

Summary
Periventricular heterotopia (PVH) is a congenital malformation of human cerebral cortex frequently associated with Filamin-A (FLN-A) mutations but the pathogenetic mechanisms remain unclear. Here we show that the MEKK4 (MAP3K4) pathway is involved in Fln-A regulation and PVH formation. MEKK4−/− mice developed PVH associated with breaches in the neuroependymal lining which were largely comprised of neurons that failed to reach the cortical plate. RNA interference (RNAi) targeting MEKK4 also impaired neuronal migration. Expression of Fln was elevated in MEKK4−/− forebrain, most notably near sites of failed neuronal migration. Importantly, recombinant-MKK4 protein precipitated a complex containing MEKK4 and Fln-A, and MKK4 mediated signaling between MEKK4 and Fln-A, suggesting that MKK4 may bridge these molecules during development. Finally, we showed that wild-type FLN-A over-expression inhibited neuronal migration. Collectively, our results demonstrate a link between MEKK4 and Fln-A that impacts neuronal migration-initiation and provides insight into the pathogenesis of human PVH.
Introduction
Cerebral cortical development is a dynamic process that requires complex but precise coordination of cell proliferation, death, migration and differentiation (Marin and Rubenstein, 2003; Rakic, 1988b). Cortical malformations are often associated with mutations in genes that regulate each of these processes (Bielas et al., 2004; Haydar et al., 1999; Rakic, 1988a; Walsh, 1999). Mitogen-activated protein kinases (MAPKs) are intracellular signal transduction molecules expressed in all eukaryotic cells that modulate these basic cellular events by responding to context-dependent extracellular signals (Morrison and Davis, 2003). MAPKs are activated via signaling cascades involving MAPK kinases (MAP2Ks) that are in turn activated by MAPK kinase kinases (MAP3Ks). There are five major subgroups of MAPKs, namely ERK1/2, JNK, p38, ERK5 and ERK7 (Johnson et al., 2005). The c-Jun NH2-terminal kinase (JNK) subgroup of MAPKs has been reported to affect region-specific cell death during early CNS development (Kuan et al., 2000). More recently, JNK was shown to regulate cytoskeletal movement and cell migration by maintaining microtubule stability (Chang et al., 2003; Huang et al., 2003). JNK binds and phosphorylates the microtubule associated protein, doublecortin (DCX) in growth cones of migrating neurons (Gdalyahu et al., 2004), and over-expression of either dominant-negative JNK or MUK (a mixed lineage kinase and regulator of JNK activity) can impede radial neuronal migration (Hirai et al., 2002; Hirai et al., 2005; Kawauchi et al., 2003). One of the MAP2Ks that activates JNK, MKK4 (also known as SEK1), can interact with Filamin-A (FLN-A), an actin-binding protein essential for cytoskeletal rearrangement and cell locomotion (Marti et al., 1997; Stossel et al., 2001). DCX and FLN-A mutations in human cause subcortical band and periventricular nodular heterotopias, respectively (Fox et al., 1998; Gleeson et al., 1998). Thus, JNK or its upstream regulators may be involved in the pathogenesis of these human disorders. How this pathway is regulated during neural development remains poorly understood.
MEKK4 (MTK1 in human) is one of ~17 MAP3Ks cloned from mammalian cells that affects the activity of downstream MAP2Ks and MAPKs including JNK and p38 (Gerwins et al., 1997; Takekawa et al., 2005). MEKK4 activity is stimulated by growth factors (e.g. EGF), inflammatory cytokines (e.g. angiotensin II) and environmental stress (e.g. MMS, UV and γ-irradiation) (Abell et al., 2005; Derbyshire et al., 2005; Fanger et al., 1997; Gerwins et al., 1997). Mice deficient in MEKK4 or possessing kinase-dead MEKK4 (MEKK4K1361R) develop CNS phenotypes including severe neural tube closure defects and massive neuroepithelial apoptosis (Abell et al., 2005; Chi et al., 2005). These mice exhibited reduced MKK4/SEK1 or MKK3/6 activity in the developing neuroepithelium but no detectable changes in JNK activity. The activity of p38, however, was altered in MEKK4K1361R fibroblasts and affected downstream targets that possibly contributed to actin cytoskeletal breakdown. Thus, MEKK4 activity may be necessary for the integrity and rearrangement of the actin cytoskeleton (Abell et al., 2005; Yuzyuk and Amberg, 2003). Notably, MEKK4−/− mice frequently develop PVH that manifest as cell accumulations lining and protruding into the lateral ventricles (Chi et al., 2005). This suggests that MEKK4 may be required for cytoskeletal stability or integrity of the ventricular surface lining the developing forebrain (FB) which could influence neuronal migration away from the proliferative zone (PZ). Because a role for MEKK4 in developing FB is unknown, we hypothesized that MEKK4 regulates migration within and/or away from the cortical PZ by regulating key modulators of the actin cytoskeleton suspected to be essential during this process.
Here we show that MEKK4 is expressed in embryonic mouse and human FB. PVH in MEKK4−/− mice arise from breaches in the ventricular surface lining and are largely comprised of post-mitotic neurons. Cells within PVH fail to leave the VZ surface and migrate into the CP. Likewise; in utero electroporation of short interference RNA (siRNA) against MEKK4 impairs neuronal migration. Surprisingly, MEKK4 suppression results in abnormally high Fln-A expression and phosphorylation at Ser2152, a residue implicated in Fln-A cleavage and modulation of actin dynamics. We show that MKK4/SEK1 can interact with and mediate signaling between MEKK4 and Fln-A. Lastly, over-expression of FLN-A can inhibit neuronal migration. Together our results indicate that MEKK4 contributes to the integrity of the VZ surface, regulates the amount of Fln and mediates cell migration during FB development.
Results
MEKK4 is expressed in progenitors and neurons in fetal and adult murine and human forebrain
MTK1 mRNA is ubiquitously expressed in fetal and adult CNS (Chan-Hui and Weaver, 1998; Takekawa et al., 1997), although its expression pattern in developing FB has not been well characterized. In vitro studies have localized MEKK4 to the cytoplasm and in peri-nuclear vesicles (Gerwins et al., 1997; Halfter et al., 2004; Takekawa and Saito, 1998). MEKK4 expression during mouse development was recently reported to be enriched throughout the entire neural tube as early as embryonic day (E) 8.5 (Abell et al., 2005; Chi et al., 2005). To better localize MEKK4 signal in both developing and adult FB, we probed E16.5 and adult FB sections using in-situ hybridization. MEKK4 expression in the embryo spanned the telencephalic wall (Supplementary Figure S1A) as well as all neocortical lamina of the adult FB (Supplementary Figure S1B, C). Next we performed western blot analysis to determine whether MEKK4 protein is expressed in developing human FB. We found that MEKK4 was expressed in human fetal FB during peak ages of neurogenesis and migration (Supplementary Figure S1D). These data indicate that MEKK4 is expressed in neural progenitors and may play multiple roles throughout development and adulthood.
MEKK4−/− mice develop PVH largely comprised of differentiated neurons
MEKK4 mutation results in highly penetrant neural tube defects and ~50% of MEKK4−/− mice exhibited cranial exencephaly (Chi et al., 2005). Among these affected mice, ~70% display highly degenerated FB (“small FB”) due to massive apoptosis beginning ~E9.5–10.5, while ~30% display relatively normal size to slightly enlarged FB (“big FB”) compared to MEKK4+/+ or +/− mice (Chi et al., 2005). Big FB MEKK4 mutants exhibited bilateral PVH that were characterized by focal disruptions of the VZ/SVZ and cell expansion into the ventricular space (Figure 1A). The intermediate zone (IZ) was typically thinner than controls. Remarkably, a relatively organized cortical plate (CP) overlay PVHs, although we periodically observed subpial ectopias (Figure 1A, B) and polymicrogyria (Figure 1C). It was also not uncommon to observe an overall decrease in CP thickness in big FB mutants (Figure 1E, H). PVH and ectopias were present in small FB phenotype as well (see below). In both phenotypes, disruptions of the VZ surface were more prevalent than ectopias. These observations suggest that despite a dramatic difference in size and morphology, both small and big FB MEKK4 mutants may share common molecular mechanisms that contribute to the disruptions of the ventricular and pial surfaces.
Figure 1. Cortical malformations in MEKK4−/− mice.
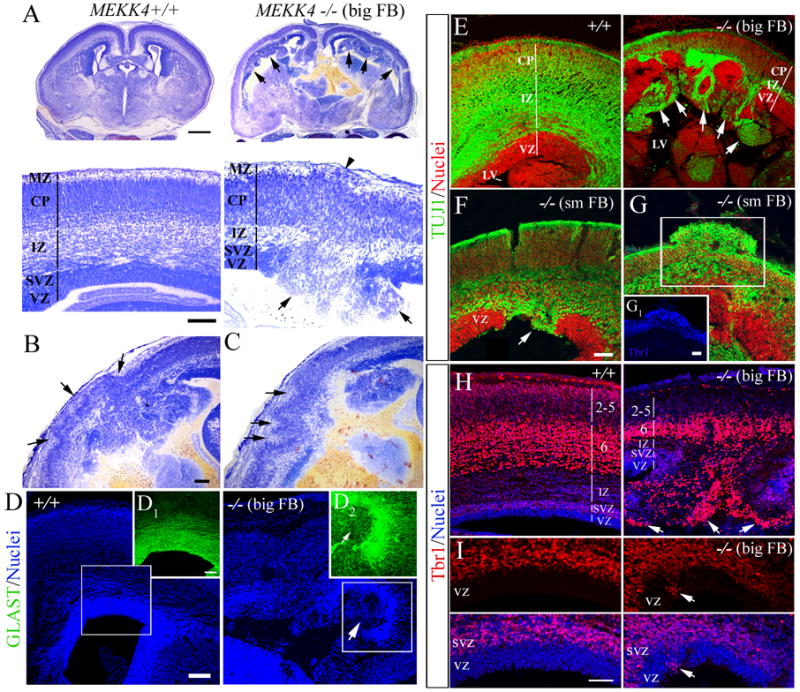
(A) Nissl staining of E17.5 MEKK4+/+ and MEKK4−/− (big FB phenotype) coronal forebrain sections. Lower magnification (upper panels) revealed prominent bilateral PVH (arrows) that disrupted the continuity of the VZ/SVZ. Higher magnification of the telencephalic wall (lower panels) shows an example of a focal disruption of the VZ surface (arrows). Also observed was reduced thickness of the intermediate zone (IZ) in MEKK4−/− compared to control. The arrowhead points to a small subpial ectopia in the marginal zone (MZ) (arrowhead). Bars (μm) in A (upper)= 500, A (lower)=100.
(B) Example of a large subpial ectopia (arrows) overlying a heterotopic area in MEKK4−/− big FB. Bar in B=100μm.
(C) Example of polymicrogyria (arrows) overlying a heterotopic area in MEKK4−/− big FB.
(D) GLAST immunoreactivity (green) and nuclear staining (blue) of MEKK4+/+ and −/− (big FB). Insets of boxed areas of nuclei (blue) show strong GLAST expression in control VZ (D1) compared to low expression in a PVH (D2, arrow) of the mutant. Bars (μm) in D = 100, D1 = 50.
(E) TUJ1 immunoreactivity showed that PVH protruding into the lateral ventricle (LV) in MEKK4−/− big FB were TUJ1+ (arrows) compared to MEKK4+/+ (left panel) where the VZ lacked TUJ1. Propidium iodide (PI) was used to label nuclei (red).
(F and G) TUJ1 immunoreactivity (green) and nuclei (red) in MEKK4−/− small FB phenotype.
(F) Example of a PVH (arrow) that disrupted the VZ continuity and strongly expressed TUJ1.
(G) An ectopia disrupting the pial surface that was was positive for TUJ1 and Tbr1 (G1). Bars in G, G1= 50μm.
(H and I) Tbr1 immunoreactivity (red) and TO-PRO-3-labeled nuclei (blue) in MEKK4+/+ and −/− big FB.
(H) Low magnification revealed many intensely labeled Tbr1+ cells in layer 6 of +/+ (left panel) and −/− (right panel). Note the numerous heavily labeled Tbr1+ cells (arrows) in −/− PVH.
(I) High magnification of the VZ revealed a small cluster of Tbr1+ cells in the mutant VZ (right panels, arrows) compared to +/+ (left panels) where Tbr1 was normally absent. Bar= 50μm.
To determine the cellular composition of PVHs, we immunostained E18.5 FB with glial (GLAST) and neuronal (TUJ1) markers. PVHs weakly expressed GLAST (Figure 1D) but showed strong TUJ1 expression in both phenotypes (Figure 1E, F). Ectopias in small FB phenotype also expressed TUJ1 (Figure 1G) and Tbr1 (Figure 1G1), a transcription factor expressed by early-born glutamatergic cortical progenitors soon after differentiation and strongly expressed by layer 6 cells (Hevner et al., 2001). Tbr1 immunostaining in big FB phenotypes revealed that PVHs contained many intensely labeled Tbr1+ cells suggesting many cells failed to reach layer 6 (Figure 1H). In addition to large PVH, we found Tbr1+ cells within “normal” areas of the VZ (where Tbr1 is normally absent) in both big (Figure 1I) and small FB (see below) phenotypes. These results suggest that small or widespread disruptions of the VZ surface in MEKK4 mutants may result in ectopic differentiation and/or inhibited migration of cortical neurons.
Failure of BrdU-positive cells to leave the ventricular surface and PVH in MEKK4−/− embryos
The presence of PVH suggests that some cells are unable to migrate away from the VZ in MEKK4−/− FB. To test this, we exposed mice to bromodeoxyuridine (BrdU), which is incorporated into cells during S-phase and examined the fates of BrdU+ cells. Short exposure (1h) at E14.5 revealed comparable levels of S-phase cells along the VZ of MEKK4−/− and MEKK4+/+ mice (data not shown). Longer exposure (96h) at E18.5 in MEKK4−/− mice with big FB showed heavily labeled BrdU+ cells within PVH (Figure 2A). BrdU+ cells within PVH were TUJ1+, indicating that these neurons failed to migrate away from the PVH (Figure 2A). Consistent with these findings, short exposure (1h) of BrdU to E18.5 mice with big FB revealed that PVH were largely BrdU− (data not shown), suggesting that PVH are largely differentiated structures. Additionally, after 96h we observed many heavily labeled BrdU+ cells at the VZ surface in MEKK4−/− with small FB phenotype compared to MEKK4+/+ mice (Figure 2B). These cells were located in abnormally TUJ1+ regions of the VZ (data not shown). Thus, Tbr1 immunostaining (Figure 1) combined with the BrdU data suggests that PVH form during early-mid neurogenesis (i.e. E14.5 or earlier) in MEKK4−/− mice with either FB phenotype. The presence of BrdU+/TUJ1+ cells at the VZ surface suggests that certain neural progenitors in MEKK4−/− FB have a marked defect in their migratory capacity to exit the VZ surface. While the most severe defects were largely restricted to areas of VZ surface disruption, we cannot rule out more subtle migration defects that may affect non-heterotopic regions of MEKK4−/− FB.
Figure 2. Failure of BrdU-positive cells to leave the ventricular surface and PVH in MEKK4-deficient forebrain.
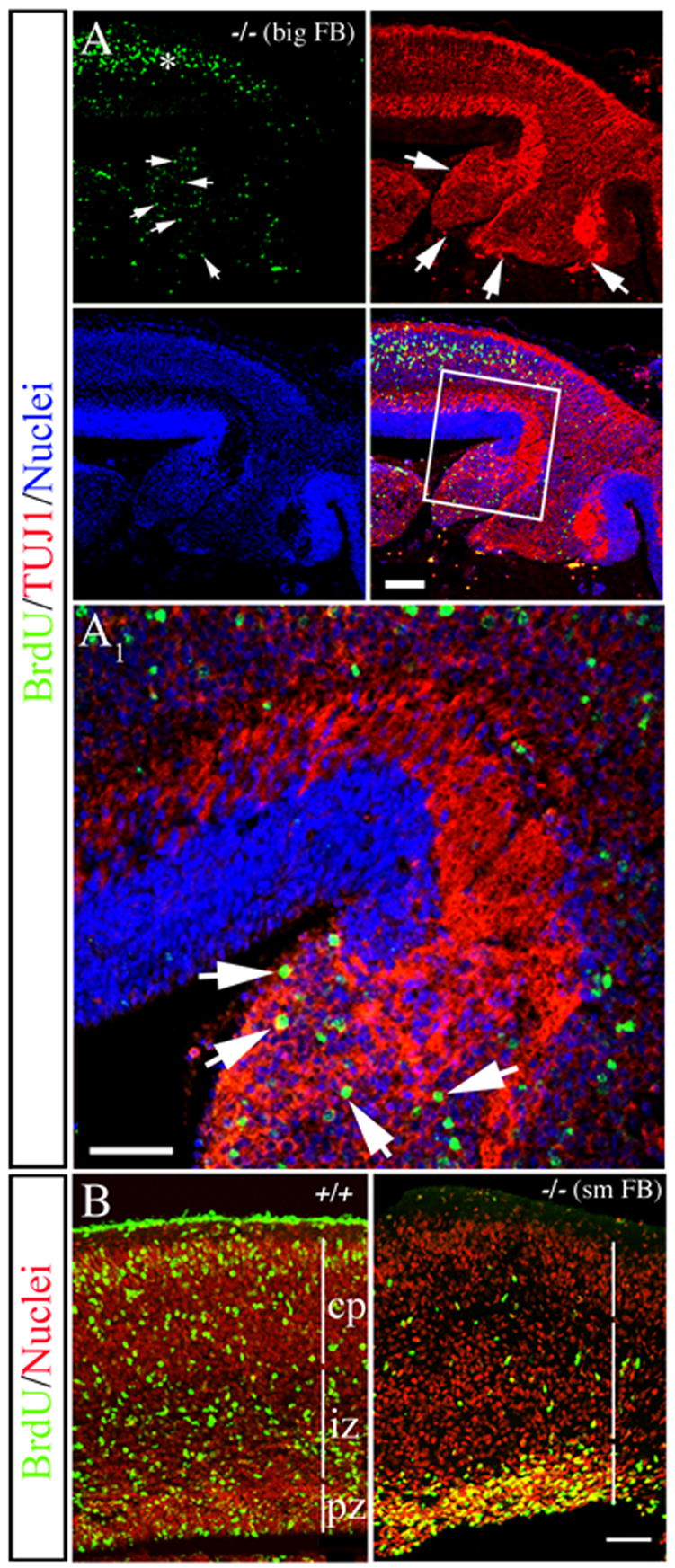
(A and B) Dams were exposed to BrdU at E14.5 and the forebrain (FB) was analyzed at E18.5.
(A) Analysis of a PVH from a MEKK4−/− “big FB” phenotype stained for BrdU (green), TUJ1 (red) and TO-PRO-3 (blue). Compared to adjacent neocortex (asterisk), heavily labeled BrdU+ cells (arrows in BrdU) co-localized with TUJ1+ (arrows) in the PVH. (A1) Higher magnification of the boxed region in A shows heavily labeled BrdU+/TUJ1+ cells (arrows) within the heterotopia. Bars = 50μm.
(B) Example of a MEKK4+/+ and MEKK4−/− with a “small FB” phenotype (sm FB) where BrdU+ cells (green) failed to leave the VZ surface compared to +/+. Nuclei were labeled with propidium iodide (red). Bar =100 μm.
In vitro and in vivo RNAi-mediated knockdown of MEKK4
Deleting the MEKK4 gene on a pure C57BL/6 mouse background caused neurulation defects ~E9.0 that spanned the FB and hindbrain neuroepithelium (Chi et al., 2005). PVH formation and subsequent migration defects could be secondary to failure of the neural tube to fuse properly. To circumvent this confounding issue, we designed siRNA against MEKK4 mRNA to assess whether depletion of MEKK4 in normal mouse FB neuroepithelium affected cell migration. We screened for effective siRNA constructs using the mU6pro vector (Yu et al., 2002) and transfected NIH3T3 cells with plasmids expressing red fluorescent protein (RFP), full-length FLAG-MEKK4 and either mU6pro vector alone or containing siRNA#555, siRNA#555 with 4 point mutations (siRNA#5554pt), or siRNA#555 scrambled (siRNA#555SCR) sequences (see Experimental Procedures for sequence details). Immunostaining for FLAG revealed that siRNA#555 dramatically reduced FLAG-MEKK4 expression compared to mU6pro alone (not shown) or siRNA#5554pt and siRNA#555SCR (Supplemental Figure S2A). Western blot analysis confirmed the knockdown of both FLAG-MEKK4 (Supplemental Figure S2B) and endogenous MEKK4 (Supplemental Figure S2C). In addition, we found that 48h post-in utero electroporation of E14.5 FB, siRNA#555 dramatically reduced MEKK4 mRNA compared to the contralateral hemisphere or siRNA#555SCR (Supplemental Figure S2D). Thus, siRNA#555 can be used to knockdown MEKK4 expression both in vitro and in vivo.
MEKK4 RNAi disrupts the migration of neocortical progenitors
We hypothesized that knocking down MEKK4 in vivo might inhibit cell migration in the developing FB. To test this, we used in-utero electroporation to deliver a 3:1 mixture of MEKK4 siRNA to RFP. Because the siRNA-containing mU6pro vector does not express a reporter gene, we performed control experiments which showed that co-electroporation of RFP and GFP results in ~90% co-transfection (data not shown) which was similar to other reports (Bai et al., 2003). We electroporated E14.5 FB with RFP alone (n=7), siRNA control (siRNA#5554pt (n=2) or siRNA#555SCR (n=2)) plus RFP, or siRNA#555 and RFP (n=7) and analyzed the fates of RFP+ cells at P0. In controls, we found that the majority of RFP+ cells were located within the CP (Figure 3A). In contrast, electroporation of siRNA#555 resulted in dramatically more RFP+ cells within the corpus callosum (CC) and PZ, and considerably fewer cells within the CP (Figure 3A). In one E15.5 dam, we electroporated siRNA#555SCR plus GFP into the left hemisphere and siRNA#555 plus RFP into the right hemisphere. Analysis at P0 revealed that the majority of RFP+ cells failed to enter the CP compared to GFP+ cells (Figure 3B). Quantification of RFP+ somas throughout the FB showed that compared to controls, siRNA#555 resulted in ~50% fewer cells reaching the CP and significantly more cells within the CC (~40% compared to ~3% in controls) and PZ (~3% compared to ~0.5% in controls) (Figure 3C). Inhibited migration by siRNA#555 was also observed after 72h (data not shown). In additional experiments, we exposed electroporated mice to BrdU 24h after electroporation (E15.5). We reasoned that at electroporated sites, since nearly all cells in the VZ would be RFP+, exposure to BrdU should label a significant percentage of RFP+ cells. Analysis of BrdU and RFP at P0, after transfection with MEKK4 siRNA#555, revealed many RFP+/BrdU+ cells in the CC compared to control (RFP alone) where most RFP+/BrdU+ cells were located in the CP (Supplementary Figure S3). In siRNA#555-transfected brains, there appeared to be fewer BrdU+ cells in the CP region located above the heterotopia compared to adjacent neocortical areas or comparable regions of the opposite hemisphere (Figure 3D). In separate experiments, we tested whether some cells arrested at the VZ surface were neuronal by co-electroporating siRNA#555 with Tα1 Venus-GFP (a neuronal-specific promoter (Gloster et al., 1999)). Six days after co-electroporation, many GFP+ cells were stuck at the VZ surface in siRNA#555-transfected brains compared to control (Figure 3E). Although examination of radial glial morphology at a range of post-electroporation timepoints (e.g., 24, 72 and 96h) did not show grossly affected morphology (data not shown), we cannot rule out that siRNA#555 resulted in subtle radial glial defects affecting migration. In any event, these results suggest that MEKK4 is important for neuronal progenitor cells to migrate away from both the VZ and IZ.
Figure 3. MEKK4 siRNA impairs radial migration of neocortical neurons.
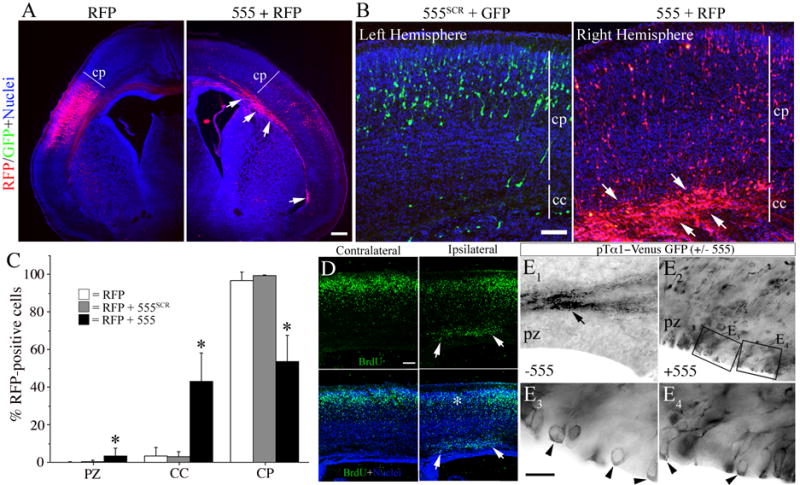
(A, C, D and E) In utero electroporation was performed at E14.5 and forebrains were examined at P0.
(A) Transfection with RFP alone (left) resulted in the majority of cells reaching the cortical plate (CP). In contrast, RFP+ cells co-transfected with siRNA#555 (arrows, right) were largely stuck in the corpus callosum (CC). Bar = 200μm.
(B) Example of an E15.5 brain electroporated with siRNA#555SCR and GFP into the left hemisphere and siRNA#555 and RFP into the right hemisphere. At P0, siRNA#555 caused the majority of RFP+ cells to arrest in the CC whereas most GFP+ cells reached the CP. Bar= 50μm.
(C) Quantification of the percentage of RFP+ cells in the proliferative zone (PZ), CC or CP from mice electroporated at E14.5 and sacrificed at P0. siRNA#555 (black bars) caused ~50% reduction of cells in the CP and significantly more cells to accumulate in the CC and PZ compared to RFP alone (white bars) or siRNA#555SCR (grey bars). *p<0.01 (ANOVA).
(D) In utero electroporation of fetuses at E14.5, exposure of dams to BrdU at E15.5, and analysis at P0. Example of a brain electroporated with siRNA#555 shows a heterotopia (arrows) beneath the CP (ipsilateral) that contained many BrdU+ cells (arrows). In the ipsilateral area of CP (corresponding to the asterisk in lower panel) reduced BrdU immunostaining is observed over the heterotopia compared to the adjacent CP or opposite hemisphere (contralateral). Bar =100 μm.
(E) Electroporation of pTα1-Venus-GFP alone (E1) or with siRNA#555 (E2–4) and immunostaining for GFP. (E1) Electroporation of pTα1-VenusGFP alone (−555) resulted in most GFP+ cells reaching the cortical plane (not visible in picture) with their axonal projections (arrow). (E2) Co-electroporation with siRNA#555 (+555) resulted in many GFP+ cells at the VZ surface. (E3 and E4) Higher magnifications of the boxed areas in E2 revealed cell bodies that remained at the VZ surface (arrowheads).
Deleting MEKK4 in mice results in enhanced apoptosis ~E8.5–9.0 and frequent FB degeneration (Chi et al., 2005). We therefore hypothesized that MEKK4 RNAi may enhance cell death if MEKK4 additionally plays an anti-apoptotic role. Analysis of the morphology of RFP+ cells at P0 in brains transfected with siRNA #555 revealed many unhealthy, apoptotic profiles in the CP, CC and VZ/SVZ (data not shown). We stained for apoptotic nuclei using the TUNEL method and found that compared to RFP alone (or 555SCR), siRNA#555 resulted in many TUNEL+/RFP+ cells in the VZ/SVZ, CC and CP (Supplementary Figure S4). Together, the results suggest that in addition to playing a role in neuronal migration, MEKK4 may also have an anti-apoptotic function that ensures the survival of proliferating, migrating and differentiating neurons.
Compromised integrity of the VZ surface may predispose MEKK4 mutants to PVH
Radial glial endfeet form an adhesive lining at both the VZ and pial surface (Levitt and Rakic, 1980), and defects in these structures could predispose mice to PVH. Integrins are involved in the assembly of the basal lamina and radial glial end-feet anchoring. Defects in integrin receptors, integrin-linked kinases as well as dystroglycan complexes have been implicated in the pathogenesis of breeches at the outer cortical surface (Moore et al., 2002; Niewmierzycka et al., 2005; Schmid et al., 2004). We therefore examined whether there were extracellular matrix abnormalities in MEKK4−/− FB by probing for laminin, a glycoprotein that binds cell membranes via integrin receptors and the dystroglycan complex (Campos et al., 2004; Powell and Kleinman, 1997). Consistent with previous studies, laminin was detected in the pia, blood vessels and along the VZ surface of MEKK4+/+ FB (Figure 4A). Co-immunostaining with RC2, a radial glial marker, revealed laminin and RC2-labeled radial glial endfeet at the VZ surface (Figure 4A). In contrast, we found areas of severe discontinuity in laminin staining at both the VZ and pial surface of MEKK4−/− small (Figure 4B, D) and big FB (Figure 4C) phenotypes. The morphology and organization of RC2+ fibers was markedly disrupted at sites where laminin was disrupted (Figure 4B). Remarkably, RC2+ fibers adjacent to PVH appeared to navigate around PVH areas to establish a normal pattern of radial glial scaffolding in the upper regions of the CP even when the underlying areas were severely disrupted (Supplementary Figure S5). Additionally, at sites of ectopias, RC2+ fibers were observed extending beyond the pia mater (Figure 4D). These observations might explain how a relatively organized CP forms over PVHs. Furthermore, staining of MEKK4−/− big FB with the F-actin-binding probe, phalloidin, appeared to show disruptions of the adherens junctions complexes along the VZ surface (Figure 4E). Notably, these disruptions also showed invasion of nuclei into the ventricular space (Figure 4E). Together, these results suggest that the loss of MEKK4 may lead to defects in the VZ surface integrity that promote the formation of PVH.
Figure 4. Defects along the ventricular and pial surface in MEKK4−/− brain.
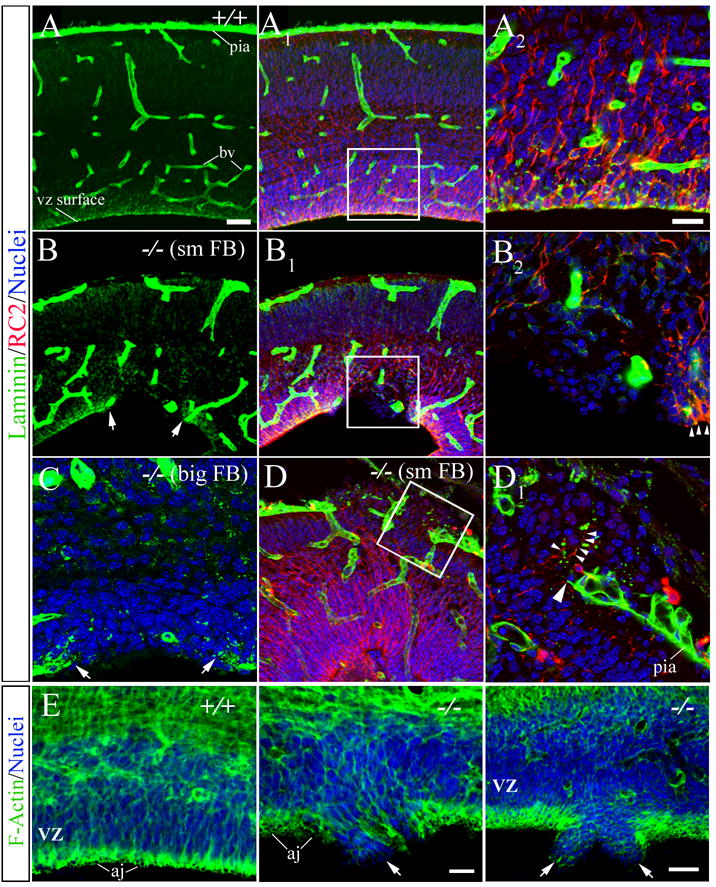
(A thru D) Laminin (green) and RC2 (red) immunostaining and TO-PRO-3-labeled nuclei (blue) in E17.5–18.5 wildtype (+/+) and MEKK4−/− small (sm) and big FB.
(A) Laminin expression in +/+ was observed at the pial surface, blood vessels (bv) and along the VZ surface. (A1) RC2+ fibers appeared normal in +/+ connecting the VZ and pial surface. (A2) Higher magnification of the boxed region in A1 shows RC2+ radial glial endfeet and laminin coincided along the VZ surface. Bar in A (also for A1, B, B1 and D) = 50μm, A2 (also for B2, C, and D1)= 20μm.
(B) Laminin staining in −/− sm FB showed an area of disrupted VZ labeling (arrows) at the site of a heterotopia. The lack of pial surface labeling was due to the meninges being separated away from the section. (B1) RC2+ fibers are disrupted at the heterotopic region (boxed area). (B2) Higher magnification of the boxed area in B1 shows abrupt ending of radial glial endfeet (arrowheads) that also corresponded to a lack of laminin immunoreactivity.
(C) Discontinuous laminin staining (arrows) along the VZ surface in −/− big FB mutants.
(D) An example of an ectopia in −/− sm FB that was associated with both a breech in the pial surface and extension of RC2+ fibers into the ectopia. (D1) Higher magnification of the boxed region in D shows disrupted pia label (large arrowhead) and extension of RC2+ fibers into the ectopia (small arrowheads).
(E) Phalloidin-labeling of F-actin (green) revealed defects in the continuity of the adherens junctions complexes (aj) along the VZ surface in MEKK4−/− big FB (arrows in middle, right panels) compared to +/+. Nuclei (blue) appeared to be invading the ventricular space at these disrupted sites (arrows). Bars (μm) representing middle (and left) panel = 20 and right = 50.
Elevated Fln expression in MEKK4−/− mice
Because bilateral PVH are common in patients with loss-of-function FLN-A mutations (Fox et al., 1998; Sheen et al., 2001), we used western blot and immunostaining to analyze Fln levels in MEKK4 +/+, +/− and −/− FB. Surprisingly, western blot analysis at E15.5 (Figure 5A,B) or E16.5 (Figure 5C) revealed that MEKK4−/− FB (regardless of small or big phenotype) showed increased Fln compared to MEKK4+/− or +/+ FB. Fln-A and Fln-B are the two major isoforms expressed in the developing brain (Sheen et al., 2002). To determine which isoform contributed to increased Fln in MEKK4−/− FB, we probed blots with Fln-A and Fln-B-specific antibodies. We found that MEKK4-deficient FB showed increased Fln-A (Figure 5B) and Fln-B (Figure 5A). To determine where these changes occurred in MEKK4−/− FB, we immunostained E17.5 FB with phospho-Fln-A, Fln-A and Fln-B antibodies. In controls, all Fln antibodies showed comparable immunostaining patterns with the strongest signal located in the IZ and CP (Figure 5D, E and Supplemental Figure S6). Blood vessel staining was also detected with some of the Fln antibodies (Figure 5E, Supplemental Figure S6). In contrast to MEKK4+/+ or +/− mice, we found that phosphorylated Fln-A was expressed within PVH of MEKK4−/− FB (Figure 5D) that were also positive for Fln-A and Fln-B (Supplemental Figure S6). In addition, we observed cells along the VZ surface (particularly at sites where the integrity of the VZ surface appeared disrupted) and in the IZ (not shown) that appeared to express abnormally high levels of Fln-A and phospho-Fln-A compared to MEKK4+/+ (Figure 5E–H). Double immunostaining with Tbr1 confirmed that these highly Fln-A positive cells were neuronal (Figure 5F–H).
Figure 5. Enhanced expression of Fln in MEKK4−/− forebrain.
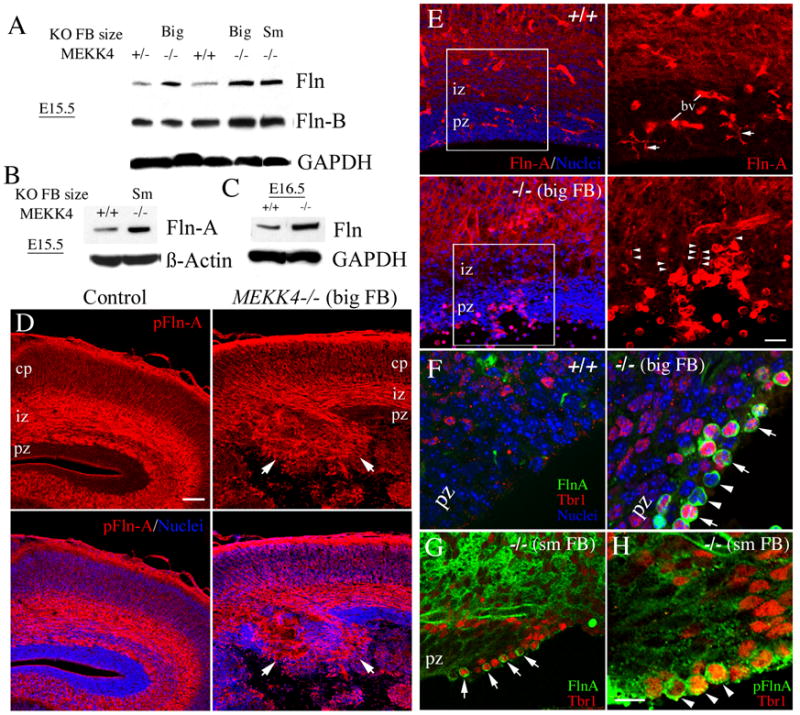
(A) E15.5 MEKK4+/+, +/− and −/− FB extracts. Compared to +/+ and +/−, MEKK4−/− with big or small FB had elevated Fln. For two of the MEKK4−/− (big and small (Sm) FB), probing the same blots with a FLN-B specific antibody showed that Fln consisted of elevated Fln-B. GAPDH was a loading control.
(B) Western blot of E15.5 FB extract showed elevated Fln-A in a MEKK4−/− with a small FB.
(C) Western blot of E16.5 FB showed elevated total Fln in the MEKK4−/−. β-actin was a loading control.
(D) Immunostaining of E17.5 FB for phospho-Fln-A (pFln-A) using mAb(p2152FLN-A). In control, p-Fln-A was highly expressed in the IZ and CP compared to the MEKK4−/− where p-Fln-A persisted to the proliferative zone (PZ) surface at sites of PVHs (arrows). Bar = 100μm.
(E) Immunostaining of Fln-A (mAb(4-4)) showed a similar pattern to pFln-A. Magnified views of Fln-A expression (red) from left hand panels are displayed to the right. In MEKK4+/+ (upper panels), Fln-A expression was low in the PZ and higher in the IZ. Microglia (arrows) and blood vessels (BV) were also labeled. In contrast, MEKK4−/− (big FB) ectopically showed areas of PZ that appeared highly positive for Fln-A. Lower panels show Fln-A positive cells with radially oriented fibers (arrowheads in magnified view) could be found within the PZ where the surface of the PZ was disrupted. Bar = 20μm.
(F) Fln-A (green) and Tbr1 (red) immunostaining showed that in MEKK4−/− big FB mutants (right panel), heavily labeled Fln-A cells were both Tbr1+ (arrows) and Tbr1- (arrowheads). No Tbr1 was observed along the VZ surface in +/+ (left panel).
(G) Example of Fln-A+/Tbr1+ (arrows) cells at the VZ surface in MEKK4−/− small FB.
(H) An adjacent section to that in G showed Tbr1+ cells also stained for pFlnA (arrowheads). Bar= 10μm.
Over-expression of Fln-A in mouse FB can restrict migrating cell polarity to a more radial orientation as cells pass through the IZ (Nagano et al., 2004). Similarly, closer analysis of TUJ1 immunoreactivity in MEKK4-deficient FB (big or small phenotype) in non-heterotopic regions showed many radially oriented TUJ1+ processes entering the IZ compared to control (Supplemental Figure S7). Together, these results suggest that MEKK4 deletion may lead to enhanced or dysregulated Fln expression that subsequently impairs the migration and morphology of many neocortical progenitors.
GST-SEK1 can precipitate MEKK4 and Fln-A
MKK4/SEK1 is a MAP2K reported to bind FLN-A in melanoma cells and serves as one substrate for MEKK4 (Gerwins et al., 1997; Marti et al., 1997). Therefore, MKK4/SEK1 could serve as a mechanism that brings MEKK4 and Fln-A together during neocortical development. To test this we incubated COS-7 or E17.5 FB cell lysates with GST-MKK4/SEK1 and probed blots for endogenous MEKK4 and Fln-A. Compared to glutathione-agarose beads or beads coated with GST, GST-MKK4/SEK1 precipitated endogenous MEKK4 and Fln-A (Figure 6A) from the same extracts. These results suggest that MEKK4 may be part of an intracellular complex with Fln-A via MKK4/SEK1. During development this signaling pathway could play a role in coordinating neuronal migration.
Figure 6. MEKK4 interaction and regulation of Fln-A phosphorylation via MKK4/SEK1.
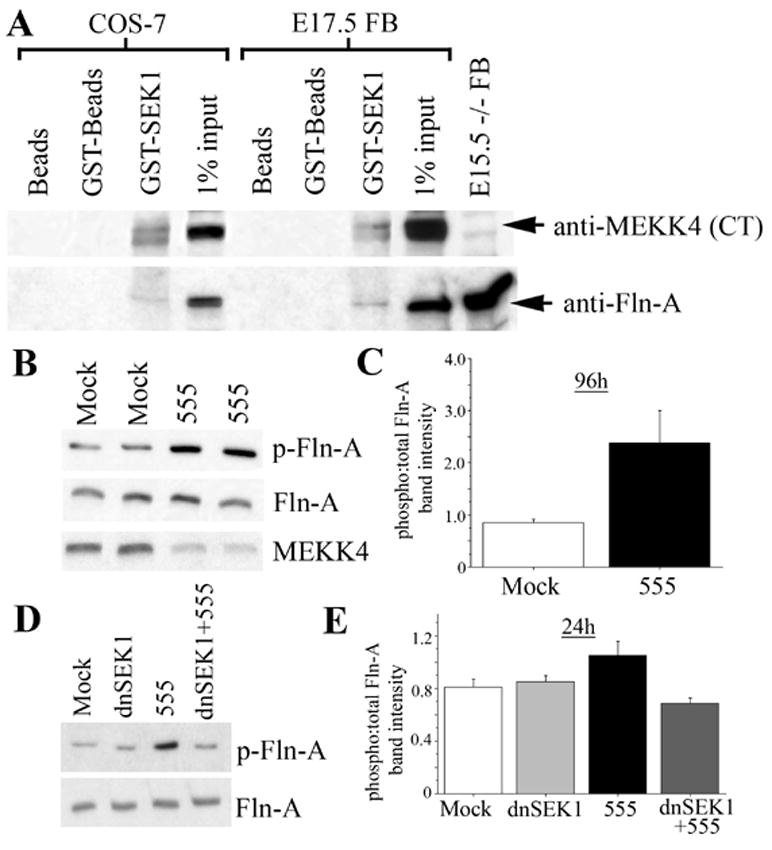
(A) Ten μg of GST-MKK4/SEK1 (GST-SEK1) was incubated in either ~500μg of COS-7 lysate or ~1mg E17.5 mouse forebrain (FB) lysate. As controls, equivalent amounts of COS-7 or FB lysates were incubated in glutathione-agarose beads (Beads) or beads pre-incubated in GST (GST-beads). One percent of lysates (1% input) were run for COS-7 or FB lysates. The upper blot was probed with anti-MEKK4 (CT) antibody and shows GST-MKK4/SEK1 precipitated endogenous MEKK4 in both COS-7 and FB lysate. Twenty μg of E15.5 MEKK4−/− FB lysate was run as a control. A doublet band is precipitated in both COS-7 and FB lysate with the upper band corresponding to MEKK4. The lower blot was probed using an anti-Fln-A antibody and showed that GST-MKK4/SEK1 also precipitated endogenous Fln-A from COS-7 and FB lysates.
(B) NIH3T3 cells were mock or siRNA#555-transfected and cultured for 96h. Lysates (20μg/lane) from different mock and siRNA#555 (555)-transfected wells were western blotted and probed for phospho-Fln-A (Ser2152) (upper blot), Fln-A (middle blot), and MEKK4 (lower blot) antibodies. Increased phosphorylation of Fln-A was observed after transfection with siRNA#555.
(C) The relative phospho- to total Fln-A band intensities were quantified for five separate transfections/group which showed increased Fln-A phosphorylation in siRNA#555-transfected cultures.
(D) NIH3T3 cells were either untransfected (Mock) or transfected with dominant-negative SEK1 (dnSEK1), siRNA#555, or dnSEK1 plus siRNA#555. Lysates were collected at 24h, western blotted and probed as in C. The blot shows increased Fln-A phosphorylation (p-Fln-A) after siRNA#555 that was not observed when co-transfected with dnSEK1.
(E) Quantification (as in C) of the results in D for at least 5–6 separate transfections/group. The enhanced Fln-A phosphorylation after siRNA#555 was blocked by co-transfection of dnSEK1.
Knockdown of MEKK4 enhances Fln-A phosphorylation on Ser2152 via MKK4/SEK1
FLN-A phosphorylation on Ser2152 has been implicated in providing resistance to FLN-A cleavage and mediating actin cytoskeletal assembly (Garcia et al., 2006; Vadlamudi et al., 2002; Woo et al., 2004). To test whether MEKK4 RNAi can affect Fln-A phosphorylation, protein extracts were collected from NIH3T3 cells that were either mock or siRNA#555-transfected and cultured for 96h. Western blot analysis revealed enhanced phospho-Fln-A relative to total Fln-A signal in siRNA#555-transfected compared to control cultures (Figure 6B,C). Therefore, MEKK4 suppression by RNAi can lead to increased Fln-A phosphorylation at Ser2152.
Next, we tested whether the increased Fln-A phosphorylation at Ser2152 induced by MEKK4 RNAi could be blocked by co-transfection with a dominant-negative form of MKK4/SEK1 (dnSEK1). We found that siRNA#555 increased Fln-A phosphorylation as early as 24h post-transfection, however when co-transfected with dnMKK4/SEK1, the phosphorylation of Fln-A was decreased (Figure 6D, E). These results suggest that in the absence of MEKK4, phosphorylation of Fln-A at Ser2152 depends on MKK4/SEK1 signaling and that under normal conditions, MEKK4 could influence Fln-A phosphorylation.
Over-expression of wild-type FLN-A can disrupt radial migration
Increased Fln-A can inhibit cell migration in non-neuronal cells and alter the morphology of migrating neurons (Cunningham et al., 1992; Nagano et al., 2004). Loss of MEKK4 resulted in increased Fln-A that was associated with regions of failed migration initiation. To test whether increased Fln-A can inhibit neuronal migration, we over-expressed a full-length human wild-type (WT) FLN-A with GFP and analyzed GFP+ cell distribution within the CP and subplate (SP) after 96h. We found that WT-FLN-A over-expression resulted in significantly fewer cells in upper CP and more cells in deeper CP and SP compared to control (GFP plus empty cDNA3.1 vector) (Fig 7A, B). These data suggest that mutations leading to excessive Fln-A could impair neuronal migration.
Figure 7. FLN-A over-expression inhibits neuronal migration.
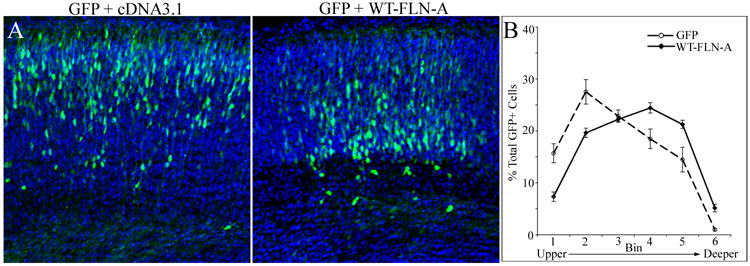
(A) E14.5 cortex was electroporated with GFP and pcDNA3.1 vector (GFP +cDNA3.1) or GFP and pcDNA3.1 expressing full-length wild-type FLN-A (WT-FLN-A). After 96h, more GFP+ cells were observed in deeper CP of WT-FLN-A (right) compared to control (left). Nuclei (blue) were stained with DAPI.
(B) Quantification of GFP+ cell distribution at 96h was determined by placing a grid (divided into six bins) spanning the upper and deeper CP and SP over the electroporated region. Significantly more GFP+ cells were located in the deeper CP and SP at 96h after WT-FLN-A (n=16 sections from 4 embryos) over-expression (solid) compared to GFP control (dashed) (n=12 sections from 3 embryos). Repeated Measures ANOVA (p<0.01).
Discussion
Malformations of human FB are often attributed to genes that regulate neuronal migration (Feng and Walsh, 2001; Rakic, 1988a). Our data suggest a relationship between MEKK4 and Fln-A that not only influences migration-initiation but also the integrity of the neuroependymal lining. Loss of MEKK4 dramatically disrupts the amount and phosphorylation of Fln-A possibly contributing to the pathogenesis of PVH.
MEKK4 expression in the mammalian brain
MEKK4 is highly expressed in the developing murine and human neural tube and persists throughout peak cortical neurogenesis into adulthood (Supplementary Figure 1 and (Abell et al., 2005; Chan-Hui and Weaver, 1998; Chi et al., 2005)). Two splice variants have been detected from mouse brain cDNA, MEKK4α and MEKK4β (Gerwins et al., 1997). MEKK4α differs from MEKK4β by an additional 52 amino acid sequence in the non-catalytic domain. The targeting strategies of both MEKK4−/− mice and siRNA would predictably result in deletion or reduction of both isoforms. Whether these two isoforms are differentially regulated in the developing or adult CNS is unknown. The widespread expression of MEKK4 in the CNS suggests its function may extend to other neural cell types and depend on local environmental stimuli.
Neuronal migration defects in MEKK4−/− and RNAi-treated brains
With the exception of FLN-A and ARFGEF2, little is known about the molecular regulators of migration initiation in developing FB (Bielas et al., 2004; Marin and Rubenstein, 2003). Our data suggest that MEKK4 is involved in this process. About 50% of MEKK4−/− mice developed bilateral PVHs that largely consisted of differentiated neurons (Figure 1). Exposure of E14.5 MEKK4−/− mice (big or small FB) to BrdU and analysis at E18.5 revealed that cells within PVH failed to leave the VZ surface (Figure 2). SiRNA#555 produced heterotopias more frequently in the CC although a significant percentage of neuronal progenitors remained at the VZ surface (Figure 3). The full effect of siRNA may be delayed until endogenous MEKK4 is completely degraded and cells have exited the VZ/SVZ. Because of these temporal and spatial differences, if migrating cells use different mechanisms to exit the VZ than to exit the CC, an RNAi-mediated strategy may differentially disrupt the signaling pathways that govern each event. In the MEKK4−/− FB, the complete loss of MEKK4 may have the most severe effect on migrating cells at sites of VZ surface abnormalities, resulting in VZ surface arrest. Notably, these were sites associated with enhanced Fln-A expression (Figure 5). Additionally, the abnormal orientation of TUJ1+ fibers in the IZ of non-heterotopic regions of neocortex suggests that many MEKK4−/− cells may be prone to subtle defects in migration (Supplementary Figure 7). Since MEKK4-deficient neurons were arrested at both the VZ surface and beneath the CP, MEKK4 may be essential for both the initial and intermediate stages of migration.
JNKs can be activated by MEKK4, and JNK can phosphorylate DCX (Gdalyahu et al., 2004; Gerwins et al., 1997). However, JNK activity was not altered in MEKK4−/− or MEKK4K1361R CNS (Abell et al., 2005; Chi et al., 2005), and we did not observe altered total or phospho-DCX by western blot (data not shown). Conceivably, insufficient MEKK4 signaling could transiently disrupt JNK signaling in turn affecting DCX activity arresting cells beneath the CP. This would be consistent with the effects of DCX siRNA (Bai et al., 2003; Ramos et al., 2006). Because MEKK4−/− mice display both affected migration and enhanced apoptosis (Figures 2, 3 and Chi et al., 2005), we propose that the MEKK4 siRNA-treated brains reflect phenomena observed in mutants with both big and small FB phenotypes. Though enhanced apoptosis may be a due to an accumulated effect of RNAi, the data further supports a pro-survival role for MEKK4 during development. Whether arrested cell migration leads to the onset of apoptosis in siRNA-transfected cells or vice versa is not clear. Although the degree to which the mechanisms underlying knockout and RNAi phenotypes are shared is unclear, taken together our data support a critical role for MEKK4 in cortical neuronal migration.
Abnormalities in the neuroependymal lining after MEKK4 deficiency
The putative cause of most PVH is a loss of function of FLN-A resulting in arrested neurons at the VZ surface (Feng and Walsh, 2004). Alternatively, PVH may arise from defects in cell adhesion or the extracellular matrix (ECM) lining the ventricular surface (Lu et al., 2006; Sheen et al., 2005). MAPK signaling is activated by β-integrins and FLN-A (Campos et al., 2004; Scott et al., 2006), suggesting that dysfunctional MAPK signaling could alter critical signaling cascades that maintain the strength of the VZ surface. Notably, a recent study suggests that FLN-A may span the plasma membrane and interact with integrins within the ECM (Bachmann et al., 2006). Therefore, mutations that alter FLN-A expression or function could impact both migrating cells and the ECM lining the lateral ventricles. Consequently, it is not clear whether PVH arise from autonomous cell migration defects, non-autonomous effects on the neuroependymal lining or both.
Here we provide evidence in MEKK4-deficient mice for a breakdown of the VZ (and pial) surface revealed by disrupted laminin staining (which binds integrins) that was associated with disorganized radial glial endfeet (Figure 4). Although not frequently observed, some siRNA#555-transfected brains at P0 did show pronounced disruption of the VZ surface at electroporated sites (Supplementary Figure S4C). Irrespective of VZ integrity, neuronal precursors were clearly stuck at the VZ surface of RNAi-exposed mice (Figure 3E) suggesting a possible intercellular signaling defect between radial glia and affected neuronal precursors. The complete deletion of MEKK4 may cause more severe, but incompletely penetrant effects on radial glial scaffolding at the VZ surface due to compensatory factors (Figure 4).
Because enhanced apoptosis was observed in both MEKK4 knockout and siRNA, death of radial glia may also compromise the integrity of the VZ surface and affect adjacent radial glial morphology and migrating cells. MEKK4 has been shown to mediate the Wnt signaling pathway (Luo et al., 2003), which when disrupted causes radial glial scaffolding defects (Zhou et al., 2004). We found that radial glial abnormalities and Fln-A upregulation typically colocalized in MEKK4−/− FB. Although Fln-A overexpression (Figure 7) did not reveal gross defects in radial glia morphology or VZ surface integrity (data not shown), we cannot rule out subtle defects in these structures that impaired the migration process. Alternatively wildtype FLN-A overexpression may preferentially disrupt migrating neurons. In any event, proper MEKK4 signaling may be required at both the inner and outer cortical limits to maintain the integrity of the neuroependymal lining which when compromised allows cells to enter the ventricular space (or breach the pial lining) contributing to PVH pathogenesis. How these defects lead to impaired migration is unclear but is likely attributable to changes in molecules that normally regulate migration initiation.
Loss of MEKK4 enhances Fln-A expression and phosphorylation
Fln-A is expressed in developing FB and regulates actin dynamics essential to cell motility (Lu et al., 2006; Nagano et al., 2002; Sheen et al., 2002; Stossel et al., 2001). Consistent with other reports, we showed that Fln expression (including phosphorylated, A and B isoforms) was low within wild-type VZ (with the exception of blood vessel and microglia staining consistent with a proposed role for FLN-A in blood vessel development (Kakita et al., 2002)) and high in the IZ and CP (Figure 5, Supplemental Figure S6). In contrast, we observed elevated Fln-A, phospho-Fln-A and Fln-B within MEKK4−/− FB. PVHs expressed high levels of Fln isoforms compared to control VZ, and ectopic sites (especially where the VZ surface was disrupted) displayed Tbr1+ neurons with excessive Fln-A (Figure 5). These results suggest that MEKK4 deletion causes ectopic increases in Fln-A and subsequent inhibition of neuronal migration. Whether or not increased Fln-A is also directly responsible for defects in the neuroependymal lining remains unclear.
Can MEKK4 influence Fln-A function? MAPK signaling complexes are connected by protein scaffolds within the cytoplasm which include Fln-A (Feng and Walsh, 2004; Morrison and Davis, 2003). We show that MKK4/SEK1 may serve to bridge MEKK4 and Fln-A as both molecules precipitated from FB (Figure 6). Further, MEKK4 siRNA enhanced Fln-A phosphorylation that was blocked by dnMKK4/SEK1. MKK4/SEK1 binds but does not appear to phosphorylate Fln-A in vitro (Marti et al., 1997), although whether MKK4/SEK1 directly phosphorylates Fln-A at Ser2152 in the brain is unknown. Thus under normal conditions, MEKK4-MKK4/SEK1 signaling may act to inhibit Fln-A phosphorylation via downstream targets. In the absence of MEKK4 these targets may over-phosphorylate Fln-A at Ser2152. These data suggest that MEKK4 signaling could influence Fln-A phosphorylation and, as discussed below, alter its expresssion.
Consequence of increased Fln-A on neuronal migration
Loss of function FLN-A is clearly associated with defective neuronal migration, but our findings are consistent with multiple lines of evidence that suggest excess Fln-A also affects migration. Non-neuronal studies have shown that migration depends on the intracellular concentration of FLN-A, and that too little or too high FLN-A can impede cell migration (Cunningham et al., 1992). Similarly, increased FLN-A binding to β-integrin can inhibit migrating cells (Calderwood et al., 2001) and since both molecules are present along the VZ surface (Campos et al., 2004; Lu et al., 2006), abnormally high Fln-A could increase Fln-A-β-integrin binding impairing migration away from the VZ surface.
Fln-A expression is controlled both genetically and molecularly. At the genetic level, mechanical force induction of β1-integrin receptors can selectively activate the p38 pathway and induce FLN-A transcription (D’Addario et al., 2002). Notably, we also observed increased p38 phosphorylation after MEKK4 RNAi (Supplemental Figure 8). Why in vitro studies did not show increased total Fln-A like our mutants was puzzling and may reflect intrinsic differences between neurons and fibroblasts or require more time for significant Fln-A accumulation to occur.
Molecular studies have shown that Fln-A phosphorylation on Ser2152 confers resistance to calpain cleavage and is required for p21-activated kinase-1-mediated actin cytoskeletal assembly (Garcia et al., 2006; Vadlamudi et al., 2002). Enhanced Fln-A phosphorylation at Ser2152 was observed in MEKK4−/− brains and after siRNA (Figures 5, 6). Therefore, enhanced Ser2152 phosphorylation may prevent Fln-A cleavage or degradation and result in Fln-A accumulation that subsequently affects actin dynamics during neuronal migration. Alternatively, FILIP (Fln-A interacting protein) is a potent degrader of Fln-A and is expressed in developing FB. FILIP is thought to maintain a gradient of Fln-A such that expression is low in the VZ and higher toward the IZ and CP (Nagano et al., 2002; Sato and Nagano, 2005). FILIP siRNA (which increases Fln-A) significantly altered the morphology of migrating neurons. Thus, future studies should examine whether FILIP activity is altered in MEKK4−/− mice or whether FILIP is itself regulated by MEKK4 (or other MAP3K pathways). In any event, mechanisms are in place in the cortical PZ to maintain low Fln-A (Sato and Nagano, 2005). Over-expression of wild-type Fln-A was reported to affect the morphology of migrating neurons (Nagano et al., 2004). Here we add to that finding and show that wild-type FLN-A over-expression can delay migration into the CP (Figure 7). Our findings suggest that MEKK4 deficiency results in an overall increase in Fln-A. Cells with excessively high Fln-A may have impaired migration initiation, while those with moderately increased Fln-A may have milder defects including arrested migration in the IZ and disrupted morphology. Therefore, mutations resulting in increased Fln-A may impair neuronal migration.
MEKK4 as a candidate PVH gene
MEKK4-deficient and MEKK4K1361R mice display a range of malformations including neural tube closure defects, skeletal anomalies and body wall closure defects (omphalocoele) (Abell et al 2005; Chi et al., 2005). Interestingly, similar abnormalities have been observed in humans with mutations in the FLN-A gene leading to FLN-A gain-of-function (Robertson et al., 2003; Zenker et al., 2004). While the majority of PVH phenotypes are caused by X-linked loss of function FLN-A mutations (Fox et al., 1998; Sheen et al., 2001), there are many cases of PVH that are not due to FLN-A (Sheen et al., 2004; Sheen et al., 2003a; Sheen et al., 2003b). Surprisingly, a recent report showed that mice lacking Fln-A do not develop PVH (Hart et al., 2006). It has been hypothesized that in the absence of FLN-A, FLN-B can compensate, but this mechanism has not been well studied. Our data suggests that genetic mutations resulting in the over-expression or enhanced phosphorylation of FLN-A may also lead to PVH. Until recently Fln-A was thought to localize intracellularly, however it may have an unappreciated role on the outer cell surface and interact with ECM proteins (Bachmann et al., 2006). Thus, the impact of MEKK4 deficiency on Fln-A could affect both neuronal migration and the ECM that stabilizes the neuroependymal lining. MEKK4 may therefore be considered a candidate autosomal gene underlying human PVH or contribute to the pathogenesis of PVH in the presence of FLN-A mutations. Exactly how MEKK4 and Fln-A maintain VZ surface integrity and regulate migration is the focus of future studies.
Supplementary Material
Acknowledgments
We thank M. Pappy for help with in-situ hybridization; J. Bao for technical assistance, S. Shapiro for providing rabbit anti-FLN-B antibody, R. Vaillancourt for providing rabbit anti-MEKK4 antibodies, C. Svendsen for providing human fetal cortex samples, J. LoTurco for RFP and GFP-expressing plasmids, T. Stossel and J. Breunig for helpful comments during manuscript preparation. This work was supported by a James Hudson Brown-Alexander Brown Coxe and Epilepsy Foundation of America fellowship (to M.R.S), an under-represented minority fellowship (to C.M.B) supplemental to U.S. Public Health Service grants (to P.R.), and a Child Health Research Grant from the Charles H. Hood Foundation, Inc. (Boston) (to H.C.). R.A.F. is an Investigator of the Howard Hughes Medical Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abell AN, Rivera-Perez JA, Cuevas BD, Uhlik MT, Sather S, Johnson NL, Minton SK, Lauder JM, Winter-Vann AM, Nakamura K, et al. Ablation of MEKK4 kinase activity causes neurulation and skeletal patterning defects in the mouse embryo. Mol Cell Biol. 2005;25:8948–8959. doi: 10.1128/MCB.25.20.8948-8959.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachmann AS, Howard JP, Vogel CW. Actin-binding protein filamin A is displayed on the surface of human neuroblastoma cells. Cancer Sci. 2006 doi: 10.1111/j.1349-7006.2006.00327.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai J, Ramos RL, Ackman JB, Thomas AM, Lee RV, LoTurco JJ. RNAi reveals doublecortin is required for radial migration in rat neocortex. Nat Neurosci. 2003;6:1277–1283. doi: 10.1038/nn1153. [DOI] [PubMed] [Google Scholar]
- Bielas S, Higginbotham H, Koizumi H, Tanaka T, Gleeson JG. Cortical neuronal migration mutants suggest separate but intersecting pathways. Annu Rev Cell Dev Biol. 2004;20:593–618. doi: 10.1146/annurev.cellbio.20.082503.103047. [DOI] [PubMed] [Google Scholar]
- Calderwood DA, Huttenlocher A, Kiosses WB, Rose DM, Woodside DG, Schwartz MA, Ginsberg MH. Increased filamin binding to beta-integrin cytoplasmic domains inhibits cell migration. Nat Cell Biol. 2001;3:1060–1068. doi: 10.1038/ncb1201-1060. [DOI] [PubMed] [Google Scholar]
- Campos LS, Leone DP, Relvas JB, Brakebusch C, Fassler R, Suter U, ffrench-Constant C. Beta1 integrins activate a MAPK signalling pathway in neural stem cells that contributes to their maintenance. Development. 2004;131:3433–3444. doi: 10.1242/dev.01199. [DOI] [PubMed] [Google Scholar]
- Chan-Hui PY, Weaver R. Human mitogen-activated protein kinase kinase kinase mediates the stress-induced activation of mitogen-activated protein kinase cascades. Biochem J . 1998;336:599–609. doi: 10.1042/bj3360599. Pt 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang L, Jones Y, Ellisman MH, Goldstein LS, Karin M. JNK1 is required for maintenance of neuronal microtubules and controls phosphorylation of microtubule-associated proteins. Dev Cell. 2003;4:521–533. doi: 10.1016/s1534-5807(03)00094-7. [DOI] [PubMed] [Google Scholar]
- Chi H, Sarkisian MR, Rakic P, Flavell RA. Loss of mitogen-activated protein kinase kinase kinase 4 (MEKK4) results in enhanced apoptosis and defective neural tube development. Proc Natl Acad Sci U S A. 2005;102:3846–3851. doi: 10.1073/pnas.0500026102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham CC, Gorlin JB, Kwiatkowski DJ, Hartwig JH, Janmey PA, Byers HR, Stossel TP. Actin-binding protein requirement for cortical stability and efficient locomotion. Science. 1992;255:325–327. doi: 10.1126/science.1549777. [DOI] [PubMed] [Google Scholar]
- D’Addario M, Arora PD, Ellen RP, McCulloch CA. Interaction of p38 and Sp1 in a mechanical force-induced, beta 1 integrin-mediated transcriptional circuit that regulates the actin-binding protein filamin-A. J Biol Chem. 2002;277:47541–47550. doi: 10.1074/jbc.M207681200. [DOI] [PubMed] [Google Scholar]
- Derbyshire ZE, Halfter UM, Heimark RL, Sy TH, Vaillancourt RR. Angiotensin II stimulated transcription of cyclooxygenase II is regulated by a novel kinase cascade involving Pyk2, MEKK4 and annexin II. Mol Cell Biochem. 2005;271:77–90. doi: 10.1007/s11010-005-5386-9. [DOI] [PubMed] [Google Scholar]
- Fanger GR, Johnson NL, Johnson GL. MEK kinases are regulated by EGF and selectively interact with Rac/Cdc42. Embo J. 1997;16:4961–4972. doi: 10.1093/emboj/16.16.4961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, Walsh CA. Protein-protein interactions, cytoskeletal regulation and neuronal migration. Nat Rev Neurosci. 2001;2:408–416. doi: 10.1038/35077559. [DOI] [PubMed] [Google Scholar]
- Feng Y, Walsh CA. The many faces of filamin: a versatile molecular scaffold for cell motility and signalling. Nat Cell Biol. 2004;6:1034–1038. doi: 10.1038/ncb1104-1034. [DOI] [PubMed] [Google Scholar]
- Fox JW, Lamperti ED, Eksioglu YZ, Hong SE, Feng Y, Graham DA, Scheffer IE, Dobyns WB, Hirsch BA, Radtke RA, et al. Mutations in filamin 1 prevent migration of cerebral cortical neurons in human periventricular heterotopia. Neuron. 1998;21:1315–1325. doi: 10.1016/s0896-6273(00)80651-0. [DOI] [PubMed] [Google Scholar]
- Garcia E, Stracher A, Jay D. Calcineurin dephosphorylates the C-terminal region of filamin in an important regulatory site: a possible mechanism for filamin mobilization and cell signaling. Arch Biochem Biophys. 2006;446:140–150. doi: 10.1016/j.abb.2005.12.006. [DOI] [PubMed] [Google Scholar]
- Gdalyahu A, Ghosh I, Levy T, Sapir T, Sapoznik S, Fishler Y, Azoulai D, Reiner O. DCX, a new mediator of the JNK pathway. Embo J. 2004;23:823–832. doi: 10.1038/sj.emboj.7600079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerwins P, Blank JL, Johnson GL. Cloning of a novel mitogen-activated protein kinase kinase kinase, MEKK4, that selectively regulates the c-Jun amino terminal kinase pathway. J Biol Chem. 1997;272:8288–8295. doi: 10.1074/jbc.272.13.8288. [DOI] [PubMed] [Google Scholar]
- Gleeson JG, Allen KM, Fox JW, Lamperti ED, Berkovic S, Scheffer I, Cooper EC, Dobyns WB, Minnerath SR, Ross ME, Walsh CA. Doublecortin, a brain-specific gene mutated in human X-linked lissencephaly and double cortex syndrome, encodes a putative signaling protein. Cell. 1998;92:63–72. doi: 10.1016/s0092-8674(00)80899-5. [DOI] [PubMed] [Google Scholar]
- Gloster A, El-Bizri H, Bamji SX, Rogers D, Miller FD. Early induction of Talpha1 alpha-tubulin transcription in neurons of the developing nervous system. J Comp Neurol. 1999;405:45–60. doi: 10.1002/(sici)1096-9861(19990301)405:1<45::aid-cne4>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- Goetsch SC, Martin CM, Embree LJ, Garry DJ. Myogenic progenitor cells express filamin C in developing and regenerating skeletal muscle. Stem Cells Dev. 2005;14:181–187. doi: 10.1089/scd.2005.14.181. [DOI] [PubMed] [Google Scholar]
- Halfter UM, Derbyshire ZE, Vaillancourt RR. Interferon gamma-dependent tyrosine phosphorylation of MEKK4 via Pyk2 is regulated by annexin II and SHP2 in keratinocytes. Biochem J. 2004 doi: 10.1042/BJ20041236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart AW, Morgan JE, Schneider J, West K, McKie L, Bhattacharya S, Jackson IJ, Cross SH. Cardiac malformations and midline skeletal defects in mice lacking filamin A. Hum Mol Genet. 2006 doi: 10.1093/hmg/ddl168. [DOI] [PubMed] [Google Scholar]
- Haydar T, Kuan CY, Flavell R, Rakic P. The role of cell death in regulating the size and shape of the mammalian forebrain. Cereb Cortex. 1999;9:621–626. doi: 10.1093/cercor/9.6.621. [DOI] [PubMed] [Google Scholar]
- Hevner RF, Shi L, Justice N, Hsueh Y, Sheng M, Smiga S, Bulfone A, Goffinet AM, Campagnoni AT, Rubenstein JL. Tbr1 regulates differentiation of the preplate and layer 6. Neuron. 2001;29:353–366. doi: 10.1016/s0896-6273(01)00211-2. [DOI] [PubMed] [Google Scholar]
- Hirai S, Kawaguchi A, Hirasawa R, Baba M, Ohnishi T, Ohno S. MAPK-upstream protein kinase (MUK) regulates the radial migration of immature neurons in telencephalon of mouse embryo. Development. 2002;129:4483–4495. doi: 10.1242/dev.129.19.4483. [DOI] [PubMed] [Google Scholar]
- Hirai S, Kawaguchi A, Suenaga J, Ono M, Cui de F, Ohno S. Expression of MUK/DLK/ZPK, an activator of the JNK pathway, in the nervous systems of the developing mouse embryo. Gene Expr Patterns. 2005;5:517–523. doi: 10.1016/j.modgep.2004.12.002. [DOI] [PubMed] [Google Scholar]
- Huang C, Rajfur Z, Borchers C, Schaller MD, Jacobson K. JNK phosphorylates paxillin and regulates cell migration. Nature. 2003;424:219–223. doi: 10.1038/nature01745. [DOI] [PubMed] [Google Scholar]
- Johnson GL, Dohlman HG, Graves LM. MAPK kinase kinases (MKKKs) as a target class for small-molecule inhibition to modulate signaling networks and gene expression. Curr Opin Chem Biol. 2005;9:325–331. doi: 10.1016/j.cbpa.2005.04.004. [DOI] [PubMed] [Google Scholar]
- Kakita A, Hayashi S, Moro F, Guerrini R, Ozawa T, Ono K, Kameyama S, Walsh CA, Takahashi H. Bilateral periventricular nodular heterotopia due to filamin 1 gene mutation: widespread glomeruloid microvascular anomaly and dysplastic cytoarchitecture in the cerebral cortex. Acta Neuropathol (Berl) 2002;104:649–657. doi: 10.1007/s00401-002-0594-9. [DOI] [PubMed] [Google Scholar]
- Kaufman MH. The Atlas of Mouse Development. San Diego: Academic Press; 1995. [Google Scholar]
- Kawauchi T, Chihama K, Nabeshima Y, Hoshino M. The in vivo roles of STEF/Tiam1, Rac1 and JNK in cortical neuronal migration. Embo J. 2003;22:4190–4201. doi: 10.1093/emboj/cdg413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuan CY, Roth KA, Flavell RA, Rakic P. Mechanisms of programmed cell death in the developing brain. Trends Neurosci. 2000;23:291–297. doi: 10.1016/s0166-2236(00)01581-2. [DOI] [PubMed] [Google Scholar]
- Levitt P, Rakic P. Immunoperoxidase localization of glial fibrillary acidic protein in radial glial cells and astrocytes of the developing rhesus monkey brain. J Comp Neurol. 1980;193:815–840. doi: 10.1002/cne.901930316. [DOI] [PubMed] [Google Scholar]
- Lu J, Tiao G, Folkerth R, Hecht J, Walsh C, Sheen V. Overlapping expression of ARFGEF2 and Filamin A in the neuroependymal lining of the lateral ventricles: insights into the cause of periventricular heterotopia. J Comp Neurol. 2006;494:476–484. doi: 10.1002/cne.20806. [DOI] [PubMed] [Google Scholar]
- Luo W, Ng WW, Jin LH, Ye Z, Han J, Lin SC. Axin utilizes distinct regions for competitive MEKK1 and MEKK4 binding and JNK activation. J Biol Chem. 2003;278:37451–37458. doi: 10.1074/jbc.M305277200. [DOI] [PubMed] [Google Scholar]
- Marin O, Rubenstein JL. Cell migration in the forebrain. Annu Rev Neurosci. 2003;26:441–483. doi: 10.1146/annurev.neuro.26.041002.131058. [DOI] [PubMed] [Google Scholar]
- Marti A, Luo Z, Cunningham C, Ohta Y, Hartwig J, Stossel TP, Kyriakis JM, Avruch J. Actin-binding protein-280 binds the stress-activated protein kinase (SAPK) activator SEK-1 and is required for tumor necrosis factor-alpha activation of SAPK in melanoma cells. J Biol Chem. 1997;272:2620–2628. doi: 10.1074/jbc.272.5.2620. [DOI] [PubMed] [Google Scholar]
- Moore SA, Saito F, Chen J, Michele DE, Henry MD, Messing A, Cohn RD, Ross-Barta SE, Westra S, Williamson RA, et al. Deletion of brain dystroglycan recapitulates aspects of congenital muscular dystrophy. Nature. 2002;418:422–425. doi: 10.1038/nature00838. [DOI] [PubMed] [Google Scholar]
- Morrison DK, Davis RJ. Regulation of MAP kinase signaling modules by scaffold proteins in mammals. Annu Rev Cell Dev Biol. 2003;19:91–118. doi: 10.1146/annurev.cellbio.19.111401.091942. [DOI] [PubMed] [Google Scholar]
- Nagano T, Morikubo S, Sato M. Filamin A and FILIP (Filamin A-Interacting Protein) regulate cell polarity and motility in neocortical subventricular and intermediate zones during radial migration. J Neurosci. 2004;24:9648–9657. doi: 10.1523/JNEUROSCI.2363-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagano T, Yoneda T, Hatanaka Y, Kubota C, Murakami F, Sato M. Filamin A-interacting protein (FILIP) regulates cortical cell migration out of the ventricular zone. Nat Cell Biol. 2002;4:495–501. doi: 10.1038/ncb808. [DOI] [PubMed] [Google Scholar]
- Nakamura F, Hartwig JH, Stossel TP, Szymanski PT. Ca2+ and calmodulin regulate the binding of filamin A to actin filaments. J Biol Chem. 2005;280:32426–32433. doi: 10.1074/jbc.M502203200. [DOI] [PubMed] [Google Scholar]
- Niewmierzycka A, Mills J, St-Arnaud R, Dedhar S, Reichardt LF. Integrin-linked kinase deletion from mouse cortex results in cortical lamination defects resembling cobblestone lissencephaly. J Neurosci. 2005;25:7022–7031. doi: 10.1523/JNEUROSCI.1695-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell SK, Kleinman HK. Neuronal laminins and their cellular receptors. Int J Biochem Cell Biol. 1997;29:401–414. doi: 10.1016/s1357-2725(96)00110-0. [DOI] [PubMed] [Google Scholar]
- Rakic P. Defects of neuronal migration and the pathogenesis of cortical malformations. Prog Brain Res. 1988a;73:15–37. doi: 10.1016/s0079-6123(08)60494-x. [DOI] [PubMed] [Google Scholar]
- Rakic P. Specification of cerebral cortical areas. Science. 1988b;241:170–176. doi: 10.1126/science.3291116. [DOI] [PubMed] [Google Scholar]
- Ramos RL, Bai J, LoTurco JJ. Heterotopia formation in rat but not mouse neocortex after RNA interference knockdown of DCX. Cereb Cortex. 2006;16:1323–1331. doi: 10.1093/cercor/bhj074. [DOI] [PubMed] [Google Scholar]
- Robertson SP, Twigg SR, Sutherland-Smith AJ, Biancalana V, Gorlin RJ, Horn D, Kenwrick SJ, Kim CA, Morava E, Newbury-Ecob R, et al. Localized mutations in the gene encoding the cytoskeletal protein filamin A cause diverse malformations in humans. Nat Genet. 2003;33:487–491. doi: 10.1038/ng1119. [DOI] [PubMed] [Google Scholar]
- Sato M, Nagano T. Involvement of filamin A and filamin A-interacting protein (FILIP) in controlling the start and cell shape of radially migrating cortical neurons. Anat Sci Int. 2005;80:19–29. doi: 10.1111/j.1447-073x.2005.00101.x. [DOI] [PubMed] [Google Scholar]
- Schmid RS, Shelton S, Stanco A, Yokota Y, Kreidberg JA, Anton ES. alpha3beta1 integrin modulates neuronal migration and placement during early stages of cerebral cortical development. Development. 2004;131:6023–6031. doi: 10.1242/dev.01532. [DOI] [PubMed] [Google Scholar]
- Scott MG, Pierotti V, Storez H, Lindberg E, Thuret A, Muntaner O, Labbe-Jullie C, Pitcher JA, Marullo S. Cooperative regulation of extracellular signal-regulated kinase activation and cell shape change by filamin A and beta-arrestins. Mol Cell Biol. 2006;26:3432–3445. doi: 10.1128/MCB.26.9.3432-3445.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheen VL, Basel-Vanagaite L, Goodman JR, Scheffer IE, Bodell A, Ganesh VS, Ravenscroft R, Hill RS, Cherry TJ, Shugart YY, et al. Etiological heterogeneity of familial periventricular heterotopia and hydrocephalus. Brain Dev. 2004;26:326–334. doi: 10.1016/j.braindev.2003.09.004. [DOI] [PubMed] [Google Scholar]
- Sheen VL, Dixon PH, Fox JW, Hong SE, Kinton L, Sisodiya SM, Duncan JS, Dubeau F, Scheffer IE, Schachter SC, et al. Mutations in the X-linked filamin 1 gene cause periventricular nodular heterotopia in males as well as in females. Hum Mol Genet. 2001;10:1775–1783. doi: 10.1093/hmg/10.17.1775. [DOI] [PubMed] [Google Scholar]
- Sheen VL, Feng Y, Graham D, Takafuta T, Shapiro SS, Walsh CA. Filamin A and Filamin B are co-expressed within neurons during periods of neuronal migration and can physically interact. Hum Mol Genet. 2002;11:2845–2854. doi: 10.1093/hmg/11.23.2845. [DOI] [PubMed] [Google Scholar]
- Sheen VL, Jansen A, Chen MH, Parrini E, Morgan T, Ravenscroft R, Ganesh V, Underwood T, Wiley J, Leventer R, et al. Filamin A mutations cause periventricular heterotopia with Ehlers-Danlos syndrome. Neurology. 2005;64:254–262. doi: 10.1212/01.WNL.0000149512.79621.DF. [DOI] [PubMed] [Google Scholar]
- Sheen VL, Topcu M, Berkovic S, Yalnizoglu D, Blatt I, Bodell A, Hill RS, Ganesh VS, Cherry TJ, Shugart YY, Walsh CA. Autosomal recessive form of periventricular heterotopia. Neurology. 2003a;60:1108–1112. doi: 10.1212/01.wnl.0000055898.00349.02. [DOI] [PubMed] [Google Scholar]
- Sheen VL, Wheless JW, Bodell A, Braverman E, Cotter PD, Rauen KA, Glenn O, Weisiger K, Packman S, Walsh CA, Sherr EH. Periventricular heterotopia associated with chromosome 5p anomalies. Neurology. 2003b;60:1033–1036. doi: 10.1212/01.wnl.0000052689.03214.61. [DOI] [PubMed] [Google Scholar]
- Stossel TP, Condeelis J, Cooley L, Hartwig JH, Noegel A, Schleicher M, Shapiro SS. Filamins as integrators of cell mechanics and signalling. Nat Rev Mol Cell Biol. 2001;2:138–145. doi: 10.1038/35052082. [DOI] [PubMed] [Google Scholar]
- Takekawa M, Posas F, Saito H. A human homolog of the yeast Ssk2/Ssk22 MAP kinase kinase kinases, MTK1, mediates stress-induced activation of the p38 and JNK pathways. Embo J. 1997;16:4973–4982. doi: 10.1093/emboj/16.16.4973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takekawa M, Saito H. A family of stress-inducible GADD45-like proteins mediate activation of the stress-responsive MTK1/MEKK4 MAPKKK. Cell. 1998;95:521–530. doi: 10.1016/s0092-8674(00)81619-0. [DOI] [PubMed] [Google Scholar]
- Takekawa M, Tatebayashi K, Saito H. Conserved docking site is essential for activation of mammalian MAP kinase kinases by specific MAP kinase kinase kinases. Mol Cell. 2005;18:295–306. doi: 10.1016/j.molcel.2005.04.001. [DOI] [PubMed] [Google Scholar]
- Vadlamudi RK, Li F, Adam L, Nguyen D, Ohta Y, Stossel TP, Kumar R. Filamin is essential in actin cytoskeletal assembly mediated by p21-activated kinase 1. Nat Cell Biol. 2002;4:681–690. doi: 10.1038/ncb838. [DOI] [PubMed] [Google Scholar]
- Walsh CA. Genetic malformations of the human cerebral cortex. Neuron. 1999;23:19–29. doi: 10.1016/s0896-6273(00)80749-7. [DOI] [PubMed] [Google Scholar]
- Woo MS, Ohta Y, Rabinovitz I, Stossel TP, Blenis J. Ribosomal S6 kinase (RSK) regulates phosphorylation of filamin A on an important regulatory site. Mol Cell Biol. 2004;24:3025–3035. doi: 10.1128/MCB.24.7.3025-3035.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu JY, DeRuiter SL, Turner DL. RNA interference by expression of short-interfering RNAs and hairpin RNAs in mammalian cells. Proc Natl Acad Sci U S A. 2002;99:6047–6052. doi: 10.1073/pnas.092143499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuzyuk T, Amberg DC. Actin recovery and bud emergence in osmotically stressed cells requires the conserved actin interacting mitogen-activated protein kinase kinase kinase Ssk2p/MTK1 and the scaffold protein Spa2p. Mol Biol Cell. 2003;14:3013–3026. doi: 10.1091/mbc.E02-11-0747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zenker M, Rauch A, Winterpacht A, Tagariello A, Kraus C, Rupprecht T, Sticht H, Reis A. A dual phenotype of periventricular nodular heterotopia and frontometaphyseal dysplasia in one patient caused by a single FLNA mutation leading to two functionally different aberrant transcripts. Am J Hum Genet. 2004;74:731–737. doi: 10.1086/383094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou CJ, Zhao C, Pleasure SJ. Wnt signaling mutants have decreased dentate granule cell production and radial glial scaffolding abnormalities. J Neurosci. 2004;24:121–126. doi: 10.1523/JNEUROSCI.4071-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.