at 0 hr of treatment. *p< 0.05, paired t test, compared with no parkin transfection at 12 or 24 hr of puromycin treatment.
A significant difference was found in the level of α- or β-tubulin between untransfected cells and cells transfected with wild-type parkin at 12 and 24 hr of puromycin treatment (p < 0.05, n = 4 experiments, paired t test) (Fig. 10B,C). Tubulin degradation rates were not significantly different between cells untransfected or transfected with any one of the three mutants (p > 0.05) (Fig.10B,C). The much faster rate for the degradation of α- and β-tubulin in the presence of parkin, but not its mutants that lack E3 ligase activity, shows that parkin enhances the turnover of both tubulins, most likely through increasing their ubiquitination.
Discussion
In this report, we describe the strong binding and colocalization between parkin and microtubules. In contrast to typical MAPs, which bind to microtubules primarily through electrostatic interactions, parkin cannot be dissociated from microtubules with 2m of NaCl. It suggests that this interaction is mostly hydrophobic in nature. The strong binding appears to be caused by direct interactions between parkin and tubulin α/β heterodimers, the building blocks of microtubules. In our coimmunoprecipitation experiment (Fig. 3), we pretreated ultracentrifuged brain lysates with colchicine at 4°C to ensure that microtubules were completely depolymerized into tubulin heterodimers. The large amount of α- and β-tubulins coimmunoprecipitated with parkin at this condition suggests that parkin binds to tubulin α/β heterodimers with high affinity. Even in high salt conditions, the coimmunoprecipitation could not be disrupted (data not shown), which is consistent with the tight binding between parkin and microtubules. Additional studies are necessary to purify parkin from tubulins and measure the dissociation constant and stoichiometry of the interaction between parkin and tubulin heterodimers, as well as that between parkin and pure microtubules. It would also be interesting to investigate the role of parkin in microtubule assembly, stability, and dynamic properties.
The interaction between parkin and α/β tubulin apparently enhances the ubiquitination and degradation of both tubulins. The expression of transfected parkin in HEK293 cells significantly increased the ubiquitination of α- and β-tubulins (Fig. 7A,C). This effect was shown to be dependent on the E3 ligase activity of parkin. Three point mutations found in PD patients (K161N, T240R, C431F) abolished the E3 ligase activity of parkin toward tubulins (Fig.9B) and other substrates (Fig. 9C) in the cell. These data indicate that parkin is a protein–ubiquitin E3 ligase for α- and β-tubulin. This is confirmed by the results that parkin, but not its PD-linked mutants that lack E3 ligase activity, accelerated the degradation of both α- and β-tubulins (Fig. 10).
To ensure that what we saw in HEK293 cells was not an artifact attributable to the overexpression of parkin, we examined the ubiquitination of tubulins in the rat brain. Indeed, similarly ubiquitinated α- and β-tubulins (Figs. 7A,C, 8, ∼100 kDa bands) were observed in both systems, suggesting that a fraction of α- and β-tubulins are in fact ubiquitinated in vivounder normal conditions. The size of these bands indicates that they are probably penta-ubiquitinated tubulins. The minimal length of polyubiquitin chain that can be efficiently recognized by the 26S proteasome is generally believed to be 4–5 ubiquitin molecules (Hershko and Ciechanover, 1998). This might be why we saw distinct bands of penta-ubiquitinated tubulins in the brain; additional ubiquitination of these products would promote their rapid degradation. In transfected HEK293 cells, the overexpression of parkin would produce the whole range of polyubiquitinated tubulins in addition to the penta-ubiquitinated species. The balance between ubiquitination enzymes and de-ubiquitination enzymes, as well as activity of the 26S proteasomes ultimately determines the steady-state distribution of various polyubiquitinated species. Regardless of the details of how parkin ubiquitinates α- and β-tubulin, the expression of transfected parkin in HEK293 cells accelerated the degradation of both tubulins, which further corroborates that parkin is an E3 ligase for α- and β-tubulins.
Because tubulin can exist stably in the cell only as heterodimers or as polymers of the heterodimers (i.e., microtubules), the most likely source of ubiquitinated tubulin may be misfolded tubulin. Indeed, the correct folding of each monomer and the formation of functional heterodimers require coordinated actions of a series of cellular chaperonins and tubulin-specific folding cofactors (for review, seeLewis et al., 1997). The complex and reversible nature of the folding process would inevitably result in the production of misfolded intermediates, which were believed to be quickly degraded through an unknown mechanism. Our results indicate that parkin may be responsible for the ubiquitination of misfolded α- and β-tubulin. Once tubulins are ubiquitinated, they are apparently recognized and degraded by the 26S proteasome, because our data showed that parkin also accelerated the turnover rate of both tubulins (Fig. 10).
In addition to the folding process, which would inherently produce a small amount of misfolded tubulin, sudden depolymerization of microtubules, e.g., by colchicine, results in increased ubiquitination and degradation of tubulin (Y.R. and J.F., unpublished observations). When the concentration of tubulin heterodimers is too high, the cell shuts down tubulin synthesis by degrading its mRNA being translated on polysomes (Pachter et al., 1987). However, this “autoregulation” mechanism may not be able to reduce the free tubulin concentration quickly if microtubules are suddenly depolymerized by drugs like colchicine. In such a case, parkin may play a critical role in ubiquitinating excess tubulin heterodimers so that they can be efficiently degraded by the 26S proteasome. However, it is unclear whether the E3 ligase activity of parkin has any preference toward misfolded tubulin polypeptides or properly folded heterodimers. Additional studies are necessary to resolve this issue.
MPP+ and rotenone are two neurotoxins that cause the selective death of DA neurons and PD-like symptoms in human subjects or animals (Langston et al., 1983; Betarbet et al., 2000). In addition to their widely recognized ability to inhibit complex I of the mitochondrial respiratory chain (Mizuno et al., 1987; Higgins and Greenamyre, 1996), they also strongly induce the depolymerization of microtubules in vivo and in vitro (Brinkley et al., 1974; Marshall and Himes, 1978; Cappelletti et al., 1999, 2001). On the other hand, the inhibition of complex I by either toxin leads to overproduction of reactive oxygen species, which covalently modify many cellular proteins and increase their misfolding. The combination of the two activities of these toxins would produce a significant amount of misfolded tubulin in the cell, which may be toxic (Burke et al., 1989;Weinstein and Solomon, 1990). Thus, the ability of parkin to ubiquitinate misfolded tubulin and accelerate its degradation may be crucial to the survival of any cell affected by MPP+ or rotenone. In the case of MPP+, its selective uptake by DA neurons through the DA transporter renders these cells particularly vulnerable (Przedborski and Jackson-Lewis, 1998). Although rotenone is nonselectively absorbed by many types of tissues because of its hydrophobicity, it appears to cause specific degeneration of nigral DA neurons (Betarbet et al., 2000), which suggests that these cells are particularly vulnerable to rotenone-induced toxicity.
Because microtubules play obligatory functions in the maintenance of cellular morphology and intracellular transport, their depolymerization by either toxin would cause significant damage to the cell, especially for long projection neurons such as DA cells on the nigrostriatal pathway. Among the many things that could be affected by microtubule depolymerization, the lack of sufficient transport of DA vesicles to the terminal would directly reduce DA release in the striatum. Thus, the microtubule-depolymerizing activity of MPP+ and rotenone may significantly affect the normal functions of nigral DA neurons. Parkin, through its ability to ubiquitinate and degrade misfolded tubulin, may protect neurons from the toxic accumulation of misfolded tubulin, particularly when the cells are exposed to neurotoxins such as MPP+ and rotenone.
Although it is not clear whether AR-JP patients with parkin mutations exhibit microtubule abnormalities, future studies are warranted to examine the ubiquitination of tubulins in these brains. In the rotenone model of PD, the degeneration of dopamine neurons appears to be more prominent at neuronal terminals and processes compared with the cell bodies (Betarbet et al., 2000). The low level of rotenone used in those studies (30 nm in the rat brain) may cause only a small amount of microtubule depolymerization. This may cause more harm to the terminals and processes than to the cell bodies, because tubulin synthesis can rapidly replenish damaged microtubules in the cell bodies, but not those in the terminals and distal processes, because newly formed microtubules need to be transported along old microtubules to these places (Baas, 1997, 2002).
In summary, our results show that parkin strongly binds to microtubules and tubulin α/β heterodimers. It also enhances the ubiquitination and degradation of α- and β-tubulin. This E3 ligase activity of parkin may play an important role in handling misfolded tubulins produced during their complex and reversible folding processes, as well as those produced by microtubule-depolymerizing activities of drugs that cause PD.
Footnotes
This work is supported by National Institutes of Health Grant NS41722 (J.F.) and Howard Hughes Medical Institute Biomedical Research Support Program Grant 53000261 (State University of New York at Buffalo).
Correspondence should be addressed to Dr. Jian Feng, Department of Physiology and Biophysics, State University of New York at Buffalo, 124 Sherman Hall, Buffalo, NY 14214. E-mail: jianfeng@buffalo.edu.
References
- 1.Baas PW. Microtubules and axonal growth. Curr Opin Cell Biol. 1997;9:29–36. doi: 10.1016/s0955-0674(97)80148-2. [DOI] [PubMed] [Google Scholar]
- 2.Baas PW. Microtubule transport in the axon. Int Rev Cytol. 2002;212:41–62. doi: 10.1016/s0074-7696(01)12003-6. [DOI] [PubMed] [Google Scholar]
- 3.Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT. Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nat Neurosci. 2000;3:1301–1306. doi: 10.1038/81834. [DOI] [PubMed] [Google Scholar]
- 4.Brinkley BR, Barham SS, Barranco SC, Fuller GM. Rotenone inhibition of spindle microtubule assembly in mammalian cells. Exp Cell Res. 1974;85:41–46. doi: 10.1016/0014-4827(74)90210-9. [DOI] [PubMed] [Google Scholar]
- 5.Burke D, Gasdaska P, Hartwell L. Dominant effects of tubulin overexpression in Saccharomyces cerevisiae. Mol Cell Biol. 1989;9:1049–1059. doi: 10.1128/mcb.9.3.1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cappelletti G, Maggioni MG, Maci R. Influence of MPP+ on the state of tubulin polymerisation in NGF- differentiated PC12 cells. J Neurosci Res. 1999;56:28–35. doi: 10.1002/(SICI)1097-4547(19990401)56:1<28::AID-JNR4>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 7.Cappelletti G, Pedrotti B, Maggioni MG, Maci R. Microtubule assembly is directly affected by MPP(+) in vitro. Cell Biol Int. 2001;25:981–984. doi: 10.1006/cbir.2001.0772. [DOI] [PubMed] [Google Scholar]
- 8.Chung KK, Zhang Y, Lim KL, Tanaka Y, Huang H, Gao J, Ross CA, Dawson VL, Dawson TM. Parkin ubiquitinates the alpha-synuclein-interacting protein, synphilin-1: implications for Lewy-body formation in Parkinson disease. Nat Med. 2001;7:1144–1150. doi: 10.1038/nm1001-1144. [DOI] [PubMed] [Google Scholar]
- 9.Cleveland DW. Autoregulated control of tubulin synthesis in animal cells. Curr Opin Cell Biol. 1989;1:10–14. doi: 10.1016/s0955-0674(89)80030-4. [DOI] [PubMed] [Google Scholar]
- 10.Fallon L, Moreau F, Croft BG, Labib N, Gu WJ, Fon EA. Parkin and CASK/LIN-2 associate via a PDZ-mediated interaction and are co-localized in lipid rafts and postsynaptic densities in brain. J Biol Chem. 2002;277:486–491. doi: 10.1074/jbc.M109806200. [DOI] [PubMed] [Google Scholar]
- 11.Feng J, Lin Y. Parkin interacts with MAP1A and promotes its ubiquitination. Soc Neurosci Abstr. 2001;27:195.10. [Google Scholar]
- 12.Fenteany G, Schreiber SL. Lactacystin, proteasome function, and cell fate. J Biol Chem. 1998;273:8545–8548. doi: 10.1074/jbc.273.15.8545. [DOI] [PubMed] [Google Scholar]
- 13.Galloway PG, Mulvihill P, Perry G. Filaments of Lewy bodies contain insoluble cytoskeletal elements. Am J Pathol. 1992;140:809–822. [PMC free article] [PubMed] [Google Scholar]
- 14.Giasson BI, Lee VM. Parkin and the molecular pathways of Parkinson's disease. Neuron. 2001;31:885–888. doi: 10.1016/s0896-6273(01)00439-1. [DOI] [PubMed] [Google Scholar]
- 15.Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998;67:425–479. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- 16.Higgins DS, Greenamyre JT. [H-3]dihydrorotenone binding to NADH: ubiquinone reductase (complex I) of the electron transport chain: an autoradiographic study. J Neurosci. 1996;16:3807–3816. doi: 10.1523/JNEUROSCI.16-12-03807.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Imai Y, Soda M, Inoue H, Hattori N, Mizuno Y, Takahashi R. An unfolded putative transmembrane polypeptide, which can lead to endoplasmic reticulum stress, is a substrate of Parkin. Cell. 2001;105:891–902. doi: 10.1016/s0092-8674(01)00407-x. [DOI] [PubMed] [Google Scholar]
- 18.Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:605–608. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- 19.Langston JW, Ballard P, Tetrud JW, Irwin I. Chronic parkinsonism in humans due to a product of meperidine-analog synthesis. Science. 1983;219:979–980. doi: 10.1126/science.6823561. [DOI] [PubMed] [Google Scholar]
- 20.Lewis SA, Tian GL, Cowan NJ. The alpha- and beta-tubulin folding pathways. Trends Cell Biol. 1997;7:479–484. doi: 10.1016/S0962-8924(97)01168-9. [DOI] [PubMed] [Google Scholar]
- 21.Lowe J, Blanchard A, Morrell K, Lennox G, Reynolds L, Billett M, Landon M, Mayer RJ. Ubiquitin is a common factor in intermediate filament inclusion bodies of diverse type in man, including those of Parkinson's disease, Pick's disease, and Alzheimer's disease, as well as Rosenthal fibres in cerebellar astrocytomas, cytoplasmic bodies in muscle, and mallory bodies in alcoholic liver disease. J Pathol. 1988;155:9–15. doi: 10.1002/path.1711550105. [DOI] [PubMed] [Google Scholar]
- 22.Lucking CB, Durr A, Bonifati V, Vaughan J, De Michele G, Gasser T, Harhangi BS, Meco G, Denefle P, Wood NW, Agid Y, Brice A. Association between early-onset Parkinson's disease and mutations in the parkin gene. N Engl J Med. 2000;342:1560–1567. doi: 10.1056/NEJM200005253422103. [DOI] [PubMed] [Google Scholar]
- 23.Manders EMM, Verbeek FJ, Aten JA. Measurement of co-localization of objects in dual-color confocal images. J Microsc. 1993;169:375–382. doi: 10.1111/j.1365-2818.1993.tb03313.x. [DOI] [PubMed] [Google Scholar]
- 24.Marshall LE, Himes RH. Rotenone inhibition of tubulin self-assembly. Biochim Biophys Acta. 1978;543:590–594. doi: 10.1016/0304-4165(78)90315-x. [DOI] [PubMed] [Google Scholar]
- 25.Mayer RJ, Lowe J, Lennox G, Landon M, MacLennan K, Doherty FJ. Intermediate filament-ubiquitin diseases: implications for cell sanitization. Biochem Soc Symp. 1989;55:193–201. [PubMed] [Google Scholar]
- 26.Mizuno Y, Suzuki K, Sone N, Saitoh T. Inhibition of ATP synthesis by 1-methyl-4-phenylpyridinium ion (MPP+) in isolated-mitochondria from mouse brains. Neurosci Lett. 1987;81:204–208. doi: 10.1016/0304-3940(87)90366-1. [DOI] [PubMed] [Google Scholar]
- 27.Pachter JS, Yen TJ, Cleveland DW. Autoregulation of tubulin expression is achieved through specific degradation of polysomal tubulin mRNAs. Cell. 1987;51:283–292. doi: 10.1016/0092-8674(87)90155-3. [DOI] [PubMed] [Google Scholar]
- 28.Przedborski S, Jackson-Lewis V. Mechanisms of MPTP toxicity. Mov Disord. 1998;13:35–38. [PubMed] [Google Scholar]
- 29.Scott WK, Nance MA, Watts RL, Hubble JP, Koller WC, Lyons K, Pahwa R, Stern MB, Colcher A, Hiner BC, Jankovic J, Ondo WG, Allen FH, Goetz CG, Small GW, Masterman D, Mastaglia F, Laing NG, Stajich JM, Slotterbeck B. Complete genomic screen in Parkinson disease: evidence for multiple genes. JAMA. 2001;286:2239–2244. doi: 10.1001/jama.286.18.2239. [DOI] [PubMed] [Google Scholar]
- 30.Shimura H, Hattori N, Kubo S, Mizuno Y, Asakawa S, Minoshima S, Shimizu N, Iwai K, Chiba T, Tanaka K, Suzuki T. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat Genet. 2000;25:302–305. doi: 10.1038/77060. [DOI] [PubMed] [Google Scholar]
- 31.Shimura H, Schlossmacher MG, Hattori N, Frosch MP, Trockenbacher A, Schneider R, Mizuno Y, Kosik KS, Selkoe DJ. Ubiquitination of a new form of α-synuclein by parkin from human brain: implications for Parkinson's disease. Science. 2001;293:263–269. doi: 10.1126/science.1060627. [DOI] [PubMed] [Google Scholar]
- 32.Vallee RB. Purification of brain microtubules and microtubule-associated protein 1 using taxol. In: Vallee RB, editor. Methods in enzymology. Academic; San Diego: 1986. pp. 104–115. [DOI] [PubMed] [Google Scholar]
- 33.Wang DM, Moriggl R, Stravopodis D, Carpino N, Marine JC, Teglund S, Feng J, Ihle JN. A small amphipathic alpha-helical region is required for transcriptional activities and proteasome-dependent turnover of the tyrosine-phosphorylated Stat5. EMBO J. 2000;19:392–399. doi: 10.1093/emboj/19.3.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weinstein B, Solomon F. Phenotypic consequences of tubulin overproduction in Saccharomyces cerevisiae: differences between α-tubulin and β-tubulin. Mol Cell Biol. 1990;10:5295–5304. doi: 10.1128/mcb.10.10.5295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang Y, Gao J, Chung KK, Huang H, Dawson VL, Dawson TM. Parkin functions as an E2-dependent ubiquitin- protein ligase and promotes the degradation of the synaptic vesicle-associated protein, CDCrel-1. Proc Natl Acad Sci USA. 2000;97:13354–13359. doi: 10.1073/pnas.240347797. [DOI] [PMC free article] [PubMed] [Google Scholar]