

Abstract
Metabotropic glutamate receptors mGluR1 and mGluR5 stimulate phospholipase C, leading to an increased inositol triphosphate level and to Ca2+ release from intracellular stores. Cyclothiazide (CTZ), known as a blocker of AMPA receptor desensitization, produced a non-competitive inhibition of [Ca2+]i increases induced by mGluR agonists in HEK 293 cells transfected with rat mGluR1a but had no effect on the [Ca2+]i signals in cells expressing rat mGluR5a. In cells expressing mGluR1, CTZ also inhibited phoshoinositide hydrolysis, as well as cAMP accumulation and arachidonic acid release induced by mGluR1 agonists, indicating a direct inhibition of the receptor and not of a particular signal transduction system. However, CTZ failed to antagonize cAMP inhibition stimulated by rat mGluR2, -3, -4, -6, -7 and -8 receptors confirming its selectivity for mGluR1. The use of chimeric receptors with substituted N-terminal domains showed that CTZ did not interact with the N-terminal mGluR1a domain. Instead, mutation analysis revealed that CTZ interacts with the Thr-815 and Ala-818 residues, located at the 7th transmembrane domain, similarly as the mGluR1-selective antagonist CPCCOEt. In primary cultures of cerebellar granule neurons, expressing native metabotropic and ionotropic glutamate receptors, the final outcome of CTZ effects depended on its combined ability to potentiate AMPA receptors and inhibit mGluR1a receptors.
Keywords: metabotropic glutamate receptors, cyclothiazide, non-competitive antagonist, cerebellar granule cells
1. Introduction
Metabotropic glutamate receptors (mGluRs) belong to the large family of G-protein-coupled receptors. Eight mGluRs subtypes and multiple splice variants have been identified and classified into three groups on the basis of sequence similarities, second-messenger coupling and pharmacological properties. Group I receptors (mGluR1 and -5) are coupled to the stimulation of phospholipase C (PLC), which results in the hydrolysis of membrane phosphoinositides (PI) followed by increased Ca2+ release from intracellular stores. In addition, when expressed in several cellular systems, mGluR1a can also enhance the formation of cAMP and the release of arachidonic acid (Aramori and Nakanishi, 1992). Group II (mGluR2 and -3) and III (mGluR4, -6, -7 and -8) receptors are negatively coupled to adenylyl cyclase and decrease cAMP accumulation in heterologous expression systems (Conn and Pin, 1997; De Blasi et al., 2001). All mGluRs share structural similarities, which include a large N-terminal extracellular domain, seven transmembrane (TM) spanning regions, and a variable-length C-terminal domain (Jingami et al., 2003; Bhave et al., 2003).
Group I mGluR antagonists include competitive antagonists, which bind to the N-terminal receptor domain and non-competitive antagonists interacting within the TMVII domain (Gasparini et al., 2002). The first reported selective non-competitive mGluR1 antagonist 7-hydroxyiminocyclopropan[b]chromen-1a-carboxylic acid ethyl ester (CPCCOEt), was shown to interact with the Thr-815 and Ala-818 residues, located at the extracellular surface of TMVII (Litschig et al., 1999). Another potent selective non-competitive mGluR1 antagonist (3aS,6aS)-6a-naphtalen-2-ylmethyl-5-methyliden-hexahydro-cyclopental[c]furan-1-on (BAY36-7620) was also reported to interact with the TM region of the receptor (Carroll et al., 2001). Recently a selective non-competitive mGluR1 antagonist 1-ethyl-2-methyl-6-oxo-4-(1,2,4,5-tetrahydro-benzo[d]azepin-3-yl)-1,6-dihydro-pyrimidine-5-carbonitrile (EM-TBPC) was shown to bind within the TM domains, however, residues Val-757, Trp-798, Phe-801, Tyr-805 and Thr-815 are critical determinants of the EM-TBPC binding pocket of mGluR1 (Malherbe et al., 2003). Another study showed that the binding of a new potent noncompetitive mGluR1 receptor-selective antagonist [3H]1-(3,4-dihydro-2H-pyrano[2,3-b]quinolin-7-yl)-2-phenyl-1-ethanone (R214127) was completely blocked by 2-quinoxaline-carboxamide-N-adamantan-1-yl (NPS 2390), BAY 36-7620 and CPCCOEt, but was not displaced by competitive mGluR1 ligands such as glutamate and quisqualate, suggesting that R214127, NPS 2390, BAY 36-7620, and CPCCOEt bind to the same site or to mutually exclusive sites (Lavreysen et al., 2003). A recently discovered selective non-competitive mGluR1 antagonist 6-amino-N-cyclohexyl-N,3-dimethylthiazolo[3,2-a]benzimidazole-2-carboxamide (YM-298198) has also been shown to bind to the CPCCOEt allosteric binding site (Kohara et al., 2005). In addition, a new class of non-competitive mGluR1 antagonists, 2,4-dicarboxy-pyrroles were shown to interact within the TMVII domain of the receptor (Micheli et al., 2003). In contrast, the mGluR5 selective antagonist 2-methyl-6-(phenylethynyl)pyridine (MPEP) binds to different non-conserved residues, Pro-655 and Ser-658 in TMIII and Ala-810 in TMVII.
Our findings indicate that, in addition to the mentioned above non-competitive antagonists, mGluR1 can be inhibited by cyclothiazide (CTZ). CTZ is known to enhance the activity of α-amino-3-hydroxy-5-methyl- 4-isoxazolepropionic acid (AMPA) receptors by suppressing their desensitization (Partin et al., 1993; Partin et al., 1995). Moreover, CTZ inhibits γ-aminobutyric acid type A receptors (GABAA) having no effect on GABA binding (Deng and Chen, 2003). In addition, in Xenopus oocytes injected with mRNA for group I mGluRs, CTZ reversibly blocked mGluR1a-mediated, Ca2+-dependent chloride currents but slightly potentiated mGluR5a-mediated currents (Sharp et al., 1994). The inhibition of mGluR1a receptors expressed in oocytes was non-competitive; however the site of CTZ action has not been identified.
The aim of this study was to characterize the ability of CTZ to modulate agonist-induced intracellular [Ca2+]i signals and PI hydrolysis in cells expressing rat group I mGluRs (mGluR1a and mGluR5a). Moreover, we have determined that CTZ binds to the same binding site as the selective non-competitive mGluR1 antagonist CPCCOEt.
2. Materials and Methods
2.1. Materials
Fura-2/AM and Pluronic F-127 were obtained from Molecular Probes, Inc. (Eugene, OR) and ionomycin from Calbiochem (San Diego, CA). Neurobasal culture media, B27 supplement and fetal bovine serum for neuronal cultures were purchased from Gibco-BRL (Gaithersburg, MD). All restriction enzymes were from New England Biolabs (Beverly, MD). EMEM, DMEM and fetal bovine serum for transfected cell cultures were purchased from Biofluids (Rockville, MD). L-Quisqualic acid, (1S,3R)-1-aminocyclopentane-1,3-dicarboxylic acid (ACPD), CTZ, CPCCOEt, (2S,1′S,2′S)-2-(Carboxycyclopropyl)glycine (L-CCG-I), (±)-2-Amino-4-phosphonobutyric acid (AP-4), 1-(4′-Aminophenyl)-3,5-dihydro-7,8-dimethoxy-4H-2,3-benzodiazepin-4-one (CFM2), 2,3-dihydro-6-nitro-7-sulfamoyl-benzo(f)quinoxaline (NBQX) and (RS)-4-carboxy-3-hydroxyphenylglycine (4C3HPG) were obtained from Tocris Cookson, (Ellisville, MO). All other chemicals were from Sigma (St. Louis, MO). Ionomycin, CTZ, and CPCCOEt were dissolved in 100 % DMSO and appropriate amounts of solvent were added to all controls.
2.2. Preparation of transfection vectors
Rat mGluR cDNAs were cloned into the mammalian expression vector pcDNA3.1 (Invitrogen, Carlsbad, CA) under control of the CMV IE promoter/enhancer. This vector contains neomycin resistance gene allowing stably transfected cells to be selected using G-418 (Mediatech, Herndon, VA). Chimeric molecules containing the extracellular domains of either mGluR2 or mGluR3 and the transmembrane domains and C-terminus of mGluR1a were prepared using the PCR-based overlap extension method (Horton et al., 1993). The primers, which shared complementary sequence on the strands to be joined were designed as follows: 5′-GTACATCCGCTGGGGTGAT-ATAGAATCTATCATAGCC-3′ (primer A, mGluR2-mGlur1a), 5′-GGCTATGATAGATTCTATATCACCCCAGCGGATGTAC-3′ (primer B, mGluR1a-mGluR2), and 5′-TTACATCAAATGGGAAGAC-ATAGAATCTATCATAGCC-3′ (primer C, mGluR3-mGluR1a), and 5′-GGCTATGATAGATTCTATGTCTTCCCATTT GATGTAA-3′ (primer D, mGluR1a-mGluR3). To obtain the 5′-end of the chimeric molecule, mGluR2 cDNA was PCR-amplified with T7-primer and primer B, while for the 3′-end, SP6-primer and primer A were used. To obtain the 5′-end and 3′-end of mGluR3/mGluR1a chimeric molecules, the primer pairs T7 and primer D, and SP6 and primer C were used, respectively. The reaction mixture contained, 2 mM MgCl2, 1 μM primers, 200 μM dNTPs, 5 units of Pfu DNA polymerase (Stratagene, La Jolla, CA) and 100 ng of DNA-template in a volume of 100 μl. After denaturation at 94°C for 1 min 30 cycles were performed: denaturation at 94°C for 45 sec, annealing at 50°C for 2 min, extension at 72°C for 10 min. The PCR-products were purified on LMP-agarose and amplified using PCR primers: T7 and SP6. Amplified products were digested with Hind III and Not I restriction enzymes, purified on LMP-agarose, and cloned into the pcDNA3.1 expression vector.
2.3. Cell cultures
Human Embryonic Kidney (HEK 293) cells were transiently transfected using Effectene transfection reagent (Qiagen, Valencia, CA). Cells were grown in DMEM supplemented with 10% fetal bovine serum and 2 mM glutamine in CO2 (6%) incubator. Cells were used for intracellular Ca2+ measurements 24–72 hours after transfection. Chinese hamster ovary (CHO-K1) or baby human kidney (BHK) cells were used for stable expression of mGluRs. Individual cell lines were isolated and cultured in DMEM supplemented with 10% fetal bovine serum, 1% of L-proline, 2 mM glutamine and 0.8 mg/ml G-418 in CO2 (6%) incubator. Primary cultures were prepared as described previously for rat cerebellar granule cells (Wroblewski et al., 1985) and rat cortical astrocytes (Pshenichkin and Wise, 1995). Neuronal cultures were maintained in neurobasal medium supplemented with B27 and 2 mM glutamine, 100 μg/ml gentamicin, and either 5 or 25 mM KCl. To prevent the growth of non-neuronal cells cytosine arabinoside (10μM) was added next day after plating. Astrocytes were cultured in EMEM medium supplemented with 10% fetal bovine serum.
2.4. Site-directed mutagenesis
Introduction of point mutations in mGluR1a cDNA was made using the QuikChange Site-Directed Mutagenesis Kit (Stratagene). Briefly, 20 ng of plasmid containing mGluR1a cDNA were mixed with 125 ng of two mutagenic primers, dNTPs (50 μM) and 2.5 unit of Pfu DNA polymerase in a final volume of 50 μl. Mutagenic primers (forward primer: 5′-CTACAAGATCATCACTATGTGCTTCTCGG-TGAGCCTCAGTGTG-3′) were designed to introduce a new unique restriction site DraIII for fast screening of mutants. Samples were denaturated at 95°C for 30 sec and subjected to 20 cycles: denaturation (95°C, 30 sec), annealing (55°C, 1 min), and elongation (72°C, 30 min) with a final 10 min extension. Then, 10 units of DpnI were added to digest the DNA template. After incubation at 37°C for 1 h, samples were used for transformation of Epicurian Coli XL1-Blue Supercompetent cells. Positive clones were identified by restriction analysis and the authenticity of each mutation was confirmed by DNA sequencing.
2.5. Measurements of PI hydrolysis
Cells, cultured in 96-well plates were incubated overnight with 0.625 μCi/well myo-[3H]inositol (NEN, Boston, MA) to label the cell membrane phosphoinositides. After two washes with 0.1 ml of Locke’s buffer (156 mM NaCl, 5.6 mM KCl, 3.6 mM NaHCO3, 1 mM MgCl2, 1.3 mM CaCl2, 5.6 mM glucose and 20 mM Hepes, pH 7.4) incubations with receptor ligands were carried out for 45 min at 37°C in Locke’s buffer containing 20 mM LiCl to block inositol phosphate degradation. The reaction was terminated by aspiration of media and inositol phosphates were extracted with 100 μl of 0.1 M HCl for 10 min. The separation of [3H]inositol phosphates was performed by ion-exchange chromatography on AG 1-X8 resin, 200–400 mesh (Bio-Rad, Hercules, CA). The samples were diluted 10 times with water and applied to columns equilibrated in 0.1 M formic acid. The columns were washed with 1ml of water and 1 ml of tetraborate buffer (5mM sodium tetraborate, 60 mM sodium formate). Total [3H]inositol phosphates were eluted from the columns with 0.5 ml of 0.1 M formic acid/1 M ammonium formate. The collected samples were mixed with Safety-Solve cocktail (RPI, Mount Prospect, IL) and measured by scintillation counting.
2.6. Intracellular calcium measurements
Measurements of intracellular Ca2+ concentrations were performed as described previously (Wroblewska et al., 1997). HEK 293 cells, cultured on 35 mm dishes (Nunc, Rochester, NY), were loaded for 30 min at 37°C with 5 μM Fura-2/AM in Locke’s buffer supplemented with 0.1% Pluronic F-127. Then, cells were washed and incubated for another 30 min in buffer to allow de-esterification of Fura-2/AM. The drug solutions were delivered at the rate of 2 ml/min. Ca2+ imaging experiments were performed using an Axiovert 135 fluorescence microscope with 40x/0.6 LD Achroplan objective (Zeiss, Germany). Fluorescence of Fura-2 was monitored with Attofluor fluorescence imaging system (Rockville, MD). Intracellular Ca2+ concentrations were calculated from the ratio (R) of fluorescence signals obtained at excitation wavelengths 334 nm and 380 nm, as described previously (Grynkiewicz et al., 1985) and using the Kd value of 386 nM for the Fura-2-Ca2+ complexes (Kajita and Yamaguchi, 1993). In each experiment Rmin values were measured in the cells incubating for 10 min in Ca2+-free Locke’s buffer in the presence of 10 mM EGTA and 10 μM ionomycin (free acid). Rmax signals were measured by subsequent incubation of the same cells for 10 min in Locke’s buffer containing 6 mM Ca2+ and 10 μM Ca2+-ionomycin. The background fluorescence, measured in the presence of 10 mM MnCl2 was subtracted for each individual cell. All experiments were performed at room temperature employing a custom-made buffer flow system.
2.7. Measurements of cAMP
Cells, cultured on 96-well plates, were washed twice and preincubated 10 min at room temperature in Locke’s buffer containing 300 μM isobutylmethylxantine (IBMX) to inhibit the activity of phosphodiesterases. Then 10 μM forskolin alone, or in combination with mGluR ligands, was added and the incubation was continued for 6 min. After medium aspiration, cAMP was extracted for 10 min with 0.1 M HCl and measured by radioimmunoassay using a magnetic Amerlex RIA kit (Amersham). Cells expressing mGluR1a, cultured on 24-well plates, were incubated for 40 min at 37°C.
2.8. Measurements of arachidonic acid release
Experiments were performed as described previously (Lazarewicz et al., 1990) Cells, cultured in 24 well plates, were labeled for 24 hr with 0.5 μCi [3H]Arachidonic acid (NEN, Boston, MA) s. After washing with Locke’s buffer agonists were added and cells were incubated for 30 min at 37°C in the same buffer. Incubation media were collected and processed for measurement of radioactivity.
2.9. Measurements of cell viability
Cell viability was measured using the colorimetric MTT (thiazolyl blue tetrazolium) assay (Mosmann, 1983). MTT is converted into color product formazan by active mitochondria. After 1 h incubation of cells with MTT (0.2 mg/ml of medium) in a CO2 incubator the produced formazan was extracted in DMSO and absorbance at 570 nm was measured. Alive cells with normal mitochondrial function generated an intense purple color, which was weaker in dying cells.
2.10. Data analysis
The data were analyzed using one-way ANOVA, and the differences between the groups were assessed using the Student’s t-test. The results were considered statistically significant when p < 0.05. IC50 values were obtained from dose-response curves by nonlinear regression, fitting the data from individual experiments to a logistic equation using SigmaPlot software.
3. Results
3.1. CTZ inhibits [Ca2+]i signals in HEK 293 cells expressing mGluR1a but not mGluR5a
HEK 293 cells, transiently expressing rat mGuR1a or mGluR5a receptors, were used to study the effects of CTZ on [Ca2+]i signals mediated by the transfected receptors. Cells expressing mGluR1a showed a fast transient [Ca2+]i increases in response to application of mGluR agonists glutamate, quisqualate and ACPD (Fig. 1A). The response was quickly desensitizing with [Ca2+]i levels returning to basal values before removal of the agonists. Addition of CTZ (100 μM) almost completely inhibited the increase of [Ca2+]i induced by 100 μM ACPD. Similar traces were obtained for 60–90 cells in each of four separate experiments. The removal of CTZ restored a full response to ACPD indicating that the inhibition produced by CTZ was reversible (Fig. 1A).
Figure 1.
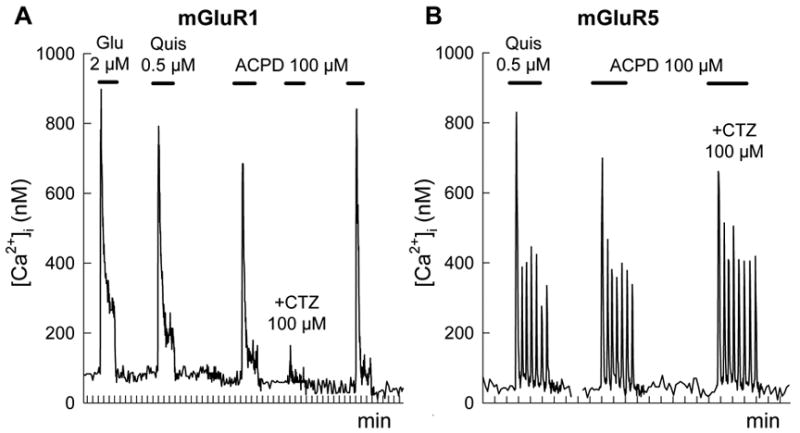
CTZ selectively inhibits [Ca2+]i responses in HEK 293 cells expressing mGluR1a but not mGluR5a. Transiently transfected cells were stimulated by applications of mGluR agonists glutamate (Glu), quisqualate (Quis) and ACPD, as indicated by the horizontal bars. CTZ (100 μM) was applied together with ACPD as indicated. Traces represent individual responses of single cells, which express mGluR1a (A) or mGluR5a (B) receptors.
Application of agonists to HEK 293 cells transfected with mGluR5a revealed a different pattern of [Ca2+]i responses. In all responding cells (124 cells in 9 experiments) agonists induced a rapid initial [Ca2+]i increase followed by a series of periodic spikes, which lasted until the agonist was present (Fig. 1B). CTZ, failed to change the amplitude of [Ca2+]i responses in the cells expressing mGluR5a (Fig. 1B). Neither a 10-fold decrease of ACPD concentration nor the addition of CTZ 1.5 min before the agonist (data not shown) were effective in revealing the inhibitory effect of CTZ on the ACPD-induced [Ca2+]i signals in cells expressing mGluR5a. These data indicate that sensitivity to CTZ inhibition is a unique feature of mGluR1 receptors.
The inhibitory effect of CTZ on mGluR1a was dose-dependent (Fig. 2A and B) and, when applied in presence of 100 μM ACPD, CTZ inhibited [Ca2+]i transients with an IC50 value of 33 ± 2.8 μM (n=16). This value is close to IC50 of 18 μM reported for the inhibition of glutamate-stimulated human mGluR1a expressed in oocytes (Sharp et al., 1994). The inhibitory action of CTZ was not overcome by high concentrations of ACPD, suggesting a non-competitive type of interaction (Fig. 2C). In fact, the EC50 values for ACPD were not affected by CTZ and were 56 ± 7.9 μM and 55 ± 6.3 μM, in the absence and presence of 40 μM CTZ, respectively.
Figure 2.
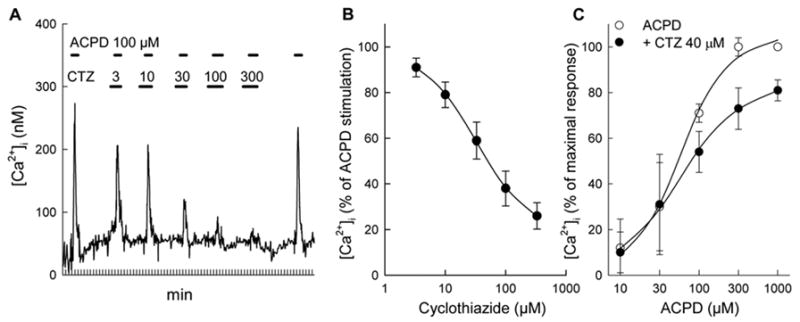
CTZ, in a dose-dependent manner, inhibits [Ca2+]i signals induced by ACPD in HEK 293 cells expressing mGluR1a. (A) Trace of a single cell response. Increasing concentrations of CTZ were added 1.5 min prior to each consecutive addition of 100 μM ACPD, as represented by horizontal bars. (B) Dose-response curve of CTZ calculated from 16 cells monitored in three separate experiments. Results from all traces were expressed as the per cent of response induced in each cell by 100 μM ACPD. (C) CTZ decreased the amplitude of [Ca2+]i responses at maximal ACPD concentrations. [Ca2+]i signals were obtained from 84 cells in 5 separate experiments. Values represent means ± S.E.M. from all the collected data.
3.2. CTZ inhibits activation of several transduction pathways induced by mGluR1a in CHO cell lines
To further characterize the pharmacological profile and specificity of CTZ we used stably transfected CHO cell lines expressing rat mGluR1a. In these cells, CTZ dose-dependently inhibited PI hydrolysis stimulated by mGluR agonists quisqualate and ACPD (Fig. 3A and B). High doses of agonists failed to overcome the inhibitory effect of CTZ indicating a non-competitive type of antagonism, as observed in the Ca2+ mobilization experiments (Fig. 2C). To ascertain that the effect of CTZ was not limited to the inhibition of PI hydrolysis and not due to a nonspecific interaction with the mGluR1 transduction system, we tested the ability of CTZ to inhibit cAMP production and arachidonic acid release, two other signal transduction mechanisms activated by mGluR1 (Aramori and Nakanishi, 1992). In CHO cells expressing mGluR1a, CTZ not only inhibited PI hydrolysis but also antagonized glutamate-induced stimulation of cAMP accumulation (Fig. 3C) and the stimulation of arachidonic acid release (Fig. 3D). Therefore, CTZ inhibited three independent transduction pathways known to be activated by mGluR1a in permanently transfected CHO cells, suggesting that the inhibitory effect of CTZ was mediated through the transfected receptors rather than due to an inhibition of the downstream transduction mechanisms. The specificity of CTZ action was further confirmed using untransfected CHO cells where PI hydrolysis was stimulated by ATP acting at endogenous purinergic receptors, but was not significantly affected by CTZ (Fig. 3E).
Figure 3.
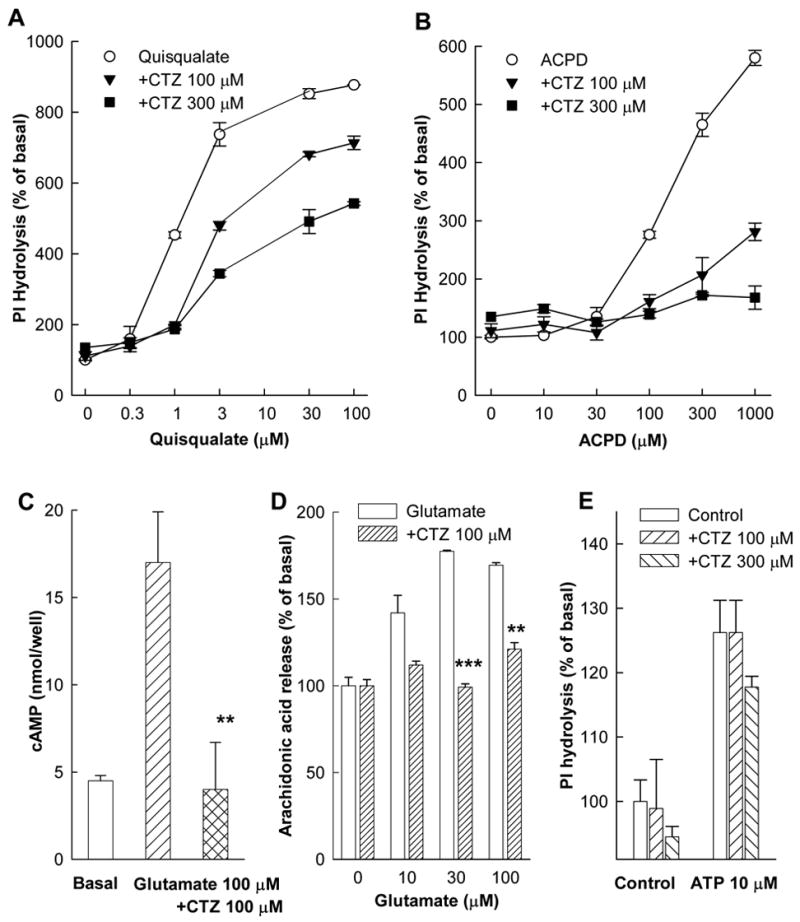
Effects of CTZ on different transduction pathways in CHO cell lines expressing mGluR1a. (A and B) Inhibition of PI hydrolysis in cells stimulated with different concentrations of quisqualate or ACPD. cAMP accumulation (C) and arachidonic acid release (D) were measured in cells stimulated by glutamate. CTZ was added together with the agonists. (E) Lack of CTZ effect on PI hydrolysis stimulated by ATP. Values represent means ± S.E.M. from at least 3 independent measurements. ** p<0.01 and *** p<0.001 by Student’s t-test as compared to respective data without CTZ.
3.3. CTZ does not inhibit group II and III mGluRs
In contrast to mGluR1a, group II and III mGluRs inhibit adenylyl cyclase, therefore, the activation of these receptors leads to the inhibition of cAMP accumulation stimulated by forskolin. We applied ACPD/glutamate and AP-4 to stimulate group II and III receptors, respectively. CTZ, even at the highest dose of 300 μM, failed to antagonize the agonist-stimulated cAMP decrease in all tested cell lines expressing rat mGluR2, -3, -4, -6, -7 and -8 (Fig. 4). In cells expressing mGluR3 and mGluR4 receptors CTZ appeared to potentiate the receptor action, however, in these cells, CTZ significantly decreased forskolin-induced cAMP accumulation in the absence of mGluR agonists. Because this effect was visible only in BHK cells (expressing mGluR3 and mGluR4) but not in CHO cells (expressing all other mGluRs) it is likely to reflect a nonspecific inhibition of cAMP production in these cells.
Figure 4.
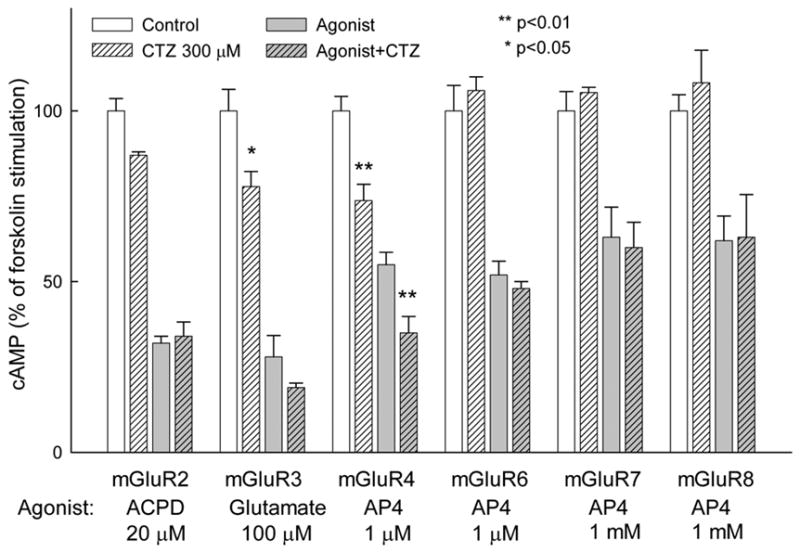
CTZ fails to antagonize agonist responses of group II and III mGluRs. cAMP accumulation was stimulated in permanently transfected CHO (mGluR2, -6, -7 and -8) or BHK (mGluR3 and -4) cells with 10 μM forskolin in presence of 300 μM phosphodiesterase inhibitor IBMX. mGluRs agonists (20 μM ACPD for mGluR2, 100 μM glutamate for mGluR3, 1 μM AP4 for mGluR4 and -6, 1 mM AP4 for mGluR7 and -8) and CTZ were added simultaneously with forskolin. cAMP accumulation is expressed as % of forskolin stimulation in individual cell lines. Values represent means ± S.E.M. from two independent experiments performed in triplicate. * p<0.05 and ** p<0.01 by Student’s t-test as compared to respective data without CTZ.
3.4. CTZ does not bind to the N-terminus of mGluR1a
The non-competitive nature of the CTZ inhibitory effects suggested the possibility that the site of CTZ action may involve receptor domains other than the agonist binding site located in the N-terminal domain. To test this hypothesis we constructed two chimeric receptors in which the extracellular N-terminal domain of mGluR1a was replaced by the corresponding domain of either mGluR2 (mGluR2/1a chimera) or mGluR3 (mGluR3/1a chimera). These chimeras possess the agonist pharmacology of group II mGluRs while retaining the coupling of mGluR1a to PLC and subsequent intracellular Ca2+ release (Takahashi et al., 1993). As shown in Fig. 4 neither mGluR2 nor mGluR3 were inhibited by CTZ, hence, if the site of CTZ action were located on the N-terminal domain of mGluR1a it would be absent in mGluR2/1a and mGluR3/1a chimeras.
The measurements of [Ca2+]i in HEK 293 cells expressing chimeric receptors showed that, similarly to mGluR1a, both receptors responded to agonist stimulation by elevation in [Ca2+]i levels seen as single transient peaks (Fig. 5A and D). In contrast, the sensitivity to agonists was typical to that of group II mGluRs. Addition of L-CCG-I, which at low concentrations (below 5 μM) is a selective agonist of group II receptors, induced strong [Ca2+]i responses (Fig. 5A and D). More importantly, 4C3HPG, which is a competitive antagonist of mGluR1a (Kingston et al., 1995) but an agonist of group II receptors (Hayashi et al., 1994), stimulated [Ca2+]i increases in cells expressing both mGluR3/1a and mGluR2/1a chimeric receptors (Fig. 5A and D). These results confirm that both chimeric receptors combine the agonist pharmacology of group II receptors with signal transduction characteristic of mGluR1a. Moreover, each of the chimeric receptors retained its unique agonist selectivity, as quisqualate (100 μM), which was reported to activate mGluR3 (EC50 = 40 μM) but not mGluR2 (EC50 > 1mM) (Pin and Duvoisin, 1995), induced [Ca2+]i increases in cells expressing mGluR3/1a but not in cells expressing mGluR2/1a (Fig. 5A and D).
Figure 5.
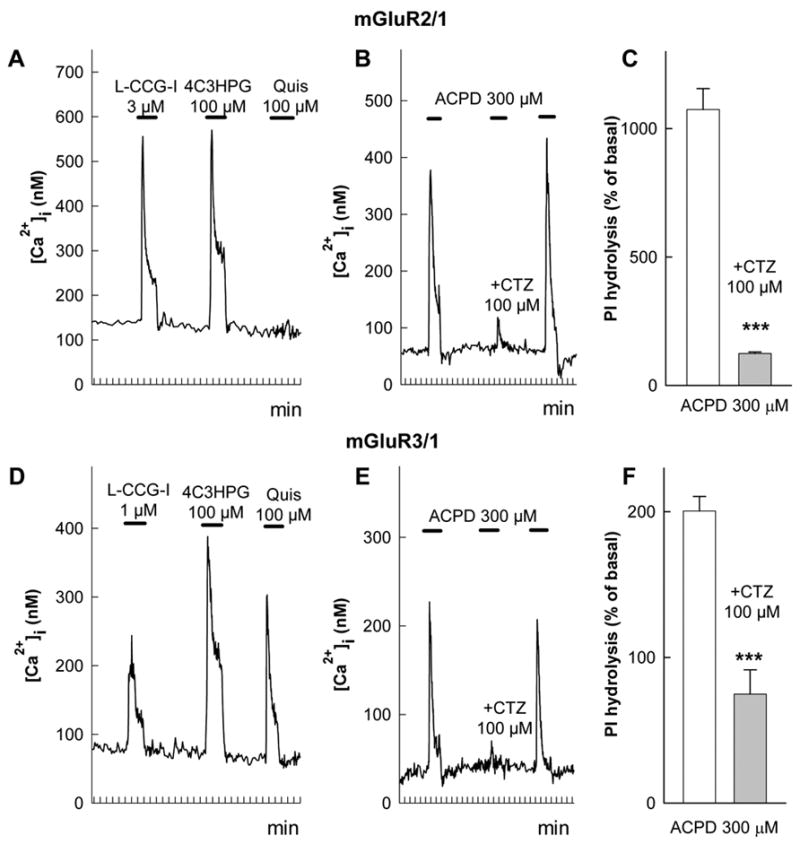
Effects of CTZ in cells expressing chimeric receptors mGluR2/1a (A, B, C) and mGluR3/1a (D, E, F). Chimeras were obtained by replacing the entire N-terminal domain of mGluR1a with homologous domains of mGluR2 or mGluR3. Pharmacological profiles of [Ca2+]i signals in HEK 293 cells expressing mGluR2/1a (A) and mGluR3/1a (D) chimeras. CTZ (100 μM) reversibly inhibited [Ca2+]i responses induced by 300 μM ACPD at both mGluR2/1a (B) and mGluR3/1a (E) receptors. Traces represent responses of individual cells stimulated as indicated by the horizontal bars. Similar results were obtained from 70 cells in 9 experiments for mGluR2/1a and from 33 cells in 3 experiments for mGluR3/1a. CTZ inhibits ACPD-stimulated PI hydrolysis in CHO cells expressing mGluR2/1a (C) and mGluR3/1a (F) chimeras. Values represent means ± S.E.M. from 3 independent experiments *** p<0.001 by Student’s t-test as compared to data without CTZ.
CTZ, as shown by traces in Fig. 5B and 5E, suppressed the ACPD-induced [Ca2+]i increases in cells expressing both chimeric receptors. The average inhibition of ACPD-induced [Ca2+]i responses by 100 μM CTZ was 80 ± 2% (70 cells in 9 experiments) in cells expressing mGluR2/1a and 88 ± 2% (33 cells in 3 experiments) in cells expressing mGluR3/1a. The inhibition was totally reversed by washing out the antagonist. An additional confirmation of the inhibitory effect of CTZ in chimeric receptors was obtained in CHO cells transfected with these receptors. The stimulation of PI hydrolysis induced by ACPD was fully inhibited by CTZ in cells expressing either mGluR2/1a (Fig. 3C) or mGluR3/1a (Fig. 3F) chimeric receptors. These results indicate that the inhibition of mGluR1a by CTZ was not determined by an interaction with the N-terminal extracellular domain of the receptor, which was absent in the chimeric receptors.
3.5. Determination of the residues involved in the inhibitory effect of CTZ
The observations that CTZ does not interact with the N-terminal domain of the receptor and displays inhibitory properties selective to mGluR1 and similar to CPCCOEt and other non-competitive antagonists, prompted us to construct a mGluR1a mutant (T815M, A818S) which, as shown before (Litschig et al., 1999), is insensitive to CPCCOEt inhibition. In this mutant, two residues Thr815 and Ala818 of mGluR1 were substituted with the homologous amino acids Met802 and Ser805 of mGluR5. As shown in Fig. 6A and B the wild-type mGluR1a was inhibited by both CPCCOEt and CTZ but not by the mGluR5-selective antagonist MPEP. In contrast, mGluR5a was strongly inhibited only by its selective antagonist MPEP. The mutated mGluR1a (T815M, A818S), retained its sensitivity to agonists, but was completely insensitive to both CPCCOEt and CTZ (Fig. 6C) indicating that the amino acids Thr815 and Ala818 of mGluR1 are necessary to mediate subtype-selecive action of both CPCCOEt and CTZ.
Figure 6.
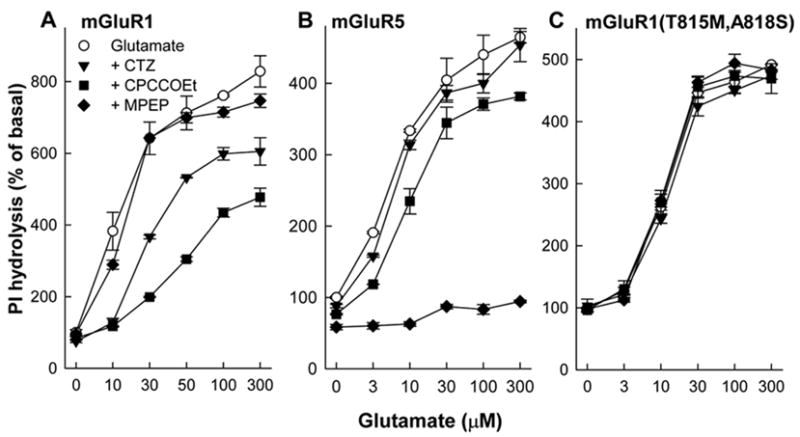
CTZ and CPCCOEt fail to inhibit PI hydrolysis in cells expressing the mutant mGluR1a(T815M,A818S). CHO cells, permanently transfected with mGluR1a (A), mGluR5a (B) or mGluR1a(T815M,A818S) (C), were stimulated with glutamate in presence of mGluR1-selective antagonist CPCCOEt (100 μM), mGluR5-selective antagonist MPEP (10 μM) and CTZ (100 μM). Values represent means ± S.E.M. from 5 independent experiments.
3.6. CTZ differently regulates PI hydrolysis in rat granule neurons and cortical astrocytes
Because neuronal and glial cells may express several types of native receptors sensitive to CTZ, the final outcome of the drug effect may be more complex compared to cell lines expressing only the individual receptors. We used primary cultures of rat cerebellar granule cells and rat cortical astrocytes to study the effects of CTZ in cells expressing various native glutamate receptors. Cerebellar granule neurons express a heterogenous population of glutamate receptors including mGluRs, NMDA and AMPA/Kainate receptors (Audinat et al., 1994; Hack et al., 1995; Santi et al., 1994). Expression of mGluR1a in these neurons strictly depends on culture conditions. Thus, at physiological (5 mM) K+ concentrations (K5) in the medium the neurons express predominantly mGluR1a (Santi et al., 1994). As expected, under these conditions, CPCCOEt completely inhibited mGluR1a-mediated PI hydrolysis (data not shown). CTZ, in K5 cells, also inhibited both ACPD and quisqualate-stimulated PI hydrolysis in a non-competitive manner (Fig. 7A and B).
Figure 7.
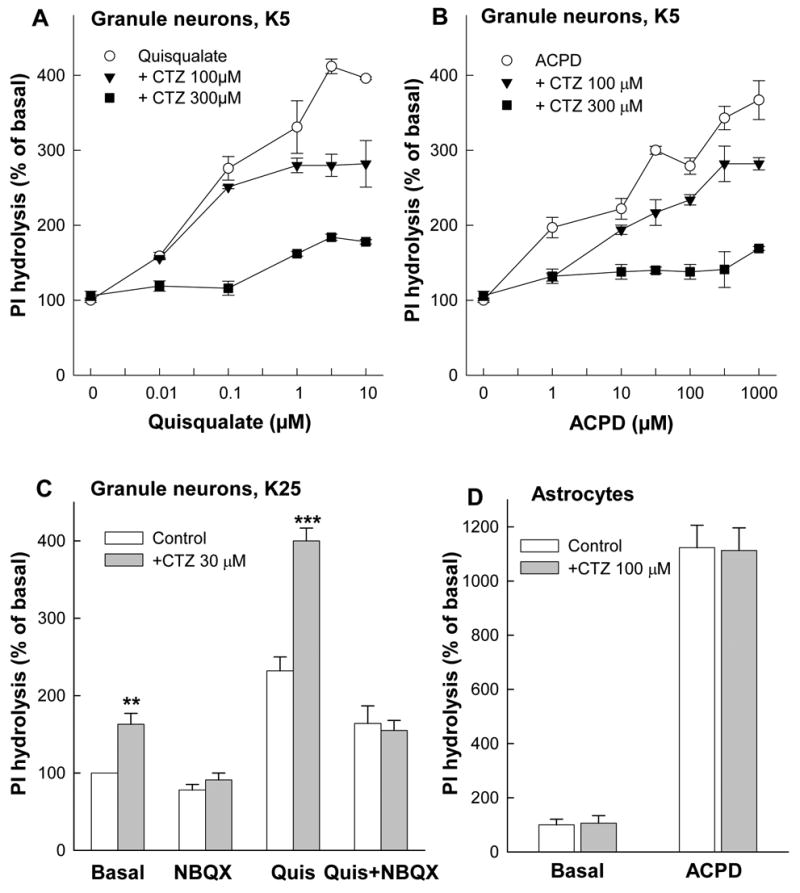
Effects of CTZ on PI hydrolysis in rat cerebellar granule neurons and in rat cortical astrocytes. CTZ inhibits PI hydrolysis in granule cells cultured in 5 mM K+ (K5) medium stimulated by quisqualate (A) or by ACPD (B). CTZ increases PI hydrolysis in granule cells cultured in 25 mM K+ (K25) where mGluR1 expression is very low; Quis 1 μM, NBQX 10 μM (C). CTZ had no effect on PI hydrolysis stimulated by ACPD (30 μM) in rat cortical astrocytes, which express mGluR5 but not mGluR1. Values represent means ± S.E.M. from 3 independent experiments performed in triplicate. ** p<0.01 and *** p<0.001 by Student’s t-test as compared to respective data without CTZ.
In granule cells, cultured in high (25 mM) K+ medium (K25), in which the expression of mGluR1a remains very low (Santi et al., 1994) the picture is more complex. Under these conditions, ACPD failed to activate PI hydrolysis (data not shown). In contrast, the non-selective mGluR/AMPA/Kainate receptor agonist quisqualate stimulated PI hydrolysis (Fig. 7C). This stimulation was abolished by the AMPA/Kainate receptor antagonist NBQX. CTZ, used in concentrations known to potentiate AMPA receptors (30 μM), strongly enhanced the effect of quisqualate. This combined stimulatory effect of CTZ and quisqualate was completely inhibited by NBQX, confirming the involvement of AMPA receptors. A small, NBQX-sensitive, stimulatory effect of CTZ on basal PI hydrolysis seen in Fig. 7C was probably due to AMPA receptor activation by glutamate released during the incubation. In contrast to granule neurons, CTZ failed to inhibit PI hydrolysis stimulated by ACPD in cortical astrocytes (Fig. 7D), which express predominantly mGluR5 receptors (Miller et al., 1995).
4. Discussion
Untransfected HEK 293 and CHO cells do not express endogenous metabotropic or ionotropic glutamate receptors functionally coupled to PLC and intracellular Ca2+ mobilization (Gabellini et al., 1994; Kawabata et al., 1996; Daggett et al., 1995). In transfected cells activation of mGluR1a and mGluR5a elicits different patterns of intracellular Ca2+ responses suggesting that these two receptors play distinct roles in intracellular Ca2+ homeostasis. HEK 293 cells transfected with mGluR1a revealed, upon stimulation, fast transient [Ca2+]i increases. Similar responses have been reported in HEK 293 cells expressing mGluR1a (Kawabata et al., 1996; Gomeza et al., 1996; Flor et al., 1996) and in CHO cells transfected with mGluR1b (Knopfel et al., 1995). In contrast, the analysis of Ca2+ responses in HEK 293 cells expressing mGluR5a revealed [Ca2+]i oscillations in almost all cells that were able to respond to agonists. This implies that pharmacological agents which selectively modulate one, but not the other group I receptor may have different physiological effects. It has been shown before that CTZ inhibited non-competitively glutamate-induced Ca2+-dependent chloride currents in oocytes expressing human mGluR1 while it slightly potentiated currents in oocytes expressing mGluR5 (Sharp et al., 1994). We failed to detect a stimulatory effect of CTZ on mGluR5a-induced [Ca2+]i signals in HEK 293 cells and PI hydrolysis in CHO cell lines. It cannot be excluded that the potentiating effect of CTZ on responses in oocytes expressing mGluR5a was not due to a direct action of the antagonist at the receptor but, instead, to an amplification of agonist action at the level of signal transduction mechanisms. Such amplification would not be visible in oocytes expressing mGluR1a where it would be masked by the inhibitory effect of CTZ. However, our results indicate that CTZ selectively inhibits [Ca2+]i responses and PI hydrolysis induced by mGluR1a in a reversible and non-competitive manner.
We have demonstrated that among mGluRs, the inhibitory effect of CTZ was selective to mGluR1 receptors. Neither mGluR5 nor any of group II or III mGluRs were inhibited. CTZ reduced mGluR1-mediated PI hydrolysis, intracellular Ca2+ mobilization, as well as cAMP increase and arachidonic acid release, two other signal transduction systems activate by mGluR1 in CHO cells (Aramori and Nakanishi, 1992) confirming the action of CTZ directly at the receptor, as opposed to a particular signaling cascade. CTZ also failed to inhibit PI hydrolysis stimulated by ATP acting at endogenous purinergic receptors in untransfected CHO cells. Our results also allowed us to conclude that CTZ was a noncompetitive antagonist of mGluR1 receptors. Such noncompetitive properties were visible from agonist dose-response curves obtained by measurements of Ca2+ signals and of PI hydrolysis in cells expressing transfected mGluR1 receptors, as well as in granule neurons cultured in low K+ conditions, expressing high levels of native mGluR1 receptors.
In order to determine which receptor domains were involved in CTZ action we constructed two chimeric receptors in which the N-terminal extracellular domain of mGluR1a was replaced by the corresponding extracellular sequences of either mGluR2 or mGluR3 which were not inhibited by CTZ. It has been shown previously that about two-thirds of the initial N-terminal amino acid sequence determines mGluR agonist selectivity (Takahashi et al., 1993), while the specificity of G-protein coupling is controlled by intracellular domains (Pin et al., 1994; Gomeza et al., 1996). As expected, the chimeric receptors, while retaining coupling with PLC and producing a pattern of [Ca2+]i responses similar to mGluR1a, showed pharmacological responses characteristic to group II mGluRs. The activation of both chimeric receptors was blocked by CTZ. Because in these chimeras the N-terminal portion of mGluR1a was replaced by the homologous portions of CTZ-insensitive receptors, it may be concluded that CTZ inhibitory action at mGluR1a is not determined by its binding to the N-terminal domain but rather by an interaction with the TM domain, which is conserved in chimeric receptors. In a more detailed analysis, using point mutations of mGluR1a, we have shown that CTZ interacts with Thr815 and Ala818 at the extracellular surface of TMVII. It has been shown previously that substitution of those amino acids with homologous amino acids of mGluR5 eliminates the inhibitory effect of the mGluR1-selective noncompetitive antagonist CPCCOEt (Litschig et al., 1999). Neither of these amino acid residues is present in other mGluRs which are all insensitive to both CTZ and CPCCOEt.
To study the effects of CTZ in neurons expressing native receptors we used cerebellar granule cells, known to express mGluR1a (Santi et al., 1994). Levels of mGluR1a expression as well as the survival in these cultures depend on K+ concentration in the culture medium. In low K+ medium the expression of mGluR1 receptors is high but cell viability is low, while under 25 mM K+ a high viability is accompanied by low mGluR1 expression. Hence, cerebellar neurons cultured in presence of either 5 mM or 25 mM K+ display a different pharmacology of PI hydrolysis stimulation, apparently, depending on mGluR1a expression. Granule cells in culture also express ionotropic non-NMDA receptors sensitive to AMPA, kainate and quisqualate. The activation of these receptors leads to increased sodium influx followed by increased entry of calcium and indirect activation of PI hydrolysis (Wroblewski et al., 1987; Raulli et al., 1991). Therefore, in cerebellar granule cells some agonists, for example glutamate and quisqualate, and some modulators, such as CTZ may affect both metabotropic and ionotropic glutamate receptors simultaneously. For example, quisqualate enhances PI hydrolysis in both K5 and K25 cultures, while ACPD acts potently only in K5 cells, where mGluR1a expression is more pronounced. We report here that CTZ inhibited the stimulation of PI hydrolysis induced by both quisqualate and ACPD in K5 cells. Under K25 conditions, where mGluR1a expression is low, CTZ at low concentrations (30 μM) potently enhanced PI hydrolysis stimulated by quisqualate. This effect of CTZ was inhibited by NBQX, a selective antagonist of non-NMDA ionotropic receptors. These results suggest that in K25 granule cells the stimulation of PI hydrolysis by quisqualate is mediated mostly by ionotropic AMPA receptors. In contrast, in K5 the contribution of the ionotropic AMPA receptors to PI hydrolysis appears to be masked by metabotropic receptors.
At similar concentrations (100 μM) CTZ has been shown to inhibit GABAA receptors in hippocampal cultures (Deng and Chen, 2003). Because CTZ is widely used as a positive modulator of AMPA receptors, special care should be taken in studies conducted on neuronal cells where multiple receptors may be present. This should include the use of AMPA receptor antagonists to confirm the specificity of CTZ effects (Vignes, 2001). Considering the abundance of glutamatergic and GABAergic neurotransmission, applications of CTZ may greatly affect the excitatory-inhibitory balance observed under specific experimental conditions (Deng and Chen, 2003). An example may be the interpretation of the observed ability of CTZ to block DHPG-induced depolarization of rat spinal dorsal horn neurons (Zhong et al., 2000). While the potency of CTZ to inhibit mGluR1 receptors is lower than that described for CPCCOEt, our results demonstrate the existence of a new lead compound which may prove useful for the design of selective inhibitors of mGluR1 receptors.
In conclusion, our data demonstrate that among PLC-coupled mGluRs, mGluR1 but not mGluR5 receptors posses an allosteric modulatory site sensitive to CTZ. This site appears to correspond to the site sensitive to CPCCOEt which is located at the extracellular surface of TMVII. The ability of CTZ to selectively inhibit mGluR1, but not mGluR5 responses further stresses the functional heterogeneity of these two receptors which, although are both coupled to the activation of PLC, induce different patterns of intracellular Ca2+ release and, thus, may play distinct roles in intracellular Ca2+ homeostasis and in the physiology of neuronal and glial cells.
Acknowledgments
This work was supported by a grant from National Institutes of Health NS37436
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aramori I, Nakanishi S. Signal Transduction and Pharmacological Characteristics of a Metabotropic Glutamate Receptor, MGluR1, in Transfected CHO Cells. Neuron. 1992;8:757–65. doi: 10.1016/0896-6273(92)90096-v. [DOI] [PubMed] [Google Scholar]
- Audinat E, Lambolez B, Rossier J, Crepel F. Activity-Dependent Regulation of N-Methyl-D-Aspartate Receptor Subunit Expression in Rat Cerebellar Granule Cells. Eur J Neurosci. 1994;6:1792–800. doi: 10.1111/j.1460-9568.1994.tb00572.x. [DOI] [PubMed] [Google Scholar]
- Bhave G, Nadin BM, Brasier DJ, Glauner KS, Shah RD, Heinemann SF, Karim F, Gereau RW., 4th Membrane Topology of a Metabotropic Glutamate Receptor. J Biol Chem. 2003;278:30294–301. doi: 10.1074/jbc.M303258200. [DOI] [PubMed] [Google Scholar]
- Carroll FY, Stolle A, Beart PM, Voerste A, Brabet I, Mauler F, Joly C, Antonicek H, Bockaert J, Muller T, Pin JP, Prezeau L. BAY36-7620: a Potent Non-Competitive MGlu1 Receptor Antagonist With Inverse Agonist Activity. Mol Pharmacol. 2001;59:965–73. [PMC free article] [PubMed] [Google Scholar]
- Conn PJ, Pin JP. Pharmacology and Functions of Metabotropic Glutamate Receptors. Annu Rev Pharmacol Toxicol. 1997;37:205–37. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- Daggett LP, Sacaan AI, Akong M, Rao SP, Hess SD, Liaw C, Urrutia A, Jachec C, Ellis SB, Dreessen J, et al. Molecular and Functional Characterization of Recombinant Human Metabotropic Glutamate Receptor Subtype 5. Neuropharmacology. 1995;34:871–86. doi: 10.1016/0028-3908(95)00085-k. [DOI] [PubMed] [Google Scholar]
- De Blasi A, Conn PJ, Pin J, Nicoletti F. Molecular Determinants of Metabotropic Glutamate Receptor Signaling. Trends Pharmacol Sci. 2001;22:114–20. doi: 10.1016/s0165-6147(00)01635-7. [DOI] [PubMed] [Google Scholar]
- Deng L, Chen G. Cyclothiazide Potently Inhibits Gamma-Aminobutyric Acid Type A Receptors in Addition to Enhancing Glutamate Responses. Proc Natl Acad Sci U S A. 2003;100:13025–9. doi: 10.1073/pnas.2133370100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flor PJ, Gomeza J, Tones MA, Kuhn R, Pin JP, Knopfel T. The C-Terminal Domain of the MGluR1 Metabotropic Glutamate Receptor Affects Sensitivity to Agonists. J Neurochem. 1996;67:58–63. doi: 10.1046/j.1471-4159.1996.67010058.x. [DOI] [PubMed] [Google Scholar]
- Gabellini N, Manev RM, Manev H. Is the Heterologous Expression of Metabotropic Glutamate Receptors (MGluRs) an Appropriate Method to Study the MGluR Function? Experience With Human Embryonic Kidney 293 Cells Transfected With MGluR1. Neurochem Int. 1994;24:533–9. doi: 10.1016/0197-0186(94)90004-3. [DOI] [PubMed] [Google Scholar]
- Gasparini F, Kuhn R, Pin JP. Allosteric Modulators of Group I Metabotropic Glutamate Receptors: Novel Subtype-Selective Ligands and Therapeutic Perspectives. Curr Opin Pharmacol. 2002;2:43–9. doi: 10.1016/s1471-4892(01)00119-9. [DOI] [PubMed] [Google Scholar]
- Gomeza J, Joly C, Kuhn R, Knopfel T, Bockaert J, Pin JP. The Second Intracellular Loop of Metabotropic Glutamate Receptor 1 Cooperates With the Other Intracellular Domains to Control Coupling to G-Proteins. J Biol Chem. 1996;271:2199–205. doi: 10.1074/jbc.271.4.2199. [DOI] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A New Generation of Ca2+ Indicators With Greatly Improved Fluorescence Properties. J Biol Chem. 1985;260:3440–50. [PubMed] [Google Scholar]
- Hack NJ, Sluiter AA, Balazs R. AMPA Receptors in Cerebellar Granule Cells During Development in Culture. Brain Res Dev Brain Res. 1995;87:55–61. doi: 10.1016/0165-3806(95)00054-h. [DOI] [PubMed] [Google Scholar]
- Hayashi Y, Sekiyama N, Nakanishi S, Jane DE, Sunter DC, Birse EF, Udvarhelyi PM, Watkins JC. Analysis of Agonist and Antagonist Activities of Phenylglycine Derivatives for Different Cloned Metabotropic Glutamate Receptor Subtypes. J Neurosci. 1994;14:3370–7. doi: 10.1523/JNEUROSCI.14-05-03370.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton RM, Ho SN, Pullen JK, Hunt HD, Cai Z, Pease LR. Gene Splicing by Overlap Extension. Methods Enzymol. 1993;217:270–9. doi: 10.1016/0076-6879(93)17067-f. [DOI] [PubMed] [Google Scholar]
- Jingami H, Nakanishi S, Morikawa K. Structure of the Metabotropic Glutamate Receptor. Curr Opin Neurobiol. 2003;13:271–8. doi: 10.1016/s0959-4388(03)00067-9. [DOI] [PubMed] [Google Scholar]
- Kajita J, Yamaguchi H. Calcium Mobilization by Muscarinic Cholinergic Stimulation in Bovine Single Airway Smooth Muscle. Am J Physiol. 1993;264:L496–503. doi: 10.1152/ajplung.1993.264.5.L496. [DOI] [PubMed] [Google Scholar]
- Kawabata S, Tsutsumi R, Kohara A, Yamaguchi T, Nakanishi S, Okada M. Control of Calcium Oscillations by Phosphorylation of Metabotropic Glutamate Receptors. Nature. 1996;383:89–92. doi: 10.1038/383089a0. [DOI] [PubMed] [Google Scholar]
- Kingston AE, Burnett JP, Mayne NG, Lodge D. Pharmacological Analysis of 4-Carboxyphenylglycine Derivatives: Comparison of Effects on MGluR1 Alpha and MGluR5a Subtypes. Neuropharmacology. 1995;34:887–94. doi: 10.1016/0028-3908(95)00069-i. [DOI] [PubMed] [Google Scholar]
- Knopfel T, Lukic S, Leonard T, Flor PJ, Kuhn R, Gasparini F. Pharmacological Characterization of MCCG and MAP4 at the MGluR1b, MGluR2 and MGluR4a Human Metabotropic Glutamate Receptor Subtypes. Neuropharmacology. 1995;34:1099–102. doi: 10.1016/0028-3908(95)00111-i. [DOI] [PubMed] [Google Scholar]
- Kohara A, Toya T, Tamura S, Watabiki T, Nagakura Y, Shitaka Y, Hayashibe S, Kawabata S, Okada M. Radioligand Binding Properties and Pharmacological Characterization of 6-Amino-N-Cyclohexyl-N,3-Dimethylthiazolo[3,2-a]Benzimidazole-2-Carboxamid e (YM-298198), a High-Affinity, Selective, and Noncompetitive Antagonist of Metabotropic Glutamate Receptor Type 1. J Pharmacol Exp Ther. 2005;315:163–9. doi: 10.1124/jpet.105.087171. [DOI] [PubMed] [Google Scholar]
- Lavreysen H, Janssen C, Bischoff F, Langlois X, Leysen JE, Lesage AS. [3H]R214127: a Novel High-Affinity Radioligand for the MGlu1 Receptor Reveals a Common Binding Site Shared by Multiple Allosteric Antagonists. Mol Pharmacol. 2003;63:1082–93. doi: 10.1124/mol.63.5.1082. [DOI] [PubMed] [Google Scholar]
- Lazarewicz JW, Wroblewski JT, Costa E. N-Methyl-D-Aspartate-Sensitive Glutamate Receptors Induce Calcium- Mediated Arachidonic Acid Release in Primary Cultures of Cerebellar Granule Cells. J Neurochem. 1990;55:1875–81. doi: 10.1111/j.1471-4159.1990.tb05771.x. [DOI] [PubMed] [Google Scholar]
- Litschig S, Gasparini F, Rueegg D, Stoehr N, Flor PJ, Vranesic I, Prezeau L, Pin JP, Thomsen C, Kuhn R. CPCCOEt, a Noncompetitive Metabotropic Glutamate Receptor 1 Antagonist, Inhibits Receptor Signaling Without Affecting Glutamate Binding. Mol Pharmacol. 1999;55:453–61. [PubMed] [Google Scholar]
- Malherbe P, Kratochwil N, Knoflach F, Zenner MT, Kew JN, Kratzeisen C, Maerki HP, Adam G, Mutel V. Mutational Analysis and Molecular Modeling of the Allosteric Binding Site of a Novel, Selective, Noncompetitive Antagonist of the Metabotropic Glutamate 1 Receptor. J Biol Chem. 2003;278:8340–7. doi: 10.1074/jbc.M211759200. [DOI] [PubMed] [Google Scholar]
- Micheli F, Fabio RD, Cavanni P, Rimland JM, Capelli AM, Chiamulera C, Corsi M, Corti C, Donati D, Feriani A, Ferraguti F, Maffeis M, Missio A, Ratti E, Paio A, Pachera R, Quartaroli M, Reggiani A, Sabbatini FM, Trist DG, Ugolini A, Vitulli G. Synthesis and Pharmacological Characterisation of 2,4-Dicarboxy-Pyrroles As Selective Non-Competitive MGluR1 Antagonists. Bioorg Med Chem. 2003;11:171–83. doi: 10.1016/s0968-0896(02)00424-8. [DOI] [PubMed] [Google Scholar]
- Miller S, Romano C, Cotman CW. Growth Factor Upregulation of a Phosphoinositide-Coupled Metabotropic Glutamate Receptor in Cortical Astrocytes. J Neurosci. 1995;15:6103–9. doi: 10.1523/JNEUROSCI.15-09-06103.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosmann T. Rapid Colorimetric Assay for Cellular Growth and Survival: Application to Proliferation and Cytotoxicity Assays. J Immunol Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- Partin KM, Bowie D, Mayer ML. Structural Determinants of Allosteric Regulation in Alternatively Spliced AMPA Receptors. Neuron. 1995;14:833–43. doi: 10.1016/0896-6273(95)90227-9. [DOI] [PubMed] [Google Scholar]
- Partin KM, Patneau DK, Winters CA, Mayer ML, Buonanno A. Selective Modulation of Desensitization at AMPA Versus Kainate Receptors by Cyclothiazide and Concanavalin A. Neuron. 1993;11:1069–82. doi: 10.1016/0896-6273(93)90220-l. [DOI] [PubMed] [Google Scholar]
- Pin JP, Duvoisin R. The Metabotropic Glutamate Receptors: Structure and Functions. Neuropharmacology. 1995;34:1–26. doi: 10.1016/0028-3908(94)00129-g. [DOI] [PubMed] [Google Scholar]
- Pin JP, Joly C, Heinemann SF, Bockaert J. Domains Involved in the Specificity of G Protein Activation in Phospholipase C-Coupled Metabotropic Glutamate Receptors. EMBO J. 1994;13:342–8. doi: 10.1002/j.1460-2075.1994.tb06267.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pshenichkin SP, Wise BC. Okadaic Acid Increases Nerve Growth Factor Secretion, MRNA Stability, and Gene Transcription in Primary Cultures of Cortical Astrocytes. J Biol Chem. 1995;270:5994–9. doi: 10.1074/jbc.270.11.5994. [DOI] [PubMed] [Google Scholar]
- Raulli R, Danysz W, Wroblewski JT. Pretreatment of Cerebellar Granule Cells With Concanavalin A Potentiates Quisqualate-Stimulated Phosphoinositide Hydrolysis. J Neurochem. 1991;56:2116–24. doi: 10.1111/j.1471-4159.1991.tb03474.x. [DOI] [PubMed] [Google Scholar]
- Santi MR, Ikonomovic S, Wroblewski JT, Grayson DR. Temporal and Depolarization-Induced Changes in the Absolute Amounts of MRNAs Encoding Metabotropic Glutamate Receptors in Cerebellar Granule Neurons in Vitro. J Neurochem. 1994;63:1207–17. doi: 10.1046/j.1471-4159.1994.63041207.x. [DOI] [PubMed] [Google Scholar]
- Sharp RL, Mayne NG, Burnett JP. Cyclothiazide Differentially Modulates Human Metabotropic Glutamate Receptors Linked to Phosphoinositide Hydrolysis Stimulation in Oocytes. Eur J Pharmacol. 1994;269:R5–7. doi: 10.1016/0922-4106(94)90049-3. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Tsuchida K, Tanabe Y, Masu M, Nakanishi S. Role of the Large Extracellular Domain of Metabotropic Glutamate Receptors in Agonist Selectivity Determination. J Biol Chem. 1993;268:19341–5. [PubMed] [Google Scholar]
- Vignes M. Regulation of Spontaneous Inhibitory Synaptic Transmission by Endogenous Glutamate Via Non-NMDA Receptors in Cultured Rat Hippocampal Neurons. Neuropharmacology. 2001;40:737–48. doi: 10.1016/s0028-3908(00)00213-6. [DOI] [PubMed] [Google Scholar]
- Wroblewska B, Wroblewski JT, Pshenichkin S, Surin A, Sullivan SE, Neale JH. N-Acetylaspartylglutamate Selectively Activates MGluR3 Receptors in Transfected Cells. J Neurochem. 1997;69:174–81. doi: 10.1046/j.1471-4159.1997.69010174.x. [DOI] [PubMed] [Google Scholar]
- Wroblewski JT, Nicoletti F, Costa E. Different Coupling of Excitatory Amino Acid Receptors With Ca2+ Channels in Primary Cultures of Cerebellar Granule Cells. Neuropharmacology. 1985;24:919–21. doi: 10.1016/0028-3908(85)90046-2. [DOI] [PubMed] [Google Scholar]
- Wroblewski JT, Nicoletti F, Fadda E, Costa E. Phencyclidine Is a Negative Allosteric Modulator of Signal Transduction at Two Subclasses of Excitatory Amino Acid Receptors. Proc Natl Acad Sci U S A. 1987;84:5068–72. doi: 10.1073/pnas.84.14.5068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong J, Gerber G, Kojic L, Randic M. Dual Modulation of Excitatory Synaptic Transmission by Agonists at Group I Metabotropic Glutamate Receptors in the Rat Spinal Dorsal Horn. Brain Res. 2000;887:359–77. doi: 10.1016/s0006-8993(00)03066-3. [DOI] [PubMed] [Google Scholar]