

Summary
p193/CUL7 is an E3 ubiquitin ligase initially identified as an SV40 Large T Antigen binding protein. Expression of a dominant interfering variant of mouse p193/CUL7 (designated 1152stop) conferred resistance to MG132- and etoposide-induced apoptosis in U2OS cells. Immune precipitation/Western analyses revealed that endogenous p193/CUL7 formed a complex with Parc (a recently identified parkin-like ubiquitin ligase) and p53. Apoptosis resistance did not result from 1152stop-mediated disruption of the endogenous p193/CUL7 binding partners. Moreover, 1152stop molecule did not directly bind to endogenous p193/CUL7, Parc or p53. These data suggested a role for p193/CUL7 in the regulation of apoptosis independently of p53 and Parc activity.
Keywords: E3 ligase, BH-3 only, SV40 T-Antigen
Introduction
The balance between cell proliferation and apoptosis is critical for normal development and for the maintenance of homeostasis in adult organisms. Disruption of this balance has been implicated in a large number of disease processes, ranging from autoimmunity and neurodegenerative disorders to cancer. Although apoptosis can be triggered by many diverse intracellular and extracellular stimuli, once initiated a conserved set of molecules are recruited for the execution of the cell death program which is ultimately dependent upon the activation of the caspase family of cysteine proteases [1, 2].
In order to study the interplay between cell proliferation and apoptosis in myocardium, we have previously generated transgenic mice that expressed either wild-type or mutant versions of the SV40 Large T Antigen oncoprotein under the regulation of cardiomyocyte restricted promoters [3–5]. Biochemical analyses of cardiomyocyte cell lines derived from these mice identified three prominent T Antigen binding proteins, namely p53, p107 and p193 [6]. Subsequent studies revealed that transient expression of p193 (also known as CUL7, see below) induced apoptosis in NIH-3T3 cells [7]. p193/CUL7-induced apoptosis was dependent upon the presence of a short sequence motif with striking homology to the Bcl-2 homology domain 3, and furthermore was antagonized by co-expression of Bcl-xL. These data suggested that p193/CUL7 might be a new member of the BH3 only pro-apoptosis protein family.
Structure-function experiments revealed that introduction of a stop codon at amino acid residue #1153 of p193/CUL7 resulted in a molecule with apparent dominant interfering activity [8]. Expression of this molecule, designated 1152stop, enhanced cell growth in NIH-3T3 cells. A similar phenotype was observed with expression of a p193/CUL7 anti-sense construct. Expression of 1152stop rendered NIH-3T3 cells resistant to methyl methanesulfonate-induced apoptosis, an agent that promotes DNA damage and induces apoptosis via both p53-dependent and p53-independent pathways [9, 10]. Similarly, co-expression of 1152stop and dominant interfering p53 was sufficient to block adenoviral E1A oncoprotein-induced apoptosis in embryonic stem cell-derived cardiomyocytes [8]. Collectively, these data suggested that p193/CUL7 might play a role in the regulation of the balance between cell cycle activity and cell death in cardiomyocytes.
Ubiquitin-mediated proteolysis plays an important role in many biological processes, including the regulation of apoptosis [11, 12]. Ubiquitination of a protein is dependent upon the activities of a ubiquitin-activating enzyme (E1), a ubiquitin-conjugating enzyme (E2) and a multi-protein adaptor protein complex (E3). The E3 ubiquitin ligase complexes are responsible for directing the E2 activity to specific protein targets. Polyubiquitinated proteins are then recognized and degraded by the 26S proteasome [12, 13]. Recent studies revealed that p193/CUL7 shared homology with members of the cullin family, which are components of E3 ubiquitin ligases [14–16]. Moreover, p193/CUL7 was found to assemble an SCF-ROC1-like E3 ligase complex, consisting of Skp1, p193/CUL7, Fbx29 (also called Fbw8), and the ROC1 RING finger protein (also called Rbx1 or Hrt1) [16, 17]. Targets of the p193/CUL7 E3 ligase activity are currently under investigation.
In this report, the effect of dominant interfering 1152stop expression on MG132-and etoposide-induced apoptosis was examined. Transgene expression rendered U2OS cells resistant to both compounds. MG132 and etoposide induce apoptosis through markedly different mechanisms than MMS. Specifically, MG132 inhibits the 26S proteasome, leading to p53-independent apoptosis. Etoposide is a commonly used chemotherapeutic agent that has been shown to cause G2 cell cycle arrest, which in turn induces apoptosis largely via a p53-dependent pathway. In an effort to identify the mechanistic basis of apoptosis-resistance, immune precipitation (IP)/Western blot analyses were performed to determine if 1152stop expression altered the expression and/or binding activities of the endogenous p193/CUL7 complex. Endogenous p193/CUL7 was observed to complex with Parc (a recently identified parkin-like ubiquitin ligase) and p53. Expression of 1152stop did not alter this complex. Moreover, 1152stop molecule did not bind to either p53 or Parc. These data suggested a role for p193/CUL7 in the regulation of apoptosis independently of p53 activity.
Materials and Methods
Cell culture conditions and generation of stable cell lines
U2OS, MG-63 and SK-N-AS cells (American Type Culture Collection, Washington, D.C.) were cultured in DMEM containing 10% fetal calf serum and antibiotics. Clonal U2OS cell lines stably expressing mouse 1152stop-myc (U2OS-1152stop) or nuclear localized β-galactosidase (U2OS-nLAC cells) were generated by transfection of expression constructs using Transit reagent according to the manfacturer’s protocol (Mirus, Houston, TX), followed by G418 selection (300 μg/mL).
MG132 treatment, Etoposide treatment, and apoptosis assay
Cells were treated with 0.25 μM etoposide for 8 hr, washed twice with PBS, and supplemented with fresh DMEM with 10% fetal bovine serum described previously [18, 19]. Cells were treated with the 26S proteasome inhibitor N-carbobenzoxyl-Leu-Leu-leucinal (MG132, Calbiochem, San Diego, CA) at 10 μM for 24 hrs as described previously [20]. To quantitate apoptosis, cells were harvested, stained with FITC-Annexin V and propidium iodide using a Molecular Probes (Eugene, OR) Apoptosis Assay Kit as per manufacturer’s instructions, and analyzed by flow cytometry. At least three trials were performed per group for each drug treatment.
Generation of mouse monoclonal antibodies against p193/CUL7
Sequences encoding p193/CUL7 amino acid residues 805 through 1689 were subcloned into a pGEX bacterial expression vector (Amersham Biosciences, Piscataway, NJ). Recombinant protein was separated by size on polyacrylamide gels, and isolated from gel slices via electroporation as described [21]. Purified protein was submitted to the Custom Hybridoma Development Service (St. Louis University Health Sciences Center, St. Louis MO), and two anti-p193/CUL7 monoclonal antibodies (AB13 and AB38), were generated using standard hybridoma fusion protocols. To identify the p193/CUL7 epitopes recognized by these antibodies, a nested array of overlapping 13-mer synthetic peptides representing p193/CUL7 amino acid residues 805 through 1689 were spotted onto a nitrocellulose filter (Jerini BiopTools, Berlin, Germany) and reacted individually with AB13 or AB38. Signal was then developed using HRP-conjugated anti-mouse antibody and ECL (Amersham Biosciences, Piscataway, NJ).
Immune precipitation and Western blot
All immune precipitation reactions were repeated a minimum of three times, with comparable results. Proteins were extracted directly from tissues or cells using NP40 buffer as previously described [22, 23]. In some experiments, protein was prepared from p193/CUL7-null mice (H. Nakajima and L.J. Field, in preparation). Protein content was quantitated using Coomassie Blue method (Pierce, Rockford, IL). For total protein Western blots, the filters were stained with 0.1% napthol blue-black in 45% methanol, 10% acetic acid to assess the efficiency of transfer. For immune precipitations, lysate was preadsorbed with protein A-agarose beads (100 μg/ml lysate, Roche, Indianapolis, IN) for 45 min at 4°C. The beads were subsequently removed by centrifugation. 100 μg of preadsorbed lysate was then reacted with 0.1 μg of primary antibody in a final adjusted volume of 1 ml (with PBS) for 1 hr at 4°C with rocking. 100 μl protein A beads (90 mg/ml) were added and the incubation was continued for an additional hour. Immune complexes were collected by centrifugation, washed three times with ice-cold PBS, and eluted in sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) loading buffer for 5 min at 95°C. Samples were resolved on 7 or 10% SDS-PAGE gels. Fractionated proteins were then electrotransferred from the gel to nitrocellulose (Amersham) filters in Towbin buffer at 200-mA constant current and analyzed by Western blot. Antibodies used were anti-p53 DO-1 and anti-c-myc (both from Santa Cruz Biotech, Santa Cruz, CA), anti-Flag, anti-ubiquitin and pan anti-phospho-serine (from Sigma, St. Louis MO), anti-SV40 large T antigen Ab-1 and anti-p53 PC35 (both from Oncogene, Cambridge, MA), and pan anti-phospho-threonine, anti-phospho-p53 (ser 392) and anti-phospo-p53 (ser 46) (from Cell Signaling, Danvers MA). Signal was visualized by the ECL method according to the manufacturer’s protocol (Amersham). For calf intestinal phosphatase (CIP) experiments, 150 μg of total cell protein lysate was incubated with 20U CIP (New England Biolabs) for 20 min at 30°C prior to immune precipitation as previously described [24].
Yeast two-hybrid assay
Plasmids pGBKT7-53 and pGADT7-T (Clontech, Palo Alto, CA) were used as positive controls. Full length p193/CUL7 and p193/CUL7-1152stop were subcloned into the pGAD7 vector using standard approaches [25]. To quantitate protein-protein interactions, Saccaromyces cerevisiae Y187 (MATα, ura3–52, his3–200,ade2–101, trp1–901, leu2–3,112, gal4Δ, met−, gal80Δ, URA3::GAL1UAS-GAL1TATA-lacZ, MEL1) was co-transfected with pGBKT7-53 and with pGADT7-T, pGADT7-p193/CUL7 or pGADT7-1152stop using the LiOAC method [26]. Transformants were selected on synthesis define medium (SD) at 30°C for 6 days. Four independent colonies for each transfection pair were individually grown in 50 ml of SD broth at 30°C with 150 rpm shaking overnight. Cells were harvested, washed with PBS, and resuspended in Z buffer. β-galactosidase activity was measured by filter and broth assays per manufacturer protocols (Clontech).
Ablation of endogenous p193/CUL7, Parc, or p53 by RNAi
U2OS cells were maintained in DMEM supplemented with 10% fetal bovine serum. The RNAi-mediated knockdown of endogenous p193/CUL7, Parc, or p53 was performed essentially as described previously [18, 27] and manufacturers’ protocol (Dharmacon, Lafayette, CO and Invitrogen). 21-nucleotide siRNA duplexes with 3′ dTdT overhangs corresponding to p193/CUL7 mRNA (AACACUGAUCGAGAGGUGCUC) or Parc mRNA (AAGCUUUCCUCGAGAUCCAGG) were synthesized (Dharmacon). The Parc mRNA sequence in the inverted orientation (AAGGACCUAGAGCUCCUUUCG) was used as a nonspecific RNAi control. Dharmacon p53 SMARTpool was used for RNAi-mediated p53 ablation. RNAi transfections were performed using Oligofectamine Reagent or Lipofectamine 2000 (Invitrogen). 24 hr before transfection, about 1 million cells were plated on a 10 cm dish. Cells were transfected using manufacturer’s protocol for 1–3 times with 24 hr intervals at a concentration of 100 nM.
Anti-p53 immune cytology
Cells were fixed (acetone/methanol, 1:1, 2 min at room temperature) and reacted with anti-p53 monoclonal antibody (Ab-1, Oncogene, San Diego CA) over night at 4°C, rinsed and then reacted with biotinylated anti-mouse IgG (Boehringer, Indianapolis IN) for 1hr. Samples were then reacted with Streptavidin Texas Red (Zymed, San Francisco CA) for 1hr and imaged by fluorescence microscopy.
Statistical Analyses
The statistical significance was determined by ANOVA with Tukey multiple comparisons test for treated cells among groups. P values < 0.05 were considered statistically significant. All results are presented as the mean ± SEM (standard error of the mean).
Results
Antagonization of p193/CUL7 activity rendered U2OS cells resistant to apoptosis induced by 26S proteasome inhibition or etoposide treatment
The observations that p193/CUL7 exhibited E3 ubiquitin ligase activity [16] and that p193/CUL7 transfection induced apoptosis [7] suggested a potential relationship between p193/CUL7-mediated ubiquitination and cell death. To test this possibility, U2OS, U2OS-nLAC (a cell line expressing a nuclear-localized beta-galactosidase reporter), U2OS-1152stop (a cell line expressing a c-terminal truncation of mouse p193/CUL7 with apparent dominant interfering activity) [8], and MG-63 (a cell line lacking p53) [28] were studied. Cells were cultured in absence or presence of MG132, a chemical inhibitor of the 26S proteasome. To monitor apoptosis, cells were harvested, fixed, and co-stained with FITC-conjugated Annexin V and propidium iodide. Flow cytometry was used to identify cells in early stages of apoptosis (i.e., annexin-positive and propidium iodide negative; Figure 1A, lower right quadrants). U2OS, U2OS-nLAC, and MG-63 cells exhibited high levels of apoptosis following MG132 treatment (Figure 1B). In contrast, U2OS-1152stop cells were resistant to MG132-induced apoptosis. MG132 treatment resulted in similar levels of ubiquitinated proteins in U2OS-nLAC and U2OS-1152stop cells as evidenced by Western blots using an anti-ubiquitin antibody (data not shown). Thus the pro-survival effect that resulted from 1152stop expression was not due simply to global effects of transgene expression on ubiquitination.
Figure 1.
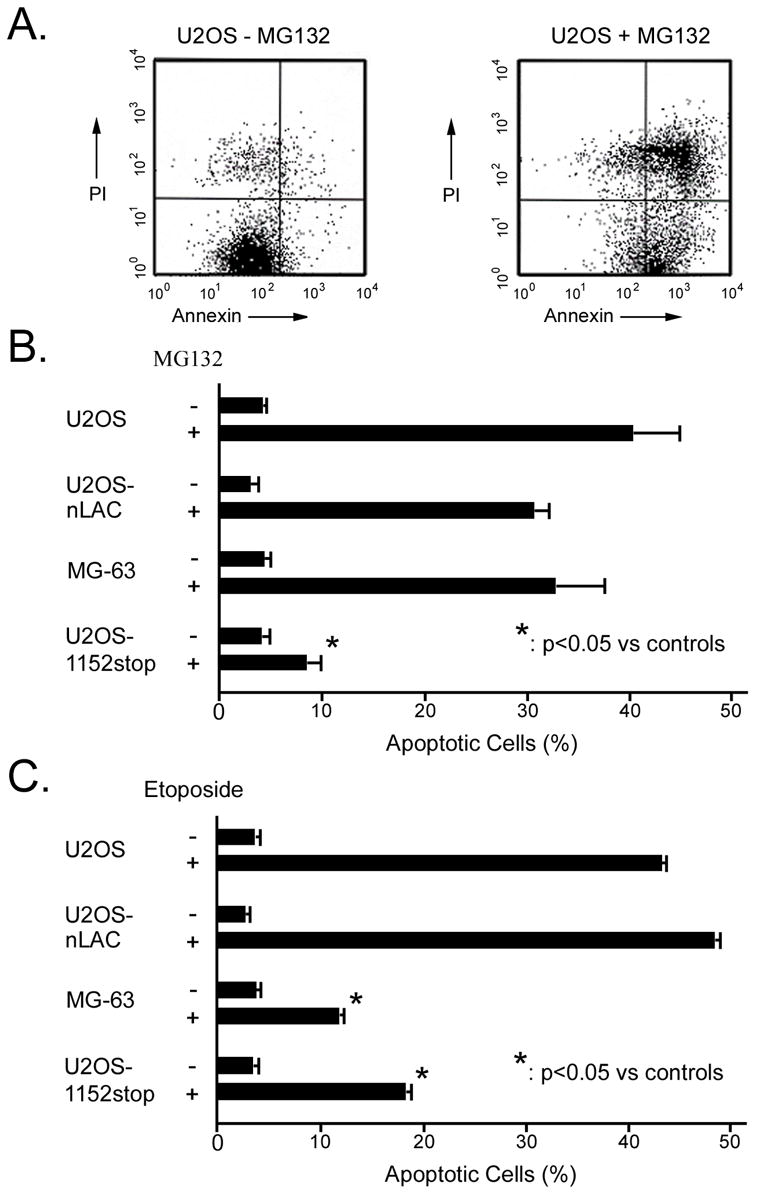
Expression of 1152stop protein rendered U2OS cells resistant to MG132-and etoposide-induced apoptosis. (A) Flow cytometric analysis for cells in early stages of apoptosis (FITC-Annexin V positive, PI negative; lower right quadrant); analysis of control (left panel) and MG132-treated (right panel) U2OS cells were shown. (B) Quantitation of cells at the early stage of apoptosis in the absence and presence of MG-132 is shown. Asterisk indicated p<0.01 vs. MG132-treated U2OS, U2OS-nLAC, and MG-63 cells. (C) Quantitation of cells at the early stage of apoptosis in the absence and presence of etoposide is shown. Asterisk indicated p<0.01 vs. etoposide-treated U2OS and U2OS-nLAC cells.
The role of p193/CUL7 in etoposide-induced apoptosis was also examined. U2OS, U2OS-nLAC, MG-63 and U2OS-1152stop cells were cultured in the presence or absence of etoposide and apoptosis was monitored as described above. Apoptosis was apparent in non-transfected U2OS and U2OS-nLAC cells after exposure to 25 μM of drug for 8 hours (Figure 1C). In contrast, apoptosis levels were significantly reduced in both MG-63 and U2OS-1152stop cells treated with the same dose of etoposide. At least three trials were performed per group for each drug treatment (MG132 or etoposide).
Generation and validation of anti- p193/CUL7 antibodies
In an effort to identify the molecular interactions leading to apoptosis resistance in cells expressing 1152-stop, monoclonal antibodies were raised against recombinant protein encoding p193/CUL7 amino acid residues 805 through 1689. Two antibodies, AB13 and AB38, were generated. To identify the epitopes recognized by these antibodies, a nested array of overlapping 13-amino acid residue synthetic peptides representing p193/CUL7 amino acid residues 805 through 1689 was spotted onto a nitrocellulose filter and reacted individually with AB13 or AB38. Signal was then developed using HRP-conjugated anti-mouse antibody and ECL (Figure 2A). These analyses revealed that AB13 recognized an epitope between p193/CUL7 amino acid residues 1665 and 1672, while AB38 recognized an epitope between amino acid residues 835 and 842.
Figure 2.
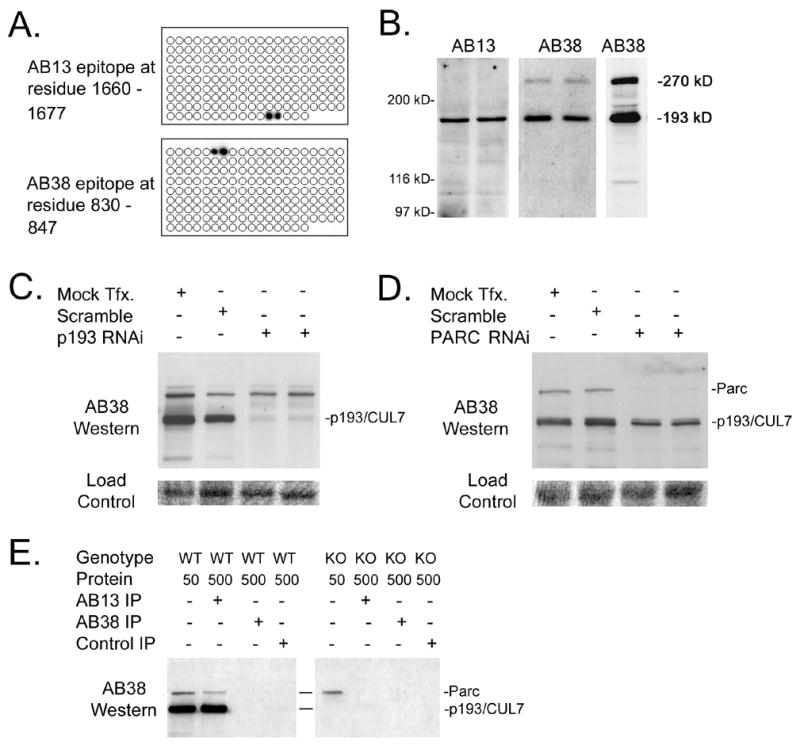
Validation of p193/CUL7 monoclonal antibodies. (A) Identification of the AB13 and AB38 epitope binding sites on p193/CUL7. Antibodies were reacted with a nitrocellulose filter containing a nested array of overlapping short synthetic peptides representing p193/CUL7 amino acid residues #805 through #1689. AB13 reacted with peptides containing amino acid residues 1660–1677; AB38 reacted with peptides containing amino acid residues 830–847. (B) Western blot analysis of fetal mouse hearts using AB13 (left panel) and AB38 (middle panel). Right panel shows Western blot analysis of fetal mouse brain using AB38. (C) Western blot analysis of U2OS cells following mock transfection, scrambled RNAi transfection, and anti-p193/CUL7 RNAi transfection. The lower panel shows a protein load control. (D) Western blot analysis of U2OS cells following mock transfection, scrambled RNAi transfection, and anti-Parc RNAi transfection. The lower panel shows a protein load control. (E) Protein from wild-type (WT) or p193/CUL7 knockout (KO) fetal mouse brain was reacted with either AB13, AB38, or an IgG subtype-matched nonspecific antibody, and the resulting immune complex was analyzed by Western blotting using AB38 antibody.
Western blot analyses with protein prepared from fetal mouse hearts revealed that both AB13 and AB38 detected a protein of approximately 193 kD (Figure 2B). AB38 detected a second protein with a mass of approximately 270 kD; this species was also detected in protein prepared from fetal mouse brain (Figure 2B). RNA interference (RNAi)/Western blot analyses were performed to confirm that the 193 kD protein detected by the monoclonal antibodies was p193/CUL7. The 193 kD protein was readily detected by AB38 in mock-transfected U2OS cells (a human osteosarcoma cell line), and in U2OS cells transfected with siRNA encoding a scrambled p193/CUL7 sequence. In contrast, the level of the 193 kD protein was markedly decreased in U2OS cells transfected with siRNA encoding sequences complementary to p193/CUL7 (Figure 2C). Similar results were obtained with AB13 (not shown). These data confirmed that the 193 kD protein recognized by AB38 and AB13 was p193/CUL7.
To establish the identity of the 270 kD protein recognized by AB38, the NCBI database was searched for large proteins with perfect homology to the AB38 amino acid epitope but lacking the AB13 epitope. A single candidate was identified, the UbcH7 associated E3 ubiquitin ligase protein H7-AP1 [29, 30], which was subsequently identified as a cytoplasmic p53 binding protein and renamed Parc [18]. RNAi/Western blot analyses were performed to determine if the 270 kD protein detected by AB38 was Parc. The 270 kD protein was readily detected by AB38 in mock-transfected U2OS cells, and in U2OS cells transfected with siRNA encoding a scrambled Parc sequence. In contrast, the level of the 270 kD protein was markedly decreased in cells transfected with siRNA complementary to sequences present in Parc (Figure 2D). These data confirmed that the 270 kD protein recognized by AB38 was Parc.
To determine if the antibodies were suitable for immune precipitation, protein from fetal mouse brain was reacted with AB13, and the resulting immune precipitate was analyzed by Western blot with AB38. Both p193/CUL7 and Parc were detected in AB13 immune precipitate (Figure 2E), but not in control immune precipitate generated with an IgG subtype-matched nonspecific monoclonal antibody. Given that AB13 failed to recognize Parc in Western blots of whole tissue lysate, these data suggest that p193/CUL7 and Parc formed a complex. In support of this, Parc was readily detected in whole tissue lysate from p193/CUL7-deficient fetal brain, but was not detected in anti-AB13 immune precipitate (Figure 2E). These data confirm the recent observation that p193/CUL7 and Parc form a complex in vivo [31]. Neither p193/CUL7 nor Parc were detected in immune precipitate generated with AB38, suggesting that the epitope recognized by this antibody was inaccessible under non-denaturing conditions.
1152stop protein does not alter the endogenous p193/CUL7 complex in U2OS cells
IP/Western blot analyses were performed to determine if 1152stop binds to and/or alters the composition of endogenous p193/CUL7 complexes in vivo. In lysate prepared from control non-transfected U2OS cells, p193/CUL7, Parc, and p53 were readily detected in total protein samples (Figure 3A, left panel). Both p193/CUL7 and Parc were detected by AB38 in immune precipitate generated with AB13, indicating that p193/CUL7 and Parc bound one another in human cells. In addition, p193/CUL7 and Parc were also detected by AB38 in immune precipitate generated with an anti-p53 antibody (DO-1). Similarly, p53 was detected by anti-p53 antibody AB-7 in immune precipitate generated with AB13. These data indicated that p193/CUL7, Parc, and p53 formed either binary or tertiary complexes.
Figure 3.
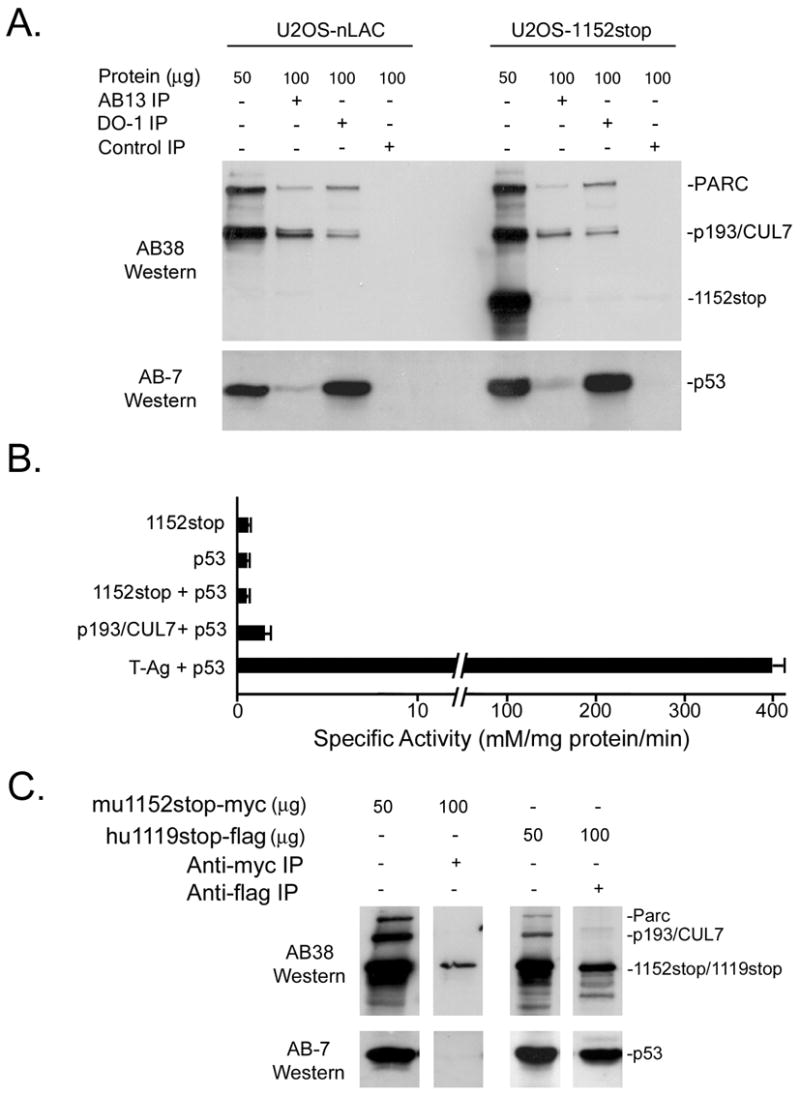
p193/CUL7 interactions in U2OS-nLAC and U2OS-1152stop cells. (A) Protein from U2OS-nLAC and U2OS-1152stop cells was reacted with either AB13, anti-p53 DO-1, or IgG subtype-matched nonspecific antibodies, and the resulting immune complexes were analyzed by Western blotting using AB38 and anti-p53 Ab-7. (B) Beta-galactosidase activity from yeast two-hybrid assays in cells transfected with the indicated constructs. (C) Protein from U2OS cells transiently expressing myc-tagged mouse 1152stop molecule (mu1152stop-myc) or flag-tagged human 1119stop (hu1119stop-flag) was reacted with anti-myc or anti-flag antibodies. The resulting immune complexes were analyzed by Western blotting using AB38 and Ab-7 antibodies.
Similar analysis of lysate prepared from U2OS-1152stop cells revealed that expression of 1152stop did not alter steady state levels of endogenous p193/CUL7, Parc, or p53 in total protein samples (Figure 3A; right panel; note that due to the c-terminal truncation, 1152stop protein is recognized by AB38 but not by AB13). p193/CUL7, Parc, and p53 were readily detected in immune precipitate from U2OS-nLAC and U2OS-1152stop generated with AB13 or with DO-1. In contrast, 1152stop protein was not detected in either AB13 or DO-1 immune precipitate from U2OS-1152stop cells, indicating that the dominant interfering p193/CUL7 molecule did not bind to endogenous p193/CUL7 or p53.
The presence of p53 in AB13 immune precipitate from U2OS cells (Figure 3A) was inconsistent with the absence of 1152stop/p53 binding in U2OS-1152stop cells. Yeast two-hybrid assay was used to further examine interactions between 1152stop and p53. The “prey” vector encoded a chimeric protein containing the GAL4 transactivation domain fused to 1152stop. The “bait” vector encoded a chimeric protein containing mouse p53 fused to the GAL4 DNA binding domain. Transfection of the prey and bait vectors alone or in combination into yeast carrying a GAL4-dependent beta-galactosidase reporter gene resulted in similar albeit very low levels of reporter activity, indicating that the two proteins did not interact (Figure 3B). As a positive control, prey vector that encoded a chimeric protein comprised of the GAL4 transactivation domain fused to T-Antigen was co-transfected with the p53/GAL4 bait vector into the reporter yeast strain. This co-transfection resulted in robust beta-galactosidase activity (Figure 3B). Additional experiments demonstrated that full-length mouse p193/CUL7 also did not interact with p53 in this assay system (Figure 3B).
To determine if there might be species-specific differences in p193/CUL7 binding partners, U2OS cells were transiently transfected with a construct expressing a myc-tagged mouse p193/CUL7 1152stop molecule (designated mu1152stop-myc) or with a construct expressing a flag-tagged human p193/CUL7 cDNA with a stop codon inserted at amino acid residue 1120 (designated hu1119stop-flag). IP/Western analyses revealed that p53 was not present in anti-myc immune precipitate from cells transiently expressing mu1152stop-myc, confirming that this fragment of mouse p193/CUL7 did not bind to p53 (Figure 3C). In contrast, p53 was readily detected in anti-flag immune precipitate from cells transiently expressing hu1119stop-flag, indicating that the analogous C-terminal truncation molecule of human p193/CUL7 did bind to p53. Thus human, but not mouse, p193/CUL7 binds to p53. Interestingly, the absence of Parc in immune complexes generated with truncated, epitope-tagged human and mouse p193/CUL7 molecules indicated that the c-terminal region was required for binding to Parc (compare IP complexes with the truncated p193/CUL7 molecules in Panel C to those with the full-length molecules in Panel A).
Binding to p193/CUL7/Parc complex is not required for cytoplasmic localization of p53
Although previous studies demonstrated co-localization of Parc and p53 in the cytoplasm of SK-N-AS neuroblastoma cells [18], their physical interaction was not demonstrated in that study. Immune cytology analyses confirmed that p53 was localized in the cytoplasm in both U2OS and SK-N-AS cells (Figure 4A). Moreover, Western blot analysis of total protein samples revealed that U2OS and SK-N-AS cells expressed similar levels of p193/CUL7, Parc, and p53 (Figure 4B). In U2OS cells, p193/CUL7, Parc, and p53 were present in immune precipitate generated using either AB13 or DO-1. In contrast, p193/CUL7 and Parc were not present in DO-1 immune precipitate generated from SK-N-AS cells, and p53 was not present in AB13 immune precipitate (Figure 4B). Since Parc and p53 do not bind one another in SK-N-AS cells, interactions between these two proteins cannot explain the observed cytoplasmic p53 localization.
Figure 4.
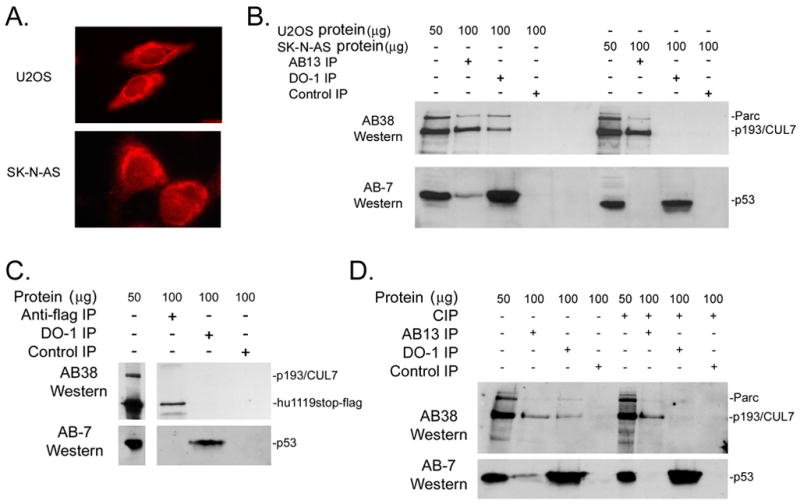
Cytoplasmic sequestration of p53 was not dependent upon interactions with p193/CUL7 or Parc. (A) Anti-p53 immune cytology (red signal) of U2OS and SK-N-AS cells. (B) Protein from U2OS or SK-N-AS was reacted with either AB13, anti-p53 DO-1, or IgG subtype-matched nonspecific antibodies, and the resulting immune complexes were analyzed by Western blotting using AB38 and anti-p53 Ab-7. (C) Protein prepared from SK-N-AS cells transiently expressing hu1119stop-flag was reacted with anti-flag, DO-1, or an IgG subtype-matched control antibody, and the resulting immune complexes were analyzed by Western blotting using AB38 and Ab-7 antibodies. (D) Protein from U2OS cells incubated in the absence or presence of calf intestinal phosphatase (CIP) was reacted with anti-flag, DO-1, or an IgG subtype-matched control antibody, and the resulting immune complexes were analyzed by Western blotting using AB38 and Ab-7 antibodies.
Since SK-N-AS cells express wild type p53, additional experiments were performed to determine if the absence of p53/p193 binding was due to acquired mutations in the endogenous p193. SK-N-AS cells were transiently transfected with hu1119stop-flag, which avidly bound p53 in U2OS cells (see above). IP/Western analyses revealed that p53 was not present in anti-flag immune complex, and that hu1119stop-flag was not present in anti-p53 DO-1 immune complex (Figure 4C). To determine if post-translational modification affected p193/CUL7 and p53 binding, IP/Western experiments were performed with control and calf intestinal phosphatase (CIP)-treated protein from U2OS cells. In the absence of CIP treatment, p53 was readily detected in immune complex generated with AB13, while p193/CUL7 and Parc were readily detected in DO-1 immune complex (Figure 4D). CIP treatment resulted in the loss of p53 in immune complex generated with AB13, and the loss of p193/CUL7 and Parc in immune complex generated with DO-1. These data suggested that post-translational modification was required for p53 to bind to p193/CUL7 and/or Parc.
To determine what phosphorylation events might give rise to p53 binding, lysate from U2OS cells was immune precipitated with AB13, DO-1, or control IgG, and the resulting immune precipitate was analyzed by Western blot. No p193/CUL7 or Parc phosphorylation was detected in Western blots using pan anti-phospho-threonine or pan anti-phospho-serine antibodies (within the limits of the sensitivity of the assay, see Figure 5A). However, p53 phosphorylation was detected with a pan anti-phospho-threonine antibody, as well as with an antibody which specifically recognizes p53 when phosphorylated at serine residue 392 (Figure 5A). The presence of phospho-p53 immune reactivity in the AB13 immune complex suggests that this species of p53 is responsible for binding activity. Similar IP-Western analyses with an antibody which specifically recognizes p53 when phosphorylated at serine residue 46 did not give rise to signal (not shown).
Figure 5.
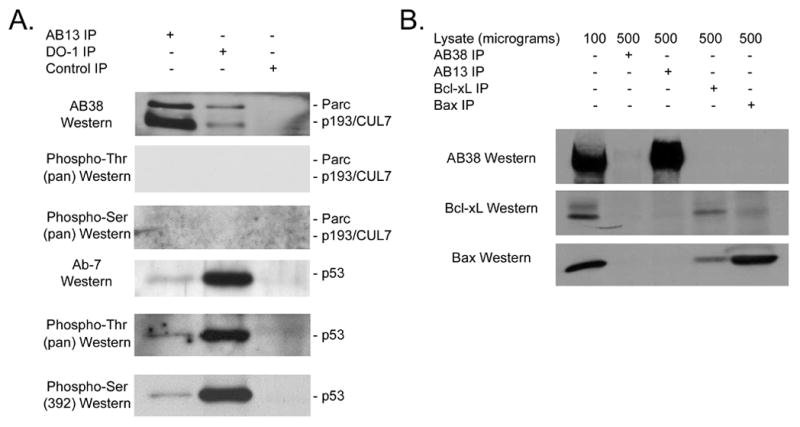
(A) p53 in anti-p93/CUL7 immune complex exhibits phospho-threonine immune reactivity. Protein from U2OS cells was reacted with either AB13, anti-p53 DO-1, or IgG subtype-matched nonspecific antibodies, and the resulting immune complexes were analyzed by Western blotting using AB38, pan anti-phospho-threonine or pan anti-phospho-serine (upper three panels), or using anti-p53 Ab-7, pan anti-phospho-threonine, or anti-p53-phospho-serine392 antibodies (lower three panels). (B) Murine p193/CUL7 does not bind to Bcl-xL or Bax. Protein from NIH-3T3 cells was reacted with either AB38, AB13, anti-Bcl-xL or anti-Bax antibody, and the resulting immune complexes were analyzed by Western blotting using AB38, anti-Bcl-xL and anti-Bax antibody.
Finally, additional IP/Western blot analyses were performed to determine if p193/CUL7 binds to Bcl-xL and/or Bax. NIH-3T3 cells were used as they are devoid of Parc, thus making identification of p193/CUL7 binding proteins less ambiguous. p193/CUL7, Bcl-xL, and Bax were readily detected in total protein samples (Figure 5B). Neither Bcl-xL nor Bax was present in immune complex generated with anti-p193/CUL7 antibody AB13. Similarly, p193/CUL7 was not present in anti-Bcl-xL or anti-Bax immune precipitate. Thus, p193/CUL7 does not bind to endogenous Bcl-xL or Bax. As a positive control, interaction between Bax and Bcl-xL was readily detected.
Discussion
The data presented here demonstrated that expression of the 1152stop protein inhibited MG132- and etoposide-induced apoptosis. The mechanism by which 1152stop inhibits apoptosis is not clear. In the case of MG132 treatment, the inability of 1152stop to bind to p53, and the presence of high levels of apoptosis in p53-null, indicated that apoptosis resistance occurred via a p53-independent pathway. This notion was supported by previous observations indicating that MG132-induced apoptosis was independent of the activities of p53 and p21 [18, 20, 29]. Given that MG132 inhibits the 26S proteasome, it is unlikely that 1152stop-induced anti-apoptotic activity resulted from simple inhibition of p193/CUL7 E3 ligase activity. In this regard it is of interest to note that p193/CUL7 was shown to induce apoptosis in NIH-3T3 cells [7]. This pro-apoptotic activity was dependent upon the presence of an intact BH3 motif in the c-terminus, and was antagonized by co-expression of Bcl-xL. Indeed, another E3 ligase with BH3-dependent pro-apoptosis activity has recently been identified [32]. Expression of 1152stop might have interfered with activation of endogenous p193/CUL7 pro-apoptotic activity, thus rendering the cells resistant to MG132. However, IP/Western blot analyses failed to demonstrate a direct interaction between p193/CUL7 and Bcl-xL or Bax.
1152stop also rendered U2OS cells resistant to etoposide-induced apoptosis. Etoposide (a commonly used chemotherapeutic agent) was previously shown to induce apoptosis largely via a p53-dependent pathway [33–35]. However, expression of 1152stop did not affect levels of p53 in the absence (Figure 3A) or presence (J. Dowell, unpublished observation) of etoposide. This, plus the observation that 1152stop did not bind to p53, suggested that 1152stop-mediated resistance to etoposide-induced apoptosis also resulted via a p53-independent mechanism. Previous studies revealed that etoposide treatment resulted in G2 cell cycle arrest, which in turn induced apoptosis [36, 37]. If G2-M transit required the presence of a protein targeted for ubiquitination by p193/CUL7, and if etoposide treatment lowered the effective concentration of that protein, it is possible that expression of 1152stop (with concomitant antagonization of endogenous p193/CUL7 activity) could restore levels to an effective concentration. The etoposide-induced G2 cell cycle block would thus be relaxed, which in turn could reduce the level of drug-induced apoptosis. In support of this scenario, expression of either 1152stop or p193/CUL7 anti-sense enhanced cell growth in an NIH-3T3 colony growth assay [8]. Similarly, targeted expression of 1152stop in the myocardium of transgenic mice was sufficient to relax the cell cycle checkpoints that otherwise prevented cardiomyocyte cell cycle reactivation following cardiac injury [38]. Definitive testing of the hypothesis however will require identification of the molecular targets of the mouse p193/CUL7 in U2OS cells.
The observation that p193/CUL7 formed a complex with Parc and p53 confirmed and extended recent observations from Skaar and colleagues [31]. These authors demonstrated the presence p193/CUL7 - Parc complex in both U2OS cells and mouse embryonic fibroblasts (MEFs). The inability of mouse p193/CUL7 to bind to p53 (Figure 3B, C) suggested that the presence of low levels of p53 in anti-Parc immune complex [31] resulted from direct interactions between p53 and Parc. Down-regulation of p193/CUL7 expression via RNAi treatment (Figure 2C) or gene targeting (Figure 2E) did not affect steady state levels of Parc. This is in contrast to the results of Skaar and colleagues, where targeting of the p193/CUL7 gene resulted in a dramatic reduction in Parc levels [31]. Given the close proximity of the p193/CUL7 and Parc genes (only 186 kb apart), it is possible that the p193/CUL7 targeting event(s) may have impacted expression of Parc. Although the significance of the binding between p193/CUL7 and Parc is presently not clear, the presence of both proteins in large (i.e., > 1 mD) complexes [18] raised the possibility that such protein-protein interactions might expand the spectrum of potential targets for p193/CUL7-mediated ubiquitination.
Gu and colleagues recently suggested that Parc functioned as an anchor protein that tethered p53 to the cytoplasm [18]. The observation that Parc and p53 bind to one another in U2OS cells, and that p53 is sequestered to the cytoplasm of those cells (Figure 4), would tend to support this view. However, the absence of co-immune precipitation of Parc with p53 in SK-N-AS cells indicates that direct binding of these two proteins is not required for sequestration of p53 to the cytoplasm. This view is further supported by the inability of truncated human p193/CUL71119stop-flag to bind to p53 following transfection into SK-N-AS cells (the observed binding of p53 to truncated human p193/CUL71119stop-flag in U2OS cells provides a compelling positive control). Although p53 binding to p193/CUL7 - Parc complex is sensitive to phosphorylation status, it is not clear if post-translational modification impacted upon sub-cellular localization in other cell types.
In summary, expression of 1152stop, a dominant interfering version of mouse p193/CUL7, conferred resistance to MG132- and etoposide-induced apoptosis in U2OS cells. The inability of the 1152stop protein to bind to p53, and the observation that 1152stop expression did not alter the endogenous p193/CUL7 - Parc - p53 complex, suggested that resistance to apoptosis occurred via a p53-independent pathway. Cytoplasmic localization of p53 in SK-N-AS cells was not dependent upon binding to Parc and/or p193/CUL7. Finally, binding of p53 to the p193/CUL7 - Parc complex appeared to be dependent upon the phosphorylation status of at least one of the proteins. These data support a role for p193/CUL7 in the regulation of apoptosis independently of p53 activity.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ashkenazi A, Dixit VM. Death receptors: signaling and modulation. Science. 1998;281:1305–8. doi: 10.1126/science.281.5381.1305. [DOI] [PubMed] [Google Scholar]
- 2.Adams JM, Cory S. The Bcl-2 protein family: arbiters of cell survival. Science. 1998;281:1322–6. doi: 10.1126/science.281.5381.1322. [DOI] [PubMed] [Google Scholar]
- 3.Field LJ. Atrial natriuretic factor-SV40 T antigen transgenes produce tumors and cardiac arrhythmias in mice. Science. 1988;239:1029–33. doi: 10.1126/science.2964082. [DOI] [PubMed] [Google Scholar]
- 4.Huh NE, Pasumarthi KB, Soonpaa MH, Jing S, Patton B, Field LJ. Functional abrogation of p53 is required for T-Ag induced proliferation in cardiomyocytes. J Mol Cell Cardiol. 2001;33:1405–19. doi: 10.1006/jmcc.2001.1403. [DOI] [PubMed] [Google Scholar]
- 5.Katz EB, Steinhelper ME, Delcarpio JB, Daud AI, Claycomb WC, Field LJ. Cardiomyocyte proliferation in mice expressing alpha-cardiac myosin heavy chain-SV40 T-antigen transgenes. Am J Physiol. 1992;262:H1867–76. doi: 10.1152/ajpheart.1992.262.6.H1867. [DOI] [PubMed] [Google Scholar]
- 6.Daud AI, Lanson NA, Jr, Claycomb WC, Field LJ. Identification of SV40 large T-antigen-associated proteins in cardiomyocytes from transgenic mice. Am J Physiol. 1993;264:H1693–700. doi: 10.1152/ajpheart.1993.264.5.H1693. [DOI] [PubMed] [Google Scholar]
- 7.Tsai SC, Pasumarthi KB, Pajak L, Franklin M, Patton B, Wang H, Henzel WJ, Stults JT, Field LJ. Simian virus 40 large T antigen binds a novel Bcl-2 homology domain 3-containing proapoptosis protein in the cytoplasm. J Biol Chem. 2000;275:3239–46. doi: 10.1074/jbc.275.5.3239. [DOI] [PubMed] [Google Scholar]
- 8.Pasumarthi KB, Tsai SC, Field LJ. Coexpression of mutant p53 and p193 renders embryonic stem cell-derived cardiomyocytes responsive to the growth-promoting activities of adenoviral E1A. Circ Res. 2001;88:1004–11. doi: 10.1161/hh1001.090878. [DOI] [PubMed] [Google Scholar]
- 9.Pandey P, Avraham S, Place A, Kumar V, Majumder PK, Cheng K, Nakazawa A, Saxena S, Kharbanda S. Bcl-xL blocks activation of related adhesion focal tyrosine kinase/proline-rich tyrosine kinase 2 and stress-activated protein kinase/c-Jun N-terminal protein kinase in the cellular response to methylmethane sulfonate. J Biol Chem. 1999;274:8618–23. doi: 10.1074/jbc.274.13.8618. [DOI] [PubMed] [Google Scholar]
- 10.Lakin ND, Jackson SP. Regulation of p53 in response to DNA damage. Oncogene. 1999;18:7644–55. doi: 10.1038/sj.onc.1203015. [DOI] [PubMed] [Google Scholar]
- 11.Orlowski RZ. The role of the ubiquitin-proteasome pathway in apoptosis. Cell Death Differ. 1999;6:303–13. doi: 10.1038/sj.cdd.4400505. [DOI] [PubMed] [Google Scholar]
- 12.Jesenberger V, Jentsch S. Deadly encounter: ubiquitin meets apoptosis. Nat Rev Mol Cell Biol. 2002;3:112–21. doi: 10.1038/nrm731. [DOI] [PubMed] [Google Scholar]
- 13.Deshaies RJ. SCF and Cullin/Ring H2-based ubiquitin ligases. Annu Rev Cell Dev Biol. 1999;15:435–67. doi: 10.1146/annurev.cellbio.15.1.435. [DOI] [PubMed] [Google Scholar]
- 14.Zachariae W, Shevchenko A, Andrews PD, Ciosk R, Galova M, Stark MJ, Mann M, Nasmyth K. Mass spectrometric analysis of the anaphase-promoting complex from yeast: identification of a subunit related to cullins. Science. 1998;279:1216–9. doi: 10.1126/science.279.5354.1216. [DOI] [PubMed] [Google Scholar]
- 15.Yu H, Peters JM, King RW, Page AM, Hieter P, Kirschner MW. Identification of a cullin homology region in a subunit of the anaphase-promoting complex. Science. 1998;279:1219–22. doi: 10.1126/science.279.5354.1219. [DOI] [PubMed] [Google Scholar]
- 16.Dias DC, Dolios G, Wang R, Pan ZQ. CUL7: A DOC domain-containing cullin selectively binds Skp1.Fbx29 to form an SCF-like complex. Proc Natl Acad Sci U S A. 2002;99:16601–6. doi: 10.1073/pnas.252646399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Arai T, Kasper JS, Skaar JR, Ali SH, Takahashi C, DeCaprio JA. Targeted disruption of p185/Cul7 gene results in abnormal vascular morphogenesis. Proc Natl Acad Sci U S A. 2003;100:9855–60. doi: 10.1073/pnas.1733908100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nikolaev AY, Li M, Puskas N, Qin J, Gu W. Parc: a cytoplasmic anchor for p53. Cell. 2003;112:29–40. doi: 10.1016/s0092-8674(02)01255-2. [DOI] [PubMed] [Google Scholar]
- 19.Luo Y, Rockow-Magnone SK, Joseph MK, Bradner J, Butler CC, Tahir SK, Han EK, Ng SC, Severin JM, Gubbins EJ, Reilly RM, Rueter A, Simmer RL, Holzman TF, Giranda VL. Abrogation of G2 checkpoint specifically sensitize p53 defective cells to cancer chemotherapeutic agents. Anticancer Res. 2001;21:23–8. [PubMed] [Google Scholar]
- 20.Drexler HC, Pebler S. Inducible p27(Kip1) expression inhibits proliferation of K562 cells and protects against apoptosis induction by proteasome inhibitors. Cell Death Differ. 2003;10:290–301. doi: 10.1038/sj.cdd.4401159. [DOI] [PubMed] [Google Scholar]
- 21.Harlow E, Lane D. Using antibodies : a laboratory manual. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, N.Y: 1999. [Google Scholar]
- 22.Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci U S A. 1979;76:4350–4. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–5. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 24.Zacchi P, Gostissa M, Uchida T, Salvagno C, Avolio F, Volinia S, Ronai Z, Blandino G, Schneider C, Del Sal G. The prolyl isomerase Pin1 reveals a mechanism to control p53 functions after genotoxic insults. Nature. 2002;419:853–7. doi: 10.1038/nature01120. [DOI] [PubMed] [Google Scholar]
- 25.Sambrook J, Fritsch EF, Maniatis T. Molecular cloning : a laboratory manual. 2. Cold Spring Harbor Laboratory; Cold Spring Harbor, N.Y: 1989. [Google Scholar]
- 26.Gietz RD, Schiestl RH. Applications of high efficiency lithium acetate transformation of intact yeast cells using single-stranded nucleic acids as carrier. Yeast. 1991;7:253–63. doi: 10.1002/yea.320070307. [DOI] [PubMed] [Google Scholar]
- 27.Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411:494–8. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- 28.Chandar N, Billig B, McMaster J, Novak J. Inactivation of p53 gene in human and murine osteosarcoma cells. Br J Cancer. 1992;65:208–14. doi: 10.1038/bjc.1992.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moynihan TP, Ardley HC, Nuber U, Rose SA, Jones PF, Markham AF, Scheffner M, Robinson PA. The ubiquitin-conjugating enzymes UbcH7 and UbcH8 interact with RING finger/IBR motif-containing domains of HHARI and H7-AP1. J Biol Chem. 1999;274:30963–8. doi: 10.1074/jbc.274.43.30963. [DOI] [PubMed] [Google Scholar]
- 30.Ardley HC, Tan NG, Rose SA, Markham AF, Robinson PA. Features of the parkin/ariadne-like ubiquitin ligase, HHARI, that regulate its interaction with the ubiquitin-conjugating enzyme, Ubch7. J Biol Chem. 2001;276:19640–7. doi: 10.1074/jbc.M011028200. [DOI] [PubMed] [Google Scholar]
- 31.Skaar JR, Arai T, Decaprio JA. Dimerization of CUL7 and PARC Is Not Required for All CUL7 Functions and Mouse Development. Mol Cell Biol. 2005;25:5579–89. doi: 10.1128/MCB.25.13.5579-5589.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhong Q, Gao W, Du F, Wang X. Mule/ARF-BP1, a BH3-Only E3 Ubiquitin Ligase, Catalyzes the Polyubiquitination of Mcl-1 and Regulates Apoptosis. Cell. 2005;121:1085–95. doi: 10.1016/j.cell.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 33.Lowe SW, Schmitt EM, Smith SW, Osborne BA, Jacks T. p53 is required for radiation-induced apoptosis in mouse thymocytes. Nature. 1993;362:847–9. doi: 10.1038/362847a0. [DOI] [PubMed] [Google Scholar]
- 34.Clarke AR, Purdie CA, Harrison DJ, Morris RG, Bird CC, Hooper ML, Wyllie AH. Thymocyte apoptosis induced by p53-dependent and independent pathways. Nature. 1993;362:849–52. doi: 10.1038/362849a0. [DOI] [PubMed] [Google Scholar]
- 35.Villunger A, Michalak EM, Coultas L, Mullauer F, Bock G, Ausserlechner MJ, Adams JM, Strasser A. p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science. 2003;302:1036–8. doi: 10.1126/science.1090072. [DOI] [PubMed] [Google Scholar]
- 36.Clifford B, Beljin M, Stark GR, Taylor WR. G2 arrest in response to topoisomerase II inhibitors: the role of p53. Cancer Res. 2003;63:4074–81. [PubMed] [Google Scholar]
- 37.Wagenknecht B, Hermisson M, Eitel K, Weller M. Proteasome inhibitors induce p53/p21-independent apoptosis in human glioma cells. Cell Physiol Biochem. 1999;9:117–25. doi: 10.1159/000016308. [DOI] [PubMed] [Google Scholar]
- 38.Nakajima H, Nakajima HO, Tsai SC, Field LJ. Expression of mutant p193 and p53 permits cardiomyocyte cell cycle reentry after myocardial infarction in transgenic mice. Circ Res. 2004;94:1606–14. doi: 10.1161/01.RES.0000132279.99249.f4. [DOI] [PubMed] [Google Scholar]