

Abstract
G protein coupled receptors (GPCRs) are remarkably versatile signaling molecules. The members of this large family of membrane proteins are activated by a spectrum of structurally diverse ligands, and have been shown to modulate the activity of different signaling pathways in a ligand specific manner. In this manuscript I will review what is known about the structure and mechanism of activation of GPCRs focusing primarily on two model systems, rhodopsin and the β2 adrenoceptor.
Keywords: GPCR, 7TM, structure, conformational changes, efficacy
INTRODUCTION
G protein coupled receptors (GPCRs) represent the largest family of membrane proteins in the human genome and the richest source of targets for the pharmaceutical industry. There has been remarkable progress in the field of GPCR biology during the past two decades. Notable milestones include the cloning of the first GPCR genes, and the sequencing of the human genome revealing the size of the GPCR family and the number of orphan GPCRs. Moreover, there is a growing appreciation that GPCR regulation and signaling is much more complex than originally envisioned, and includes signalling through G protein independent pathways [2–4]. Consequently, it has been proposed that the term GPCR be abandoned in favor of 7 transmembrane or 7TM receptors.
In spite of the remarkable advances in the biology and pharmacology of GPCRs, progress in the area of protein structure has been more limited. To date, the only high-resolution structures of a GPCR have been for bovine rhodopsin. In this manuscript I will briefly review the groundbreaking structural work on rhodopsin and discuss the challenges in obtaining high-resolution structures of other GPCRs. I will also discuss what we know about the structural changes associated with receptor activation.
GPCR STRUCTURE
COMMON STRUCTURAL FEATURS OF GPCRS
G protein coupled receptors (GPCRs) represent the single largest class of membrane proteins in the human genome. A recent and detailed analysis of the human genome reveals over 800 unique GPCRs, of which approximately 460 are predicted to be olfactory receptors [5]. Based on sequence similarity within the 7 TM segments, these receptors can be clustered into 5 families: the rhodopsin family (701 members), the adhesion family (24 members), the frizzled/taste family (24 members), the glutamate family (15 members), and the secretin family (15 members) [5]. The physiologic function of a large fraction of these 800 GPCRs is unknown; these receptors are referred to as orphan GPCRs. However, deorphanization of non-olfactory GPCRs is an ongoing process [6], as they are a promising group of targets for the pharmaceutical industry. Therefore, the actual number of orphan GPCRs continues to decline.
GPCRs share a common structural signature of seven hydrophobic transmembrane (TM) segments, with an extracellular amino terminus and an intracellular carboxyl terminus (Figure 1). GPCRs share the greatest homology within the TM segments. The most variable structures among the family of GPCRs are the carboxyl terminus, the intracellular loop spanning TM5 and TM6, and the amino terminus. The greatest diversity is observed in the amino terminus. This sequence is relatively short (10–50 amino acids) for monoamine and peptide receptors, and much larger (350–600 amino acids) for glycoprotein hormone receptors, and the glutamate family receptors. The largest amino terminal domains are observed in the adhesion family receptors.
Figure 1.

Cartoons depicting the secondary structure and the location of agonist binding sites for different GPCRs.
This structural and functional similarity of GPCRs stands in contrast to the structural diversity of the natural GPCR ligands [7]. These range from subatomic particles (a photon), to ions (H+ and Ca++), to small organic molecules, to peptides and proteins. The location of the ligand binding domains for many GPCRs has been determined [7]. While many small organic agonists bind within the TM segments, peptide hormones and proteins often bind to the amino terminus and extracellular sequences joining the TM domains. However, size of the ligand alone cannot be used to predict the location of the binding site: for instance, glycoprotein hormones, glutamate, and Ca2+ all activate their respective receptors by binding to relatively large amino terminal domains [7, 8]. It is interesting to note that it has been possible to identify small molecular weight allosteric modulators that bind within the TM domains [9–14] for many GPCRs that bind their native agonists on the extracellular loops or the amino terminus.
STRUCTURAL INSIGHTS FROM RHODOPSIN
Our understanding of GPCR structure is based largely on the high-resolution structures of the inactive state of rhodopsin. Rhodopsin is better suited for structural studies than most other GPCRs because it is possible to obtain large quantities of highly enriched protein from bovine retina. Rhodopsin is also a remarkably stable GPCR, retaining function under conditions that denature many other GPCRs.
Two-dimensional crystals
The first structures of rhodopsin came from cryoelectron microsopy of two-dimensional crystals of bovine rhodopsin from Gebhard Schertler’s group [15–19]. While the resolution of these structures was limited (ranging from 5 to 9Å), they provided the first picture of the orientation of the TM segments in a lipid environment. These structures, together with the subsequent computational work from Baldwin [20] provided the template from which most molecular models for other GPCRs were generated.
Three-dimensional crystals
More recently, three dimensional crystals structures of rhodopsin have been obtained by several groups [21–26]. Three-dimensional crystals of bovine rhodopsin have been grown using at least two different approaches. The first published crystals of rhodopsin were obtained from rhodopsin selectively solubilized from rod outer segments using a combination of alkyl(thio)glucoside detergents and divalent cations [21, 27]. The procedure used no additional purification steps. The mixture likely contained more rod out segment lipids than would be expected from protein purified by column chromatography, and it was speculated that the presence of these lipids may influence crystal formation [21]. Crystals grown from this preparation of rhodopsin initially diffracted at 2.8 Å [22] and subsequent improvements have led to diffraction at 2.2 Å [25]. All of these crystals have a P41 space group, and crystal contacts form between hydrophilic domains of the receptor.
Three-dimensional crystals have also been obtained from bovine rhodopsin solubilized from rod outer segments using the detergent lauryldimethylamine-oxide (LDAO) and subjected to lectin chromatography followed by detergent exchange into n-octyltetraoxyethylene (C8E4) followed by anion exchange chromatography [26, 28]. This more extensive purification procedure, which would be expected to remove all but tightly bound lipid, resulted in crystals diffracting at 2.6 Å [26]. These crystals have a P31 space group and crystal contacts form primarily within the transmembrane domains and the intracellular and extracellular loop structures point into solvent filled cavities. As a result, the loop structures may assume a more native structure in the P31 crystal form compared to the P41 crystals [29].
Comparison of P41 and P31 rhodopsin structures
The structures obtained from P41 and P31 crystals are very similar overall, particularly in the transmembrane, and extracellular domains (Fig. 2, for a detailed comparison of these structures see Schertler, 2005 [29]). However, there are significant differences in the cytoplasmic loop linking TM5 and TM6, which is known to be involved in G protein coupling. These differences are highlighted in red in Figure 2. The structure from the P31 crystals is in agreement with structures obtained from electron diffraction of two-dimensional crystals and with electron paramagnetic resonance spectroscopy studies [29]. As discussed above, this loop may assume a more native structure in the P31 crystals because none of the cytoplasmic domains are involved in crystal lattice contacts.
Figure 2.
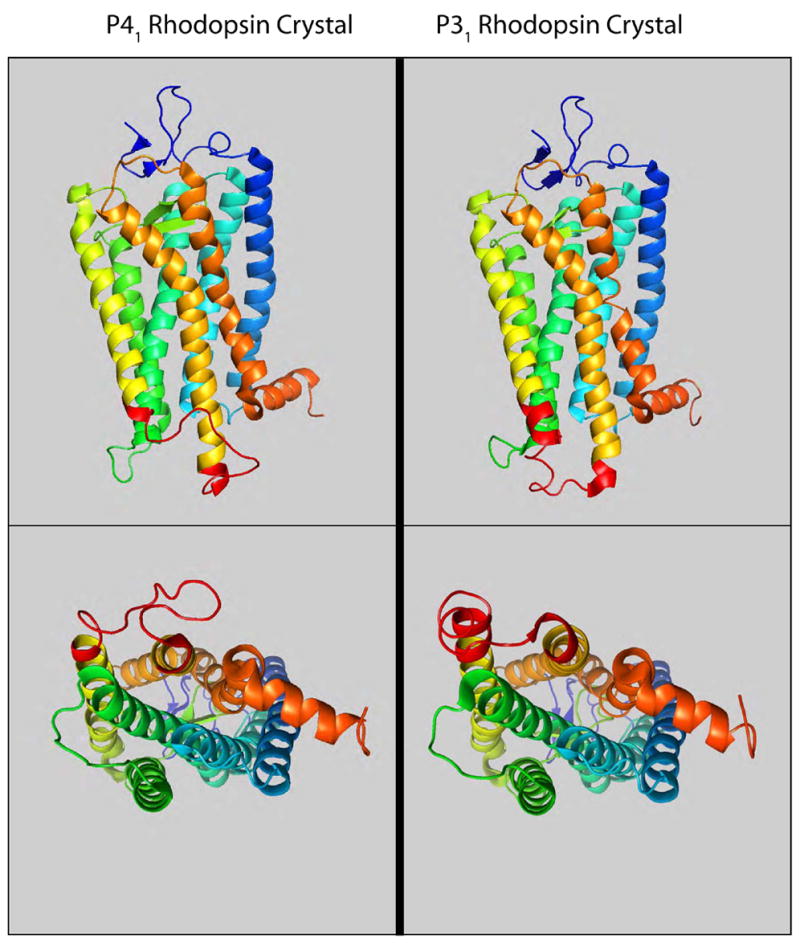
A comparison of the rhodopsin structures determined from the P41 [25] and P31 [26] crystal forms. The loop connecting TM5 and TM6 (shown in red) is the most divergent sequence.
STRUCTURE OF OTHER GPCRs
Recently the amino terminal ligand binding domain of the FSH receptor was crystallized in complex with its ligand [30]. This provides important structural insights into glycoprotein hormone binding; however, the structure does not include the transmembrane domains. With this exception, there are no other high-resolution crystal structures of GPCRS. Structure analysis of other GPCRs has largely been limited to the use of site-directed mutagenesis and cysteine scanning mutagenesis [31] to detect receptor-ligand interactions, and the use of engineered metal ion binding sites to probe intramolecular interactions [32]. While these approaches provide low-resolution structural information, this information can be used to verify and improve the accuracy of homology models based on rhodopsin. Ballesteros and Javitch found that structural insights obtained from mutagenesis data and substituted cysteine accessibility studies on monoamine receptors were consistent with the high-resolution structure of rhodopsin, suggesting that rhodopsin serves as a good template for homology modeling [33]. However, rhodopsin might not be a good template for models of more distantly related family rhodopsin family members such as the cholecystokinin CCK1 receptor [34].
Obstacles for obtaining GPCR Structures
The major obstacles to obtaining structures of other GPCRs include protein production and purification, and protein stability and homogeneity. In terms of production, it is now possible to generate sufficient quantities (tens of milligrams) of several GPCRs for crystal screening using bacterial, yeast, insect cell, and mammalian cell expression systems [35–37] [38–40]. The availability of robotic systems for preparing setups of 100 nanoliter volumes (or smaller) has enabled large parameter screens with relatively small amounts of protein. As such, protein production is no longer the major limitation for crystallography efforts.
Perhaps a greater problem is the stability of purified GPCRs in detergents compatible with crystallography. For example, the β2 adrenoceptor (β2AR) and many other GPCRs are not stable in the detergents used to obtain rhodopsin crystals; and thus far rhodopsin crystals have not been obtained in dodecylmaltoside, a detergent in which the β2AR is relatively stable. GPCRs tend to be more stable in non-ionic detergents with relatively long alkyl chains. These detergents may form larger micelles that prevent the formation of crystal contacts [41]. Another problem is the potential for both structural and conformational heterogeneity in GPCRs. By structural heterogeneity I mean heterogeneity in posttranslational modifications such as glycosylation, phosphorylation and palmitoylation. These sources of heterogeneity can often be eliminated by site directed mutagenesis of the protein, or enzymatic removal of sugars and phosphates. This source of heterogeneity is minimized if the GPCR can be expressed in bacteria.
The greatest obstacle to the formation of crystals may be conformational heterogeneity due to the inherent flexibility of GPCRs. This flexibility may be functionally important, enabling structural changes associated with agonist binding and activation of a membrane bound receptor; however, this property may lead to the existence of different protein conformations and to denaturation, particularly in detergent solutions. There is evidence that the conformational heterogentiy due to relative movements of the TM segments can be reduced by specific ligands [42]. Other sources of conformational heterogeneity are the amino and carboxyl termini, and the loops connecting the TM segments. These sequences are often longer than the homologous segments in rhodopsin and may be unstructured. In some cases the problem of conformational heterogeneity can be mitigated by structural modifications; however, identifying these poorly structured domains can be challenging. Moreover, removing hydrophilic sequences may eliminate potential crystal lattice sites.
While the challenges facing GPCR structural biologists are formidable, I believe structures will begin to appear within the next several years. The problems outlined above are solvable. This may require a highly focused effort on a specific target, where structural modifications and/or associated proteins (antibodies, arrestins, G proteins) are used to facilitate crystal formation. Another approach is to screen a large number of GCPRs to identify those with the best characteristics for structural studies. This approach has been used by a European consortium known as the Membrane Protein Network (MEPNET, http://www.mepnet.org/) consisting of academic and industrial groups. This program has made significant progress in identifying the best GPCR candidates for crystallography trials.
GPCR OLIGOMERS
There is a growing body of evidence that GPCRs exist as dimers (or oligomers) and that these dimers may be important for G protein activation for at least some GPCR families. This topic has been addressed in several excellent reviews [43–46] and is the subject of another review in this series. Therefore, GPCR dimers will be only briefly discussed here. Dimerization is clearly an important mechanism of receptor activation for the glutamate family of GPCRs [8, 47], where ligand-induced changes in the dimer interface of the amino terminal ligand binding domain has been demonstrated by crystallography [48, 49]. However, the role of dimerization in the activation of rhodopsin family members is less clear. For instance, recent cryoelectron microscopy images suggest that rhodopsin may exist as homodimers in rod outer segment membranes [50]. In addition, neutron scattering studies provide evidence that a pentameric complex forms when purified leukotriene B(4) is reconstituted with purified Gi, suggesting that a receptor homodimer is needed to complex with a heterotrimeric G protein [51]. Nevertheless, it remains to be seen if a receptor dimer is required for G protein activation. The effect of agonist binding on the formation or disruption of dimers is not consistent among the rhodopsin family members that have been examined [43]. Moreover, ligands interact with individual receptor monomers, and there is currently no evidence that ligands span the interface between receptor dimers. If changes in dimerization occur, it is likely a secondary consequence of ligand-induced changes in the arrangement of the TM segments. Evidence in support of this comes from biophysical studies on leukotriene B(4) homodimers demonstrating that ligand binding to one protomer leads to conformational changes in its partner [52]. While dimers may be important for G protein activation, it is essential to understand the agonist-induced structural changes that occur in the context of individual GPCR monomers.
GPCR ACTIVATION
ACTIVATION OF RHODOPSIN
The currently available three-dimensional, high-resolution structures of rhodopsin correspond to an inactive form of the receptor. The most detailed information about structural changes associated with activation of a GPCR comes from lower-resolution biophysical studies on rhodopsin. Rhodopsin structure and what is known about its light-induced conformational changes have been the subject of several excellent reviews [29, 53, 54] [55, 56], and some of the main points will be briefly discussed here. Electron paramagnetic resonance spectroscopy (EPR) studies provide evidence that photoactivation of rhodopsin involves a rotation and tilting of TM6 relative to TM3 [57]. Further support for motion of TM6 during rhodopsin activation was provided by chemical reactivity measurements and fluorescence spectroscopy [58], as well as by ultraviolet absorbance spectroscopy [59] and by zinc crosslinking of histidines [60]. Light-induced conformational changes have also been observed in the cytoplasmic domain spanning TM1 and TM2, and the cytoplasmic end of TM7 [61–63].
Obtaining a high-resolution structure of metarhodopsin I, the active form of rhodopsin has been hindered by the instability of rhodopsin crystals following light activation. However, there has been some progress towards this goal. Shertler’s group succeeded in obtaining two dimensional crystals and a low resolution map of metarhodopsin I [64] [29]. Metarhodopsin I is an intermediate in the process of rhodopsin activation that occurs after photoisomerization of 11-Cisretinal, but before structural changes required for transducin activation (metarhodopsin II). Electron diffraction studies reveal that the formation of metarhodopsin I is not accompanied by the large rigid-body movements in TM segments shown to be involved in rhodopsin activation [57]. However, a more subtle change, consisting in the rearrangement in the conformation of the Trp residue of the highly conserved CWxxP motif in TM6, has been detected in this intermediate. Thus, it seems that there is no gradual transformation of the inactive protein into the active form, but the activation is initiated through small scale changes in the conformation of some key residues, which will presumably trigger the larger conformational changes related to the subsequent stages of the activation process [29]. Recently, conditions for growing more light stable three-dimensional crystals of rhodopsin have been identified [65], suggesting that a high-resolution structure of metarhodopsin II may be available in the near future.
COMMON STRUCTURAL CHANGES AMONG GPCRS
In spite of the remarkable diversity of ligands and ligand binding domains in the family of GPCRs, there is also considerable evidence for a common mechanism of activation. When comparing sequences, GPCRs are most similar at the cytoplasmic ends of the transmembrane segments adjacent to the second and third cytoplasmic domains, the regions known to interact with cytoplasmic G proteins [66]. Members of the large family of GPCRs transduce signals by activating one or more members of the relatively small family of highly homologous heterotrimeric G proteins. For example, the follicle stimulating hormone (FSH) receptor is activated by a large glycoprotein hormone that binds to the amino terminus while the β2AR is activated by adrenaline (approximately the size of a single amino acid) that binds to the TM segments; yet both of these receptors activate the same G protein (Gs), indicating that the structural changes in the cytoplasmic domains of these two receptors must be very similar. Moreover, many GPCRs exhibit promiscuous coupling to more than one G protein. For example, rhodopsin preferentially couples to transducin while the β2AR preferentially couples to Gs; however, both are capable of activating Gi [67].
Additional evidence that GPCRs undergo similar conformational changes within TM segments and cytoplasmic domains comes from biophysical and biochemical studies. Fluorescence spectroscopic studies of β2AR labeled with florescent probes demonstrate movement in both TM3 and TM6 upon activation [68]. More recent studies of β2AR labeled with fluorescent probes at the cytoplasmic end of TM6 provide evidence that agonists induce a rotation or tilting movement of the cytoplasmic end of TM6 similar to that observed in rhodopsin [69, 70]. A key structural change involving the disruption of an ionic interaction between the highly conserved D(E)RY sequence at the cytoplasmic end of TM3 and an acidic residue at the cytoplasmic end of TM6 is observed upon activation of both rhodopsin and the β2AR [57, 71] Additional support for movement of TM3 and TM6 in the β2AR comes from zinc crosslinking studies [72, 73], and chemical reactivity measurements in constitutively active β2AR mutants [74, 75]. Cysteine crosslinking studies on the M3 muscarinic receptor provide evidence for the movement of the cytoplasmic ends of TM5 and TM6 toward each other upon agonist activation [76–78].
ACTIVATION BY DIFFUSABLE AGONISTS
While the structural changes associated with activation may be similar for rhodopsin and other GPCRs, the mechanism by which these changes are brought about is quite different. In inactive rhodopsin, there is virtually no activity towards the G protein transducin. Absorption of a photon of light converts covalently bound 11-Cisretinal (an inverse agonist) to all trans retinal (a full agonist) within femptoseconds. Rhodopsin then rapidly undergoes a series of conformational changes that have been characterized spectroscopically (rhodopsin > bathorohdopsin > lumirhodopsin > metarhodopsin I > metarhodopsin II). The structural changes associated with the formation of metarhodopsin II, the active form of rhodopsin, are observed within microseconds of photoactivation [79]. Photoisomerization of bound retinal is extremely efficient in using the energy of the captured photon to induce protein structure changes. It is interesting that free trans-retinal is not a very effective agonist for opsin (the unliganded form of rhodopsin) [80, 81], producing only about 14% of the response observed for light-activated rhodopsin[80].
For the vast majority of other GPCRs, activation occurs when an agonist diffuses into an unliganded receptor. In many cases the unliganded receptor has some basal (constitutive) activity towards a G protein. The term “efficacy” is used to describe the effect of a ligand on the functional properties of the receptor [For a more complete discussion of efficacy refer to [82]]. Agonists are defined as ligands that fully activate the receptor. Partial agonists induce submaximal activation of the G protein even at saturating concentrations. Inverse agonists inhibit basal activity. Antagonists have no effect on basal activity, but competitively block access of other ligands. The efficacy of a given drug may vary depending on the signaling pathway being examined [83].
A number of kinetic models have been developed to explain GPCR activation using information derived from indirect measures of receptor conformation, such as ligand binding affinity and the activation of G proteins or effector enzymes [84–87]. The simplest of them, the two-state model proposes that a receptor exists primarily in two states, the inactive state (R) and the active state (R*). In the absence of ligands, the level of basal receptor activity is determined by the equilibrium between R and R*. The efficacy of ligands reflects their ability to alter the equilibrium between these two states. Full agonists bind to and stabilize R*, while inverse agonists bind to and stabilize R. Partial agonists have some affinity for both R and R* and are therefore less effective in shifting the equilibrium towards R*.
The two-state model can describe much of the functional behavior of GPCRs and explain the spectrum of responses to ligands of different efficacy in simple experimental systems consisting of one receptor and one G protein. However, there is a growing body of experimental evidence for the existence of multiple conformational states [summarized in [83]]. Within this framework, each ligand may induce or stabilize a unique conformational state that can be distinguished by the activity of that state towards different signaling molecules (G proteins, kinases, arrestins).
β2AR AS A MODEL SYSTEM FOR STUDYING LIGAND-INDUCED CONFORMATIONAL CHANGES
With the exception of rhodopsin, the β2AR is perhaps the most extensively characterized member of the GPCR family. The β2AR is a good model system for studying agonist binding because much is known about the sites of interactions between catecholamine ligands and the receptor (Fig 3). Moreover, there is a rich source of structurally similar ligands having a spectrum of efficacies ranging from inverse agonists to full agonists (Fig 3B).
Figure 3.

Agonist binding site in the β2AR. A. Amino acids involved in forming the agonist binding site for the β2AR. The sites of interaction between catecholamines and the β2AR have been extensively characterized [100–102]. The amine nitrogen interacts with Asp 113 in TM3 [103], the catechol hydroxyls interact with serines in TM5 [100–102]. Interactions with the aromatic ring and the chiral β-hydroxyl have both been mapped to TM6 [101]. B. A panel of β2AR ligands discussed in this review. Catechol is a very weak partial agonist. Dopamine and salbutamol are partial agonists. Norepinephrine, epinephrine and isoproterenol are full agonists. ICI118,551 is an inverse agonist. D–E. Isoproterenol docked into a three-dimensional model of the β2AR. Illustrations were made with MacPyMOL software.
In the case of the β2AR it has been possible to monitor directly some of these ligand-specific states using fluorescence spectroscopy [42, 70, 71, 88, 89]. These studies reveal that for the β2AR ligand binding and activation is a kinetically and conformationally complex process. Based on these studies we have proposed a working model (Figure 4) where agonist binding and conformational changes occur through a sequence of conformational intermediates. Biophysical support for this model will be briefly summarized below.
Figure 4.
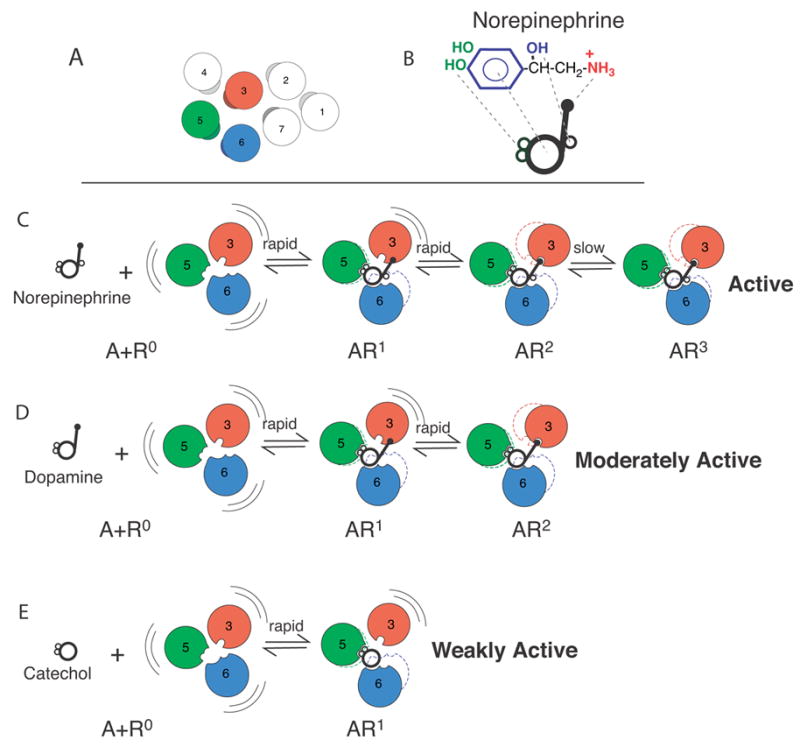
Sequential Binding Model. A. Arrangement of the TM domains of the β2AR as viewed from the extracellular surface. The agonist binding domains are shown in red (TM3), green (TM5) and blue (TM6). B. Cartoon representing structural components of norepinephrine. C–E. In the absence of ligand, the receptor (R) is conformationally flexible. Conformational state R1 is stabilized by interactions between TMs 5 and 6 and the catechol ring. The transition to state R2 occurs when Asp 113 in TM3 binds the amine nitrogen. The transitions from R to R2 are rapid. The slow transition from R2 to R3 involves interactions between the chiral β-hydroxyl and Asn293 on TM6. Adapted from Swaminath et al. [88].
To study ligand induced conformational changes, purified β2AR is labeled with relatively small fluorescent probes (less that 500 Da) at specific sites on the cytoplasmic ends of transmembrane domains involved in ligand binding and G protein coupling [42, 70, 71, 88, 89]. Selective labeling is accomplished using a modified β2AR in which the most reactive cysteines have been mutated to alanine, valine or serine [71]. The resulting minimal cysteine receptor exhibits normal ligand binding and G protein coupling behavior. A new reactive cysteine is introduced into a specific structural domain by site-directed mutagenesis. The modified β2AR is expressed in insect cells, solubilized from insect cell membranes with detergent (dodecylmaltoside), purified by affinity chromatography, and labeled with a cysteine reactive fluorescent probe. The fluorescent probes are sensitive to their local molecular environment such that changes in the β2AR structure lead to changes in the fluorescent properties of the fluorophores (intensity, lifetime, mobility). Therefore it is possible to monitor ligand-induced conformational changes in different receptor domains, and to correlate ligand structure with specific structural changes in the receptor.
Figure 3 shows the amino acid residues that form the ligand binding site for the β2AR deduced from mutagenesis studies, and illustrates the location of Cys 265, one of the labeling sites we have used for our spectroscopy studies. An environmentally sensitive fluorophore covalently bound to Cys265 is well positioned to detect agonist-induced conformational changes relevant to G protein activation. Based on homology with rhodopsin, Cys265 is located in the third intracellular loop (IC3) at the cytoplasmic end of the TM6. Mutagenesis studies have shown this region of IC3 to be important for G protein coupling [90, 91]. Moreover, TM6, along with TM3 and TM5 contain amino acids that form the agonist binding site.
Ligand-specific conformational states detected by fluorescence lifetime studies
We examined ligand-dependent changes in fluorescence lifetime of purified β2AR labeled at Cys265 with fluorescein maleimide in an effort to identify the existence of agonist-specific conformational states [42]. Fluorescence lifetime analysis can detect discrete conformational states in a population of molecules, while fluorescence intensity measurements reflect the weighted average of one or more discrete states. Fluorescence lifetime, τ, refers to the average time that a fluorophore that has absorbed a photon remains in the excited state before returning to the ground state. The lifetime of fluorescein (nanoseconds) is much faster than the predicted off-rate of the agonists we examined (μs - ms), and much shorter than the half-life of conformational states of bacteriorhodopsin (μs)[92], rhodopsin (ms) [79, 93] or of ion channels (μs - ms) [94]. Therefore, lifetime analysis of fluorescein bound to Cys265 is well suited to capture even short-lived, agonist-induced conformational states. We observed that the full agonist isoproterenol induced a conformation that was distinct from the conformations induced by the partial agonists salbutamol and dobutamine [42]. These studies also revealed the existence of an intermediate state in equilibrium with the active state for both full agonists and partial agonists. In contrast, the neutral antagonist appeared to stabilize a state that was indistinguishable from the unliganded receptor.
Intermediate conformational states detected by kinetic studies
Agonist-induced conformational changes in purified β2AR lead to an increase in the fluorescence intensity of tetramethylrhodamine bound to Cys265 [88, 89]. The increase in fluorescence intensity as a function of time following activation by the agonist norepinephrine is best fit with a two component exponential function [88] (Fig 5 A). In contrast, the response to dopamine (a partial agonist) is adequately fit by a one component exponential function. Of interest, the rapid component of norepinephrine is very similar to the response to dopamine (Fig. 5A), which suggested that the dopamine-induced conformation might represent an intermediate in the conformational response to norepinephrine (Fig. 4 D). A rapid component of fluorescence change is observed with all ligands containing a catechol ring and can be observed with catechol alone (Fig. 5B). In fact, catechol is a weak partial agonist (Fig 5C). Based on this observation we proposed that the catechol-induced conformation might represent an intermediate in the conformational response to dopamine and norepinephrine (Fig. 4 E).
Figure 5.

Agonist-induced conformational changes detected by fluorescence spectroscopy in β2AR labeled at Cys265 with tetramethylrhodamine maleimide (TMR-β2AR). A. Change in the intensity of TMR-β2AR in response to norepinephrine and dopamine. The response to norepinephrine is best fit by a two-site exponential function. The rapid and slow components of the response are illustrated by the dotted lines. B. Change in the intensity of TMR-β2AR in response to dopamine and catechol. C. Catechol and dopamine stimulated [35S]GTPγS binding to purified β2AR reconstituted with purified Gs. D. The change in the intensity of TMR-β2AR in response to the non-catechol partial agonist salbutamol followed by the addition of catechol. Catechol can induce a conformational change in TMR-β2AR bound to a saturating concentration of salbutamol, indicating that salbutamol and catechol occupy non-overlapping binding sites. E. The change in the intensity of TMR-β2AR in response to norepinephrine followed by the addition of catechol. No catechol response is observed in TMR-β2AR bound to a saturating concentration of norepinephrine indicating that these ligands share a common binding site. F. There is no significant change in the intensity of TMR-β2AR in response to the inverse agonist ICI118,551. Catechol can induce a conformational change in β2AR bound to a saturating concentration of ICI118,551, indicating that these ligands do not occupy the same binding space. G. [35S]GTPγS binding to purified β2AR reconstituted with purified Gs. Catechol weakly stimulates [35S]GTPγS binding and ICI118,551 inhibits basal [35S]GTPγS binding. Of interest, catechol can stimulate [35S]GTPγS binding in β2AR occupied by a saturating concentration of ICI118,551. The data presented here are adapted from Swaminath et al. [88, 89].
The slow component of the response to norepinephrine (Fig. 5 A) is also observed with the full agonists epinephrine and isoproterenol [88]. The only structural difference between dopamine and norepinephrine is the β-hydroxyl (Fig 3B). Thus conformational changes associated with interactions between the β-hydroxyl and the β2AR are much slower than those involving interactions with catechol ring in our experimental system. Functionally, these slower conformational changes correlate with efficient agonist-induced β2AR internalization, most likely due to interactions between the β2AR and G protein coupled receptor kinases and/or arrestins. While dopamine is a relatively good partial agonist for G protein activation (approximately 60% of isoproterenol), it is much less efficient at inducing internalization (approximately 20% of isoproterenol) [88].
Relevance of slow conformational changes
The slow component of the agonist-induced change observed by fluorescence spectroscopy in purified β2AR (Fig. 5A) is considerably slower than expected from some physiologic responses to GPCR activation, and slower than the conformational changes observed in a modified alpha 2 adrenergic receptor containing a fluorescein arsenical hairpin binder (FlAsH) site in the third intracellular loop and cyan fluorescent protein (CFP) fused to the carboxyl terminus [95, 96]. This modified receptor was expressed in HEK293 cells and agonist-induced conformational changes were detected as changes in fluorescence resonance energy transfer (FRET) between FlAsH and CFP. There are several possible explanations for these observed differences in kinetics. The use of a large fluorescence reporter such as CFP does not allow one to determine the nature of the structural change responsible for the change in FRET; therefore, these FRET experiments may be detecting a different conformational switch, possibly similar to the rapid response observed with dopamine and catechol. However, the different rates observed could also be due to the fact that the β2AR experiments use purified receptor in the absence of G protein, while the FRET experiments were performed on receptor expressed in cell membranes that contain G proteins. It is known that receptors form complexes with G proteins in the plasma membrane (precoupling), and that these complexes have a higher affinity for agonists than do receptors alone. Other factors that might contribute to differences between experiments using purified receptor and those on receptors in cells could be the influence of the pH and salt gradients across the plasma membrane in living cells, as well as the asymmetry of plasma membrane lipids. Nevertheless, while the slow component of the conformational response to agonists may be attributable to the use of purified receptor protein, we believe the structural changes are physiologically relevant because they can be linked to specific interactions between the ligand and the receptor (e.g., the β-hydroxyl of catecholamine agonists interacting with Asn293 in TM6, Fig. 3), and they correlate with specific functional behaviors stimulated by the agonists in β2AR expressed in cells (e.g., agonist-induced internalization [88]).
Catechol activates of the rotamer toggle switch
Based on what is known about the binding site for the catechol ring of catecholamines (Fig. 3), we proposed that the rapid component of the conformational response associated with catecholamine and catechol binding involves changes in the rotameric position of aromatic amino acids surrounding the highly conserved proline Pro2886.50 in TM6 (Fig. 3 C–E). This conformational change, known as a rotamer toggle switch, has been proposed to be involved in the activation of amine and opsin receptor families [97]. Upon binding, the aromatic catechol ring of catecholamines would interact directly with the aromatic residues of the rotamer toggle switch, Trp2866.48 and Phe2906.52. Molecular dynamics simulations suggest that rotamer configurations of Cys2856.47, Trp2866.48 and Phe2906.52, the residues that comprise the rotamer toggle switch, are coupled and modulate the bend angle of TM6 around the highly conserved proline kink at Pro2886.50, leading to the movement of the cytoplasmic end of TM6 upon activation[97]. This movement could be detected by tetramethylrhodamine bound to Cys265 at the cytoplasmic end of TM6.
Non-catecholamine partial agonists do not activate the rotamer toggle switch
In the same experimental system, activation of tetramethylrhodamine-labeled β2AR by salbutamol, a noncatechol partial agonist, produced only a slow monophasic increase in fluorescence intensity [89] (Figure 5 C). Catechol could induce a further increase in fluorescence in β2AR bound to saturating concentrations of salbutamol, but not in β2AR bound to norepinephrine (Fig 5 C and D). This suggests that the aromatic ring of salbutamol does not occupy the same binding space as catechol and does not activate the rotamer toggle switch [89]. Thus, the active state induced by salbutamol would be different from that induced by catecholamine agonists [89]. This is in agreement with fluorescent lifetime experiments discussed above [42].
The inverse agonist ICI118,551 does not inhibit activation of the rotamer toggle switch
The neutral agonist alprenolol and the inverse agonists ICI118,551 and timolol do not produce a detectable change in fluorescence in β2AR labeled witih tetramethylrhodamine on Cys 265 [89], although they do inhibit the response to agonists and partial agonists with one exception. In tetramethylrhodamine-labeled β2AR bound to saturating concentrations of these antagonists there is no effect on the response to catechol. This is shown in Fig. 5F for ICI118,551. The fluorescence response to catechol in β2AR bound to ICI118,551 is associated with a functional response in a G protein activation assay (Fig. 5G). This suggests that ICI118,551 does not occupy the catechol binding pocket and does not prevent activation of the rotamer toggle switch by catechol.
Activation of the ionic lock
We investigated another proposed molecular switch, the ionic lock between the Asp1303.49/Arg1313.50 pair at the cytoplasmic end of TM3 and Glu2686.30 at the cytoplasmic end of TM6 (Fig. 6A) [71]. We used a modified β2AR to introduce a single cysteine labeling site for the cysteine-reactive fluorophore monobromobimane at the cytoplasmic end of TM6 (A271C) adjacent to a Trp introduced at the cytoplasmic end of TM3 (I135W) (Fig. 6B). We took advantage of the ability of tryptophan to quench the fluorescence of bimane, with measurable effects at distances smaller than 15 Å [98], and used bimane fluorescence to monitor structural changes associated with disruption of the ionic lock between TM3 and TM6 in purified β2AR. In the three-dimensional model of the β2AR based on rhodopsin, bimane bound to Cys271 would be separated from Trp135 by the ionic lock. Disruption of the ionic lock would allow Trp to contact and quench bimane fluorescence. This is what we observed upon activation of the modified receptor by isoproterenol, which reduced the intensity of bimane by approximately 50%(Fig 6C). Our results demonstrated that the disruption of the ionic lock is an obligatory step for maximal receptor activation and is triggered by nearly all agonists, independent of efficacy (Fig. 6D). However, we found that disruption of the ionic lock is not directly coupled to the rotamer toggle switch in TM6 since catechol, which is capable of activating the rotamer toggle switch, was not able to activate the ionic lock [71]. Moreover, salbutamol which does not activate the rotamer toggle switch [89] is able to fully activate the ionic lock [71] (Fig. 6D).
Figure 6.
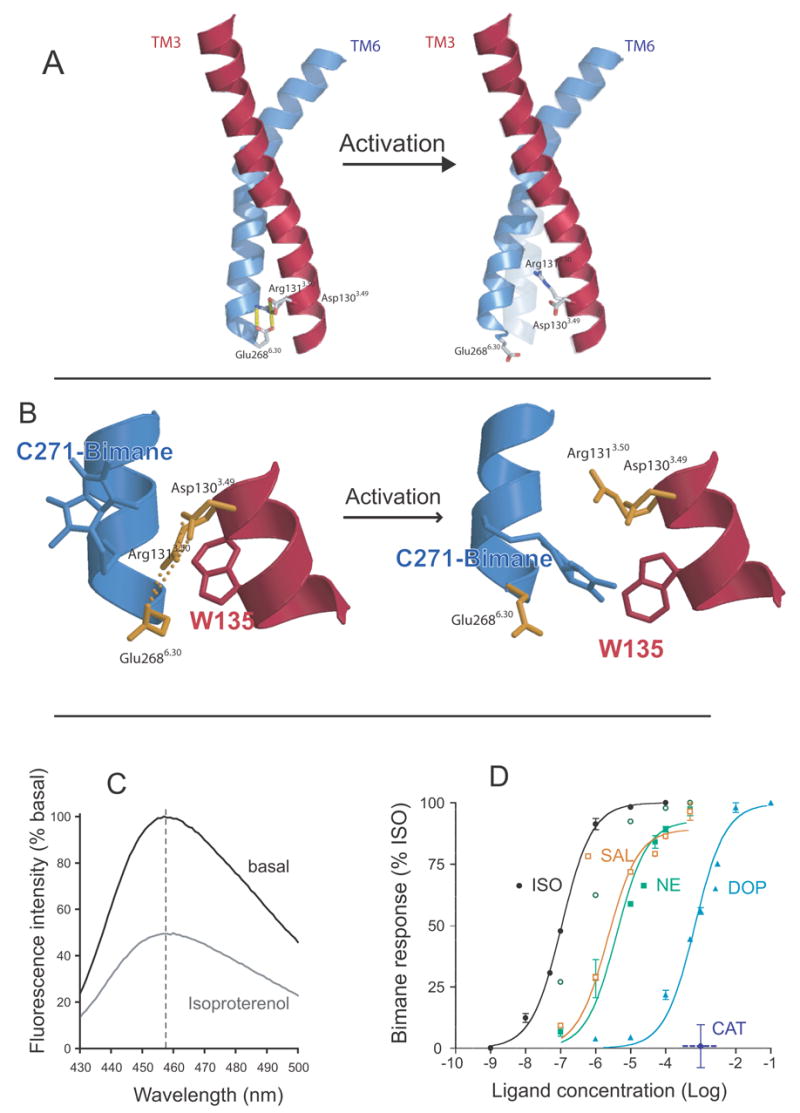
Fluorescence spectroscopy to monitor disruption of the ionic lock in the β2AR. A. Model of TM3 (red) and TM6 (blue) from the β2AR depicting the amino acids that comprise the ionic lock at the cytoplasmic end of these TM segments. B. Close up view of the ionic lock and the modifications made to monitor conformational changes in this region. Alanine 271 was mutated to cysteine (C271) and isoleucine 135 was mutated to tryptophan (W135). C271 was labeled with monobromobimane in purified β2AR. Upon activation, W135 moves closer to bimane on C271 and quenches fluorescence. C. Emission spectrum of bimane on C271 before and after activation by the agonist isoproterenol. D. Effect of different ligands on disruption of the ionic lock as determined by bimane fluorescence. The partial agonists dopamine and salbutamol are as effective at disrupting the ionic lock as the full agonists norepinephrine and isoproterenol. Only catechol has no effect on the ionic lock. These data are adapted from Yao et al. [71].
Binding energy and conformational change
It is interesting to note that catechol and dopamine have nearly the same binding affinity for purified β2AR [71]. The affinity of dopamine, as determined by competition binding, is 350 μM, while the affinity for catechol, determined by a conformational assay, is 160 μM [71]. This is surprising considering that the interaction between the primary amine of dopamine and Asp113 makes the strongest contribution to the binding energy. Since dopamine and catechol bind with the same affinity, but only dopamine disrupts the ionic lock, part of the binding energy associated with the interaction between dopamine and Asp113 may be used to offset by the energetic cost of breaking the ionic lock.
Conclusions
Based on these fluorescence studies we proposed a model where agonists stabilize partially or fully active states by using different chemical groups to activate different combinations of molecular switches, which are not necessarily interdependent. In the unliganded inactive state of a GPCR, the arrangement of TM segments is stabilized by non-covalent interactions between side chains. Structurally distinct ligands are able to break different combinations of the basal state stabilizing interactions either directly by binding to amino acids that are involved in these intramolecular interactions, or indirectly by stabilizing new intramolecular interactions. These ligand-specific conformational changes may be responsible for differential activation of the signaling cascades of the receptor. The affinity of a particular ligand will then be dependent on the energy costs and gains associated with each disrupted and created interaction, while its efficacy will be dependent on the ability to trigger the switches associated with activation. These molecular switches are normally activated by agonist binding, but will also be revealed in constitutively active mutants, where single point mutations in virtually any structural domain can lead to elevated basal activity[99]. A better understanding of the process by which ligands bind and modify GPCR structure may ultimately help in the design of more selective drugs with the appropriate efficacy for the desired physiologic function.
Abbreviations
- β2AR
beta 2 adrenoceptor
- GPCR
G protein coupled receptor
- TM
transmembrane
Footnotes
Amino acid numbering: The position of β2AR residues are followed by the Ballesteros general number [1] in superscript, in the form X.YY, where X refers to the TM segment and YY to the position relative to the most highly conserved amino acid in the TM segment, which is assigned an arbitrary position of 50.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
LITERATURE CITED
- 1.Ballesteros JA, Weinstein H. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein coupled receptors. Meth Neurosci. 1995;25:366–428. [Google Scholar]
- 2.Lefkowitz RJ, Shenoy SK. Transduction of receptor signals by beta-arrestins. Science. 2005;308(5721):512–7. doi: 10.1126/science.1109237. [DOI] [PubMed] [Google Scholar]
- 3.Luttrell LM, Lefkowitz RJ. The role of beta-arrestins in the termination and transduction of G-protein-coupled receptor signals. J Cell Sci. 2002;115(Pt 3):455–65. doi: 10.1242/jcs.115.3.455. [DOI] [PubMed] [Google Scholar]
- 4.Azzi M, Charest PG, Angers S, Rousseau G, Kohout T, Bouvier M, Pineyro G. Beta-arrestin-mediated activation of MAPK by inverse agonists reveals distinct active conformations for G protein-coupled receptors. Proc Natl Acad Sci U S A. 2003;100(20):11406–11. doi: 10.1073/pnas.1936664100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fredriksson R, Lagerstrom MC, Lundin LG, Schioth HB. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol Pharmacol. 2003;63(6):1256–72. doi: 10.1124/mol.63.6.1256. [DOI] [PubMed] [Google Scholar]
- 6.Howard AD, McAllister G, Feighner SD, Liu Q, Nargund RP, Van der Ploeg LH, Patchett AA. Orphan G-protein-coupled receptors and natural ligand discovery. Trends Pharmacol Sci. 2001;22(3):132–40. doi: 10.1016/s0165-6147(00)01636-9. [DOI] [PubMed] [Google Scholar]
- 7.Ji TH, Grossmann M, Ji I. G protein-coupled receptors. I. Diversity of receptor-ligand interactions. J Biol Chem. 1998;273(28):17299–302. doi: 10.1074/jbc.273.28.17299. [DOI] [PubMed] [Google Scholar]
- 8.Pin JP, Galvez T, Prezeau L. Evolution, structure, and activation mechanism of family 3/C G-protein-coupled receptors. Pharmacol Ther. 2003;98(3):325–54. doi: 10.1016/s0163-7258(03)00038-x. [DOI] [PubMed] [Google Scholar]
- 9.Knoflach F, Mutel V, Jolidon S, Kew JN, Malherbe P, Vieira E, Wichmann J, Kemp JA. Positive allosteric modulators of metabotropic glutamate 1 receptor: characterization, mechanism of action, and binding site. Proc Natl Acad Sci U S A. 2001;98(23):13402–7. doi: 10.1073/pnas.231358298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carroll FY, Stolle A, Beart PM, Voerste A, Brabet I, Mauler F, Joly C, Antonicek H, Bockaert J, Muller T, Pin JP, Prezeau L. BAY36-7620: a potent non-competitive mGlu1 receptor antagonist with inverse agonist activity. Mol Pharmacol. 2001;59(5):965–73. [PMC free article] [PubMed] [Google Scholar]
- 11.Pagano A, Ruegg D, Litschig S, Stoehr N, Stierlin C, Heinrich M, Floersheim P, Prezeau L, Carroll F, Pin JP, Cambria A, Vranesic I, Flor PJ, Gasparini F, Kuhn R. The non-competitive antagonists 2-methyl-6-(phenylethynyl)pyridine and 7-hydroxyiminocyclopropan[b]chromen-1a-carboxylic acid ethyl ester interact with overlapping binding pockets in the transmembrane region of group I metabotropic glutamate receptors. J Biol Chem. 2000;275(43):33750–8. doi: 10.1074/jbc.M006230200. [DOI] [PubMed] [Google Scholar]
- 12.Litschig S, Gasparini F, Rueegg D, Stoehr N, Flor PJ, Vranesic I, Prezeau L, Pin JP, Thomsen C, Kuhn R. CPCCOEt, a noncompetitive metabotropic glutamate receptor 1 antagonist, inhibits receptor signaling without affecting glutamate binding. Mol Pharmacol. 1999;55(3):453–61. [PubMed] [Google Scholar]
- 13.Ray K, Tisdale J, Dodd RH, Dauban P, Ruat M, Northup JK. Calindol, a positive allosteric modulator of the human Ca(2+) receptor, activates an extracellular ligand-binding domain-deleted rhodopsin-like seven-transmembrane structure in the absence of Ca(2+) J Biol Chem. 2005;280(44):37013–20. doi: 10.1074/jbc.M506681200. [DOI] [PubMed] [Google Scholar]
- 14.Ray K, Northup J. Evidence for distinct cation and calcimimetic compound (NPS 568) recognition domains in the transmembrane regions of the human Ca2+ receptor. J Biol Chem. 2002;277(21):18908–13. doi: 10.1074/jbc.M202113200. [DOI] [PubMed] [Google Scholar]
- 15.Krebs A, Edwards PC, Villa C, Li J, Schertler GF. The three-dimensional structure of bovine rhodopsin determined by electron cryomicroscopy. J Biol Chem. 2003 doi: 10.1074/jbc.M307995200. [DOI] [PubMed] [Google Scholar]
- 16.Krebs A, Villa C, Edwards PC, Schertler GF. Characterisation of an improved two-dimensional p22121 crystal from bovine rhodopsin. J Mol Biol. 1998;282(5):991–1003. doi: 10.1006/jmbi.1998.2070. [DOI] [PubMed] [Google Scholar]
- 17.Schertler GF. Structure of rhodopsin. Eye. 1998;12(Pt 3b):504–10. doi: 10.1038/eye.1998.138. [DOI] [PubMed] [Google Scholar]
- 18.Unger VM, Hargrave PA, Baldwin JM, Schertler GF. Arrangement of rhodopsin transmembrane alpha-helices. Nature. 1997;389(6647):203–6. doi: 10.1038/38316. [DOI] [PubMed] [Google Scholar]
- 19.Schertler GF, Villa C, Henderson R. Projection structure of rhodopsin. Nature. 1993;362(6422):770–2. doi: 10.1038/362770a0. [DOI] [PubMed] [Google Scholar]
- 20.Baldwin JM, Schertler GF, Unger VM. An alpha-carbon template for the transmembrane helices in the rhodopsin family of G-protein-coupled receptors. J Mol Biol. 1997;272(1):144–64. doi: 10.1006/jmbi.1997.1240. [DOI] [PubMed] [Google Scholar]
- 21.Okada T, Le Trong I, Fox BA, Behnke CA, Stenkamp RE, Palczewski K. X-Ray diffraction analysis of three-dimensional crystals of bovine rhodopsin obtained from mixed micelles. J Struct Biol. 2000;130(1):73–80. doi: 10.1006/jsbi.1999.4209. [DOI] [PubMed] [Google Scholar]
- 22.Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Le Trong I, Teller DC, Okada T, Stenkamp RE, Yamamoto M, Miyano M. Crystal structure of rhodopsin: A G protein-coupled receptor [see comments] Science. 2000;289(5480):739–45. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 23.Okada T, Fujiyoshi Y, Silow M, Navarro J, Landau EM, Shichida Y. Functional role of internal water molecules in rhodopsin revealed by X-ray crystallography. Proc Natl Acad Sci U S A. 2002;99(9):5982–7. doi: 10.1073/pnas.082666399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Teller DC, Okada T, Behnke CA, Palczewski K, Stenkamp RE. Advances in determination of a high-resolution three-dimensional structure of rhodopsin, a model of G-protein-coupled receptors (GPCRs) Biochemistry. 2001;40(26):7761–72. doi: 10.1021/bi0155091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Okada T, Sugihara M, Bondar AN, Elstner M, Entel P, Buss V. The retinal conformation and its environment in rhodopsin in light of a new 2.2 A crystal structure. J Mol Biol. 2004;342(2):571–83. doi: 10.1016/j.jmb.2004.07.044. [DOI] [PubMed] [Google Scholar]
- 26.Li J, Edwards PC, Burghammer M, Villa C, Schertler GF. Structure of bovine rhodopsin in a trigonal crystal form. J Mol Biol. 2004;343(5):1409–38. doi: 10.1016/j.jmb.2004.08.090. [DOI] [PubMed] [Google Scholar]
- 27.Okada T, Takeda K, Kouyama T. Highly selective separation of rhodopsin from bovine rod outer segment membranes using combination of divalent cation and alkyl(thio)glucoside. Photochem Photobiol. 1998;67(5):495–9. [PubMed] [Google Scholar]
- 28.Edwards PC, Li J, Burghammer M, McDowell JH, Villa C, Hargrave PA, Schertler GF. Crystals of native and modified bovine rhodopsins and their heavy atom derivatives. J Mol Biol. 2004;343(5):1439–50. doi: 10.1016/j.jmb.2004.08.089. [DOI] [PubMed] [Google Scholar]
- 29.Schertler GF. Structure of rhodopsin and the metarhodopsin I photointermediate. Curr Opixn Struct Biol. 2005;15(4):408–15. doi: 10.1016/j.sbi.2005.07.010. [DOI] [PubMed] [Google Scholar]
- 30.Fan QR, Hendrickson WA. Structure of human follicle-stimulating hormone in complex with its receptor. Nature. 2005;433(7023):269–77. doi: 10.1038/nature03206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shi L, Javitch JA. The binding site of aminergic G protein-coupled receptors: the transmembrane segments and second extracellular loop. Annu Rev Pharmacol Toxicol. 2002;42:437–67. doi: 10.1146/annurev.pharmtox.42.091101.144224. [DOI] [PubMed] [Google Scholar]
- 32.Elling CE, Thirstrup K, Nielsen SM, Hjorth SA, Schwartz TW. Metal-ion sites as structural and functional probes of helix-helix interactions in 7TM receptors. Ann N Y Acad Sci. 1997;814:142–51. doi: 10.1111/j.1749-6632.1997.tb46152.x. [DOI] [PubMed] [Google Scholar]
- 33.Ballesteros JA, Shi L, Javitch JA. Structural mimicry in G protein-coupled receptors: implications of the high-resolution structure of rhodopsin for structure-function analysis of rhodopsin-like receptors. Mol Pharmacol. 2001;60(1):1–19. [PubMed] [Google Scholar]
- 34.Archer E, Maigret B, Escrieut C, Pradayrol L, Fourmy D. Rhodopsin crystal: new template yielding realistic models of G-protein-coupled receptors? Trends Pharmacol Sci. 2003;24(1):36–40. doi: 10.1016/s0165-6147(02)00009-3. [DOI] [PubMed] [Google Scholar]
- 35.Tate CG, Grisshammer R. Heterologous expression of G-protein-coupled receptors. Trends Biotechnol. 1996;14(11):426–30. doi: 10.1016/0167-7799(96)10059-7. [DOI] [PubMed] [Google Scholar]
- 36.Massotte D. G protein-coupled receptor overexpression with the baculovirus-insect cell system: a tool for structural and functional studies. Biochim Biophys Acta. 2003;1610(1):77–89. doi: 10.1016/s0005-2736(02)00720-4. [DOI] [PubMed] [Google Scholar]
- 37.Sarramegna V, Talmont F, Demange P, Milon A. Heterologous expression of G-protein-coupled receptors: comparison of expression systems fron the standpoint of large-scale production and purification. Cell Mol Life Sci. 2003;60(8):1529–46. doi: 10.1007/s00018-003-3168-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grisshammer R, Averbeck P, Sohal AK. Improved purification of a rat neurotensin receptor expressed in Escherichia coli. Biochem Soc Trans. 1999;27(6):899–903. doi: 10.1042/bst0270899. [DOI] [PubMed] [Google Scholar]
- 39.Weiss HM, Grisshammer R. Purification and characterization of the human adenosine A(2a) receptor functionally expressed in Escherichia coli. Eur J Biochem. 2002;269(1):82–92. doi: 10.1046/j.0014-2956.2002.02618.x. [DOI] [PubMed] [Google Scholar]
- 40.Baneres JL, Martin A, Hullot P, Girard JP, Rossi JC, Parello J. Structure-based analysis of GPCR function: conformational adaptation of both agonist and receptor upon leukotriene B4 binding to recombinant BLT1. J Mol Biol. 2003;329(4):801–14. doi: 10.1016/s0022-2836(03)00438-8. [DOI] [PubMed] [Google Scholar]
- 41.Ostermeier C, Michel H. Crystallization of membrane proteins. Curr Opin Struct Biol. 1997;7:697–701. doi: 10.1016/s0959-440x(97)80080-2. [DOI] [PubMed] [Google Scholar]
- 42.Ghanouni P, Gryczynski Z, Steenhuis JJ, Lee TW, Farrens DL, Lakowicz JR, Kobilka BK. Functionally different agonists induce distinct conformations in the G protein coupling domain of the beta 2 adrenergic receptor. J Biol Chem. 2001;276(27):24433–6. doi: 10.1074/jbc.C100162200. [DOI] [PubMed] [Google Scholar]
- 43.Angers S, Salahpour A, Bouvier M. Dimerization: an emerging concept for G protein-coupled receptor ontogeny and function. Annu Rev Pharmacol Toxicol. 2002;42:409–35. doi: 10.1146/annurev.pharmtox.42.091701.082314. [DOI] [PubMed] [Google Scholar]
- 44.Devi LA. Heterodimerization of G-protein-coupled receptors: pharmacology, signaling and trafficking. Trends Pharmacol Sci. 2001;22(10):532–7. doi: 10.1016/s0165-6147(00)01799-5. [DOI] [PubMed] [Google Scholar]
- 45.Bulenger S, Marullo S, Bouvier M. Emerging role of homo- and heterodimerization in G-protein-coupled receptor biosynthesis and maturation. Trends Pharmacol Sci. 2005;26(3):131–7. doi: 10.1016/j.tips.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 46.Javitch JA. The ants go marching two by two: oligomeric structure of G-protein-coupled receptors. Mol Pharmacol. 2004;66(5):1077–82. doi: 10.1124/mol.104.006320. [DOI] [PubMed] [Google Scholar]
- 47.Pin JP, Kniazeff J, Goudet C, Bessis AS, Liu J, Galvez T, Acher F, Rondard P, Prezeau L. The activation mechanism of class-C G-protein coupled receptors. Biol Cell. 2004;96(5):335–42. doi: 10.1016/j.biolcel.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 48.Kunishima N, Shimada Y, Tsuji Y, Sato T, Yamamoto M, Kumasaka T, Nakanishi S, Jingami H, Morikawa K. Structural basis of glutamate recognition by a dimeric metabotropic glutamate receptor. Nature. 2000;407(6807):971–7. doi: 10.1038/35039564. [DOI] [PubMed] [Google Scholar]
- 49.Tsuchiya D, Kunishima N, Kamiya N, Jingami H, Morikawa K. Structural views of the ligand-binding cores of a metabotropic glutamate receptor complexed with an antagonist and both glutamate and Gd3+ Proc Natl Acad Sci U S A. 2002;99(5):2660–5. doi: 10.1073/pnas.052708599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liang Y, Fotiadis D, Filipek S, Saperstein DA, Palczewski K, Engel A. Organization of the G protein-coupled receptors rhodopsin and opsin in native membranes. J Biol Chem. 2003;278(24):21655–62. doi: 10.1074/jbc.M302536200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Baneres JL, Parello J. Structure-based analysis of GPCR function: evidence for a novel pentameric assembly between the dimeric leukotriene B4 receptor BLT1 and the G-protein. J Mol Biol. 2003;329(4):815–29. doi: 10.1016/s0022-2836(03)00439-x. [DOI] [PubMed] [Google Scholar]
- 52.Mesnier D, Baneres JL. Cooperative conformational changes in a G-protein-coupled receptor dimer, the leukotriene B(4) receptor BLT1. J Biol Chem. 2004;279(48):49664–70. doi: 10.1074/jbc.M404941200. [DOI] [PubMed] [Google Scholar]
- 53.Sakmar TP. Structure of rhodopsin and the superfamily of seven-helical receptors: the same and not the same. Curr Opin Cell Biol. 2002;14(2):189–95. doi: 10.1016/s0955-0674(02)00306-x. [DOI] [PubMed] [Google Scholar]
- 54.Sakmar TP, Franke RR, Khorana HG. The role of the retinylidene Schiff base counterion in rhodopsin in determining wavelength absorbance and Schiff base pKa. Proc Natl Acad Sci U S A. 1991;88(8):3079–83. doi: 10.1073/pnas.88.8.3079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ridge KD, Abdulaev NG, Sousa M, Palczewski K. Phototransduction: crystal clear. Trends Biochem Sci. 2003;28(9):479–87. doi: 10.1016/S0968-0004(03)00172-5. [DOI] [PubMed] [Google Scholar]
- 56.Hubbell WL, Altenbach C, Hubbell CM, Khorana HG. Rhodopsin structure, dynamics, and activation: a perspective from crystallography, site-directed spin labeling, sulfhydryl reactivity, and disulfide cross-linking. Adv Protein Chem. 2003;63:243–90. doi: 10.1016/s0065-3233(03)63010-x. [DOI] [PubMed] [Google Scholar]
- 57.Farrens DL, Altenbach C, Yang K, Hubbell WL, Khorana HG. Requirement of rigid-body motion of transmembrane helices for light activation of rhodopsin. Science. 1996;274(5288):768–70. doi: 10.1126/science.274.5288.768. [DOI] [PubMed] [Google Scholar]
- 58.Dunham TD, Farrens DL. Conformational changes in rhodopsin. Movement of helix f detected by site-specific chemical labeling and fluorescence spectroscopy. J Biol Chem. 1999;274(3):1683–90. doi: 10.1074/jbc.274.3.1683. [DOI] [PubMed] [Google Scholar]
- 59.Lin SW, Sakmar TP. Specific tryptophan UV-absorbance changes are probes of the transition of rhodopsin to its active state. Biochemistry. 1996;35(34):11149–59. doi: 10.1021/bi960858u. [DOI] [PubMed] [Google Scholar]
- 60.Sheikh SP, Zvyaga TA, Lichtarge O, Sakmar TP, Bourne HR. Rhodopsin activation blocked by metal-ion-binding sites linking transmembrane helices C and F. Nature. 1996;383(6598):347–50. doi: 10.1038/383347a0. [DOI] [PubMed] [Google Scholar]
- 61.Altenbach C, Klein-Seetharaman J, Cai K, Khorana HG, Hubbell WL. Structure and function in rhodopsin: mapping light-dependent changes in distance between residue 316 in helix 8 and residues in the sequence 60–75, covering the cytoplasmic end of helices TM1 and TM2 and their connection loop CL1. Biochemistry. 2001;40(51):15493–500. doi: 10.1021/bi011545o. [DOI] [PubMed] [Google Scholar]
- 62.Altenbach C, Klein-Seetharaman J, Hwa J, Khorana HG, Hubbell WL. Structural features and light-dependent changes in the sequence 59–75 connecting helices I and II in rhodopsin: a site-directed spin-labeling study. Biochemistry. 1999;38(25):7945–9. doi: 10.1021/bi990014l. [DOI] [PubMed] [Google Scholar]
- 63.Altenbach C, Cai K, Khorana HG, Hubbell WL. Structural features and light-dependent changes in the sequence 306–322 extending from helix VII to the palmitoylation sites in rhodopsin: a site-directed spin-labeling study. Biochemistry. 1999;38(25):7931–7. doi: 10.1021/bi9900121. [DOI] [PubMed] [Google Scholar]
- 64.Ruprecht JJ, Mielke T, Vogel R, Villa C, Schertler GF. Electron crystallography reveals the structure of metarhodopsin I. Embo J. 2004;23(18):3609–3620. doi: 10.1038/sj.emboj.7600374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Salom D, Le Trong I, Pohl E, Ballesteros JA, Stenkamp RE, Palczewski K, Lodowski DT. Improvements in G protein-coupled receptor purification yield light stable rhodopsin crystals. J Struct Biol. 2006 doi: 10.1016/j.jsb.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 66.Mirzadegan T, Benko G, Filipek S, Palczewski K. Sequence analyses of G-protein-coupled receptors: similarities to rhodopsin. Biochemistry. 2003;42(10):2759–67. doi: 10.1021/bi027224+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cerione RA, Staniszewski C, Benovic JL, Lefkowitz RJ, Caron MG, Gierschik P, Somers R, Spiegel AM, Codina J, Birnbaumer L. Specificity of the functional interactions of the β-adrenergic receptor and rhodopsin with guanine nucleotide regulatory proteins reconstituted in phospholipid vesicles. J Biol Chem. 1985;260(3):1493–500. [PubMed] [Google Scholar]
- 68.Gether U, Lin S, Ghanouni P, Ballesteros JA, Weinstein H, Kobilka BK. Agonists induce conformational changes in transmembrane domains III and VI of the beta2 adrenoceptor. Embo J. 1997;16(22):6737–47. doi: 10.1093/emboj/16.22.6737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jensen AD, Guarnieri F, Rasmussen SG, Asmar F, Ballesteros JA, Gether U. Agonist-induced conformational changes at the cytoplasmic side of TM 6 in the {beta}2 adrenergic receptor mapped by site-selective fluorescent labeling. J Biol Chem. 2000 doi: 10.1074/jbc.M004871200. Record as supplied by publisher. [DOI] [PubMed] [Google Scholar]
- 70.Ghanouni P, Steenhuis JJ, Farrens DL, Kobilka BK. Agonist-induced conformational changes in the G-protein-coupling domain of the beta 2 adrenergic receptor. Proc Natl Acad Sci U S A. 2001;98(11):5997–6002. doi: 10.1073/pnas.101126198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yao X, Parnot C, Deupi X, Ratnala VR, Swaminath G, Farrens D, Kobilka B. Coupling ligand structure to specific conformational switches in the beta(2)-adrenoceptor. Nat Chem Biol. 2006 doi: 10.1038/nchembio801. [DOI] [PubMed] [Google Scholar]
- 72.Sheikh SP, Vilardarga JP, Baranski TJ, Lichtarge O, Iiri T, Meng EC, Nissenson RA, Bourne HR. Similar structures and shared switch mechanisms of the beta2-adrenoceptor and the parathyroid hormone receptor. Zn(II) bridges between helices III and VI block activation. J Biol Chem. 1999;274(24):17033–41. doi: 10.1074/jbc.274.24.17033. [DOI] [PubMed] [Google Scholar]
- 73.Elling CE, Frimurer TM, Gerlach LO, Jorgensen R, Holst B, Schwartz TW. Metal ion site engineering indicates a global toggle switch model for seven-transmembrane receptor activation. J Biol Chem. 2006;281(25):17337–46. doi: 10.1074/jbc.M512510200. [DOI] [PubMed] [Google Scholar]
- 74.Javitch JA, Fu D, Liapakis G, Chen J. Constitutive activation of the beta2 adrenergic receptor alters the orientation of its sixth membrane-spanning segment. J Biol Chem. 1997;272(30):18546–9. doi: 10.1074/jbc.272.30.18546. [DOI] [PubMed] [Google Scholar]
- 75.Rasmussen SG, Jensen AD, Liapakis G, Ghanouni P, Javitch JA, Gether U. Mutation of a highly conserved aspartic acid in the beta2 adrenergic receptor: constitutive activation, structural instability, and conformational rearrangement of transmembrane segment 6. Mol Pharmacol. 1999;56(1):175–84. doi: 10.1124/mol.56.1.175. [DOI] [PubMed] [Google Scholar]
- 76.Ward SD, Hamdan FF, Bloodworth LM, Wess J. Conformational Changes That Occur during M3 Muscarinic Acetylcholine Receptor Activation Probed by the Use of an in Situ Disulfide Cross-linking Strategy. J Biol Chem. 2002;277(3):2247–57. doi: 10.1074/jbc.M107647200. [DOI] [PubMed] [Google Scholar]
- 77.Ward SD, Hamdan FF, Bloodworth LM, Siddiqui NA, Li JH, Wess J. Use of an in situ disulfide cross-linking strategy to study the dynamic properties of the cytoplasmic end of transmembrane domain VI of the M3 muscarinic acetylcholine receptor. Biochemistry. 2006;45(3):676–85. doi: 10.1021/bi051503q. [DOI] [PubMed] [Google Scholar]
- 78.Han SJ, Hamdan FF, Kim SK, Jacobson KA, Bloodworth LM, Li B, Wess J. Identification of an agonist-induced conformational change occurring adjacent to the ligand-binding pocket of the M(3) muscarinic acetylcholine receptor. J Biol Chem. 2005;280(41):34849–58. doi: 10.1074/jbc.M506711200. [DOI] [PubMed] [Google Scholar]
- 79.Farahbakhsh ZT, Hideg K, Hubbell WL. Photoactivated conformational changes in rhodopsin: a time-resolved spin label study. Science. 1993;262(5138):1416–9. doi: 10.1126/science.8248781. [DOI] [PubMed] [Google Scholar]
- 80.Han M, Lin SW, Minkova M, Smith SO, Sakmar TP. Functional interaction of transmembrane helices 3 and 6 in rhodopsin. Replacement of phenylalanine 261 by alanine causes reversion of phenotype of a glycine 121 replacement mutant. J Biol Chem. 1996;271(50):32337–42. doi: 10.1074/jbc.271.50.32337. [DOI] [PubMed] [Google Scholar]
- 81.Jager S, Palczewski K, Hofmann KP. Opsin/all-trans-retinal complex activates transducin by different mechanisms than photolyzed rhodopsin. Biochemistry. 1996;35(9):2901–8. doi: 10.1021/bi9524068. [DOI] [PubMed] [Google Scholar]
- 82.Kenakin T. Efficacy at G-protein-coupled receptors. Nat Rev Drug Discov. 2002;1(2):103–10. doi: 10.1038/nrd722. [DOI] [PubMed] [Google Scholar]
- 83.Kenakin T. Ligand-selective receptor conformations revisited: the promise and the problem. Trends Pharmacol Sci. 2003;24(7):346–54. doi: 10.1016/S0165-6147(03)00167-6. [DOI] [PubMed] [Google Scholar]
- 84.Lefkowitz RJ, Cotecchia S, Samama P, Costa T. Constitutive activity of receptors coupled to guanine nucleotide regulatory proteins. Trends Pharmacol Sci. 1993;14(8):303–7. doi: 10.1016/0165-6147(93)90048-O. [DOI] [PubMed] [Google Scholar]
- 85.Leff P. The two-state model of receptor activation [see comments] Trends Pharmacol Sci. 1995;16(3):89–97. doi: 10.1016/s0165-6147(00)88989-0. [DOI] [PubMed] [Google Scholar]
- 86.Weiss JM, Morgan PH, Lutz MW, Kenakin TP. The cubic ternary complex receptor-occupancy model. III. resurrecting efficacy. J Theor Biol. 1996;181(4):381–97. doi: 10.1006/jtbi.1996.0139. [DOI] [PubMed] [Google Scholar]
- 87.Kenakin T. Inverse, protean, and ligand-selective agonism: matters of receptor conformation. Faseb J. 2001;15(3):598–611. doi: 10.1096/fj.00-0438rev. [DOI] [PubMed] [Google Scholar]
- 88.Swaminath G, Xiang Y, Lee TW, Steenhuis J, Parnot C, Kobilka BK. Sequential Binding of Agonists to the {beta}2 Adrenoceptor: Kinetic evidence for intermediate conformational states. J Biol Chem. 2004;279(1):686–691. doi: 10.1074/jbc.M310888200. [DOI] [PubMed] [Google Scholar]
- 89.Swaminath G, Deupi X, Lee TW, Zhu W, Thian FS, Kobilka TS, Kobilka B. Probing the beta2 Adrenoceptor Binding Site with Catechol Reveals Differences in Binding and Activation by Agonists and Partial Agonists. J Biol Chem. 2005;280(23):22165–71. doi: 10.1074/jbc.M502352200. [DOI] [PubMed] [Google Scholar]
- 90.O’ Dowd BF, Hnatowich M, Regan JW, Leader WM, Caron MG, Lefkowitz RJ. Site-directed mutagenesis of the cytoplasmic domains of the human β2-adrenergic receptor. Localization of regions involved in G protein-receptor coupling. J Biol Chem. 1988;263(31):15985–92. [PubMed] [Google Scholar]
- 91.Liggett SB, Caron MG, Lefkowitz RJ, Hnatowich M. Coupling of a mutated form of the human beta 2-adrenergic receptor to Gi and Gs. Requirement for multiple cytoplasmic domains in the coupling process. J Biol Chem. 1991;266(8):4816–21. [PubMed] [Google Scholar]
- 92.Subramaniam S, Henderson R. Molecular mechanism of vectorial proton translocation by bacteriorhodopsin [see comments] Nature. 2000;406(6796):653–7. doi: 10.1038/35020614. [DOI] [PubMed] [Google Scholar]
- 93.Arnis S, Fahmy K, Hofmann KP, Sakmar TP. A conserved carboxylic acid group mediates light-dependent proton uptake and signaling by rhodopsin. J Biol Chem. 1994;269(39):23879–81. [PubMed] [Google Scholar]
- 94.Hoshi T, Zagotta WN, Aldrich RW. Shaker potassium channel gating. I: Transitions near the open state. J Gen Physiol. 1994;103(2):249–78. doi: 10.1085/jgp.103.2.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Vilardaga JP, Steinmeyer R, Harms GS, Lohse MJ. Molecular basis of inverse agonism in a G protein-coupled receptor. Nat Chem Biol. 2005;1(1):25–8. doi: 10.1038/nchembio705. [DOI] [PubMed] [Google Scholar]
- 96.Hoffmann C, Gaietta G, Bunemann M, Adams SR, Oberdorff-Maass S, Behr B, Vilardaga JP, Tsien RY, Ellisman MH, Lohse MJ. A FlAsH-based FRET approach to determine G protein-coupled receptor activation in living cells. Nat Methods. 2005;2(3):171–6. doi: 10.1038/nmeth742. [DOI] [PubMed] [Google Scholar]
- 97.Shi L, Liapakis G, Xu R, Guarnieri F, Ballesteros JA, Javitch JA. Beta2 adrenergic receptor activation. Modulation of the proline kink in transmembrane 6 by a rotamer toggle switch. J Biol Chem. 2002;277(43):40989–96. doi: 10.1074/jbc.M206801200. [DOI] [PubMed] [Google Scholar]
- 98.Mansoor SE, McHaourab HS, Farrens DL. Mapping proximity within proteins using fluorescence spectroscopy. A study of T4 lysozyme showing that tryptophan residues quench bimane fluorescence. Biochemistry. 2002;41(8):2475–84. doi: 10.1021/bi011198i. [DOI] [PubMed] [Google Scholar]
- 99.Parnot C, Miserey-Lenkei S, Bardin S, Corvol P, Clauser E. Lessons from constitutively active mutants of G protein-coupled receptors. Trends Endocrinol Metab. 2002;13(8):336–43. doi: 10.1016/s1043-2760(02)00628-8. [DOI] [PubMed] [Google Scholar]
- 100.Strader C, Candelore M, Hill W, Sigal I, Dixon R. Identification of two serine residues involved in agonist activation of the β-adrenergic receptor. J Biol Chem. 1989;264:13572–13578. [PubMed] [Google Scholar]
- 101.Wieland K, Zuurmond HM, Krasel C, Ijzerman AP, Lohse MJ. Involvement of Asn-293 in stereospecific agonist recognition and in activation of the beta 2-adrenergic receptor. Proc Natl Acad Sci U S A. 1996;93(17):9276–81. doi: 10.1073/pnas.93.17.9276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Liapakis G, Ballesteros JA, Papachristou S, Chan WC, Chen X, Javitch JA. The forgotten serine. A critical role for Ser-2035.42 in ligans binding to and activation of the beta 2-adrenergic receptor. J Biol Chem. 2000;275(48):37779–88. doi: 10.1074/jbc.M002092200. [DOI] [PubMed] [Google Scholar]
- 103.Strader CD, Candelore MR, Hill WS, Dixon RA, Sigal IS. A single amino acid substitution in the beta-adrenergic receptor promotes partial agonist activity from antagonists. J Biol Chem. 1989;264(28):16470–7. [PubMed] [Google Scholar]