

SUMMARY
Neural stem cells are retained in the postnatal subventricular zone (SVZ), a specialized neurogenic niche with unique cytoarchitecture and cell-cell contacts. Although the SVZ stem cells continuously regenerate, how they and the niche respond to local changes is unclear. Here we generated nestin-creERtm transgenic mice with inducible Cre recombinase in the SVZ, and removed Numb/Numblike, key regulators of embryonic neurogenesis from postnatal SVZ progenitors and ependymal cells. This resulted in severe damage to brain lateral ventricle integrity, and identified previously unknown roles for Numb/Numblike in regulating ependymal wall integrity and SVZ neuroblast survival. Surprisingly, the ventricular damage was eventually repaired: SVZ reconstitution and ventricular wall remodeling were mediated by progenitors that escaped Numb deletion. Our results show a self-repair mechanism in the mammalian brain, and may have implications for niche plasticity in other areas of stem cell biology, and for the therapeutic use of neural stem cells in neurodegenerative diseases.
INTRODUCTION
Neuroprogenitors in the embryonic, neonatal, and adult brain give rise to differentiated cell types including neurons, astrocytes, ependymal cells, and oligodendrocytes. Despite similarities in function, embryonic and postnatal neuroprogenitors reside in different specialized environments, or niches. During embryonic neurogenesis, neuroprogenitors (also known as radial glia) form a neuroepithelial layer adjacent to the central nervous system (CNS) ventricles (reviewed in Rakic, 2003; Kriegstein and Noctor, 2004; Gotz and Barde, 2005). Shortly after birth, this embryonic niche begins a transformation into the postnatal neural stem cell niche in the subventricular zone (SVZ) of the lateral ventricles (reviewed in Temple, 2001; Tramontin et al., 2003). Postnatally, a subpopulation of radial glia gives rise to ependymal cells that form the epithelial lining of the ventricles (Spassky et al., 2005). Other radial glial cells transform into SVZ Type B cells that have the structural properties of astrocytes (Merkle et al., 2004). These type B cells function as the primary progenitors of new neurons in the adult mouse forebrain, and generate transiently-amplifying Type C cells which in turn differentiate into neuroblasts that migrate to the olfactory bulb (reviewed in Alvarez-Buylla and Lim, 2004).
Some of the molecular programs that govern embryonic neuroprogenitor differentiation (reviewed in Bertrand et al., 2002; Campbell, 2005; Sur and Rubenstein, 2005; Tsai and Gleeson, 2005; Fuccillo et al., 2006) have also been shown to function in the postnatal SVZ niche. For example, TGFα, which activates the EGF receptor, regulates SVZ cellular proliferation and migration of neurons to the olfactory bulb (Tropepe et al., 1997). Likewise, bone morphogenetic proteins and their antagonist Noggin affect SVZ neuroprogenitor differentiation postnatally (Lim et al., 2000). The sonic hedgehog pathway is involved in maintaining the proliferative capacity of SVZ stem cells (Machold et al., 2003; Ahn and Joyner, 2005). Activated-Notch suppresses neuronal differentiation and prevent neuroblast migration to the olfactory bulb (Chambers et al., 2001), and EphB receptors can increase the proliferative capacity of SVZ progenitors in vivo (Conover et al., 2000). Our understanding of embryonic neurogenesis derives largely from in vivo analyses of mutants with corroborative findings from in vitro studies. However, experiments in the postnatal SVZ thus far have relied on in vitro differentiation, in vivo injection of constructs/growth factors, and postnatal analyses of gene deleted during embryogenesis. Though informative, without ways to genetically target the postnatal SVZ, molecular programs that control this stem cell niche cannot be studied effectively. For example, it is known that SVZ progenitors can regenerate after short periods of drug-induced depletion (Doetsch et al., 1999), but how they and the niche respond to environmental changes such as tissue damage are unclear.
Numb and Numblike (Numbl) are functionally related proteins that critically regulate progenitor differentiation and neuroepithelial integrity during embryonic neurogenesis (Petersen et al., 2002; Shen et al., 2002; Li et al., 2003; Petersen et al., 2004). They were first identified as the mammalian homologues of Drosophila numb, which functions during neural precursor asymmetric cell division to antagonize Notch function in one of the daughter cells (reviewed in Roegiers and Jan, 2004). We hypothesized that these proteins might play similarly important roles during postnatal neurogenesis, and that their deletion postnatally might reveal not only the functions of Numb/Numbl during SVZ neurogenesis, but also how this stem cell niche responds to environmental changes. We generated a tamoxifen-inducible Cre transgene that is active in the postnatal SVZ, and used this tool to reveal that Numb/Numbl maintain SVZ homeostasis by regulating ependymal integrity and survival of SVZ neuroblasts. In the process, we also discovered that the postnatal SVZ stem cell niche has remarkable plasticity, and can mediate local repair of brain ventricular wall damage.
RESULTS
Generation of nestin-creERtm mice with inducible Cre activity in postnatal SVZ neurogenic niche
To delete genes postnatally, we used a tamoxifen-inducible form of the Cre recombinase, CreERtm, that has activity in most mouse tissues including the brain (Hayashi and McMahon, 2002). To express CreERtm in neuroprogenitors, we replaced the Cre coding sequence in the Nestin-Cre DNA construct (Tronche et al., 1999), with that of CreERtm to generate nestin-creERtm mice (Figure 1A). We assayed for inducible Cre activity by crossing founders to rosa26 reporter (r26r) mice (Soriano, 1999). Of 10 lines, 3 showed inducible Cre activity in the embryonic brain and spinal cord (Figure 1B and data not shown), and of those one (line 4) showed postnatal inducible Cre activity (Figure 1C). This inducible Cre activity was tightly coupled to tamoxifen-induction (tamoxifenind), as the rate of spontaneous recombination in nestin-creERtm4, our best line, was extremely low (Figure 1B, C).
Figure 1. Inducible Cre activity of nestin-creERtm mice.
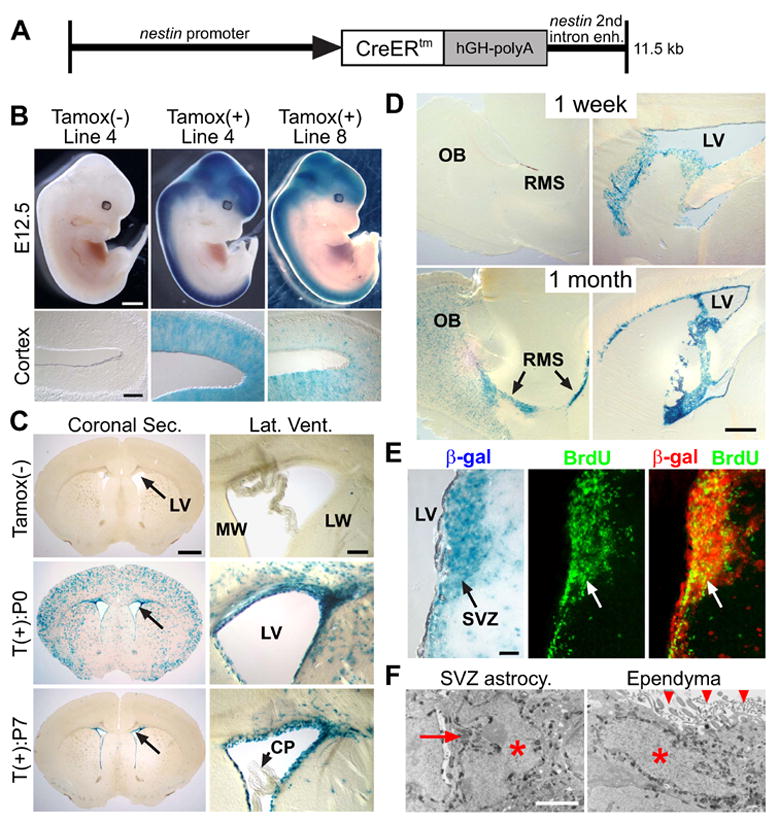
(A) Diagram of the Nestin-CreERtm construct. (B) β-gal staining of nestin-creERtm; r26r embryos: E12.5 embryos with or without E10.5 tamoxifen induction (tamoxifenind). Line 4 and 8 are 2 independent lines (Cortex, 30 μm cryostat section). Line 4 without tamoxifen showed no detectable recombination. (C, D) β-gal staining of nestin-creERtm4; r26r brain sections. (C) 5 week (wk) old nestin-creERtm4; r26r mice with or without single tamoxifenind at P0 or P7 (50 μm vibratone section). Arrows in coronal sections point to lateral ventricles (LV). Enlarged views of LV (Lat. Vent.) showing both medial (MW) and lateral wall (LW). Note the lack of β-gal+ cells in choroid plexus (CP). (D) Single dose P14-tamoxifenind in nestin-creERtm4; r26r mice assayed at 1 wk or 1 month post-injection. Note the increase in β-gal+ cells in olfactory bulb (OB) and rostral migratory stream (RMS, arrows) in 1 month post-injection sample (50 μm vibratone sagital section). (E) Co-labeling of β-gal+ and BrdU+ SVZ cells. nestin-creERtm4; r26r mice were tamoxifen induced at P14. 2 wks later, proliferating SVZ cells were labeled with 3-day BrdU pulse (arrows). 50 μm floating vibratone sections were stained for β-gal activity followed by BrdU immunohistochemistry. β-gal staining was inverted to dark-field red channel to create the merged image. (F) Electron micrographs of β-gal+ SVZ neuroprogenitor and ependymal cell from nestin-creERtm4; r26r mice injected with tamoxifen at P14 and analyzed 1 wk later. Note the electron dense β-gal enzyme granules surrounding the nuclei (red asterisks), as well as the deeply invaginated nucleus of SVZ astrocytic progenitor (red arrow) and the apical cilia of ependymal cell (red arrowheads). Scale bars: (B) embryo = 1 mm, C. Cortex = 100 μm; (C) Coronal Sec. = 1 mm, Lat. Vent./O.B. = 100 μm; (D) 500 μm; (E) 50 μm; (F) 2 μm.
To visualize postnatal Cre activity in nestin-creERtm4, we injected single dose of tamoxifen into nestin-creERtm4; r26r+/− pups at either postnatal day 0 (P0), P7, or P14, and waited until 5 weeks of age for analysis. At P0, as embryonic neuroprogenitors undergo terminal differentiation in the cerebral cortex, transient Cre activity in those progenitors would give rise to differentiated progeny that are β-gal+, which is what we observed in the P0-induced mice, along with β-gal+ cells surrounding the lateral ventricle (LV) walls (Figure 1C). When we injected tamoxifen at P7, the cortical recombination was largely abolished, which is consistent with cortical neurogenesis terminating shortly after birth in mice, but the inducible Cre activity along the LV remained robust and we did not see any recombinase activity in the choroid plexus (Figure 1C). We obtained identical results for the P14-tamoxifenind (data not shown).
SVZ neuroprogenitors continuously produce proliferating neuroblasts that migrate to the olfactory bulb (OB) via the rostral migratory stream (RMS). To test if the nestin-creERtm4; r26r+/− SVZ β-gal+ cells retained such property we injected tamoxifen at P14, and analyzed brains at either 1 week or 1 month post injection. In sagittal sections 1 week after tamoxifenind, we observed many β-gal+ cells around the LV, but only few cells within the RMS and OB (Figure 1D). However, one month post injection, in addition to the LV, many β-gal+ cells could be seen in the RMS and OB, as one would expect for postnatal SVZ neurogenesis (Figure 1D). To see if β-gal+ cells within the SVZ corresponded to proliferating progenitors and neuroblasts, we injected tamoxifen at P14, and 2 weeks later BrdU-pulsed the mice for 3 days prior to sacrifice. On these brain sections we saw BrdU co-localizing with β-gal+ cells within the SVZ neurogenic niche (Figure 1E).
To further confirm that we could induce Cre recombination in postnatal SVZ neuroprogenitors, we used electron microscopy (EM) to examine β-gal+ cells around the LV of nestin-creERtm4; r26r+/− mice induced by tamoxifen at P14. In addition to β-gal+ SVZ astrocytic neuroprogenitors, we also saw labeled transiently-amplifying C cells, neuroblasts, and ependymal cells surrounding the LV (Figure 1F and data not shown). Since β-gal+ C cells and neuroblasts were derived from r26r-recombined neuroprogenitors, Cre antibody staining showed that these cells did not retain significant CreERtm protein after differentiating from the SVZ progenitors (Figure S1). These results are consistent with postnatal expression patterns of the nestin-EGFP mice, which labels both postnatal SVZ progenitors and ependymal cells (Kawaguchi et al., 2001), and confirmed that nestin-creERtm4 transgene can inducibly recombine loxP sites within these two major cell types of the SVZ niche. These animals are healthy with no obvious phenotypes, but the females have low fertility so in all of our experiments the nestin-creERtm4 transgene came from the male mice.
Numb is expressed by postnatal SVZ neuroprogenitors and ependymal cells
Numb is highly expressed by embryonic neuroprogenitors throughout cortical development (Zhong et al., 1996). To assay whether Numb is expressed in the postnatal SVZ niche, we co-stained P0 RC2+ radial glial neuroprogenitors and P14 GFAP+ SVZ astrocytic neuroprogenitors with anti-Numb antibody, which showed that Numb continued to be expressed in postnatal SVZ progenitors (Figure S2A), as well as in transiently-amplifying C cells and neuroblasts (Figure S2B). Interestingly, we also observed increased Numb immunoreactivity in postnatal ependymal cells as they developed from immature GLAST+S100β− to mature GLAST−S100β+ ependyma (Figure S2C). To be certain that immunoreactivity in these post-mitotic epithelial cells represents Numb protein, we repeated Numb antibody staining in nestin-creERtm4; floxed Numb(F/F) mice (wild-type Numbl). Since our inducible Cre transgene had postnatal recombinase activity in ependymal cells starting at P0 (Figure 1C, F, and data not shown), deletion of floxed Numb should extinguish the antibody staining in these samples. Without tamoxifenind, nestin-creERtm4; NumbF/F mice showed identical Numb staining as controls (data not shown). However, following P0-tamoxifenind, we could not detect significant Numb antibody staining in P14 mature ependymal cells (Figure S2D), thereby verifying that Numb expression is indeed induced during postnatal ependymal maturation. This result also showed that the nestin-creERtm4 transgene can efficiently delete the floxed-Numb gene postnatally after tamoxifenind.
Postnatal deletion of Numb/Numbl results in lost of brain ventricular wall integrity
To understand if Numb/Numbl played a role in the postnatal SVZ, we used the nestin-creERtm4 transgene to delete Numb postnatally in phenotypically normal Numbl-null mutant mice. Due to lower fertility of Numbl−/ − mice, we crossed nestin-creERtm4; NumbF/F; Numbl+/− males to NumbF/F; Numbl+/− females to generate the nestin-creERtm4; NumbF/F; Numbl−/ − animals which we termed cDKO mice. Tamoxifenind of cDKO mice postnatally would remove Numb/Numbl without disturbing embryonic neurogenesis. These cDKO mice were born at expected mendelian ratios (47/385, 1 per 8.2), and appeared phenotypically normal in comparison to control littermates (data not shown). Analysis of P0 cDKO newborn brains showed no obvious developmental or structural defects when compared to control littermates (Figure S3A). To postnatally delete floxed-Numb in the cDKO mice, we injected the entire litter each with a single dose of tamoxifen at P0. Since we showed earlier that ependymal Numb expression was subsided by P14 in P0-tamoxifenind mice, we analyzed these mice at 1 and 2 weeks after P0 injection. At P7, we saw consistent LV enlargement in the induced cDKO mice when compared to uninduced cDKO and induced littermate controls (Figure 2A, asterisk, and Figure S3B). By P14, the induced cDKO pups showed growth retardation, and the LV enlargement had become quite severe with many structural abnormalities along the LV wall linings (Figure 2A, asterisk). We did observe slight LV enlargement in the uninduced cDKO mice (Figure S3B), but we never observed any LV wall abnormalities in these uninduced cDKO mice (Figure S3B).
Figure 2. Ventricular wall defects in postnatal tamoxifen-induced Numb/Numbl mutant mice.
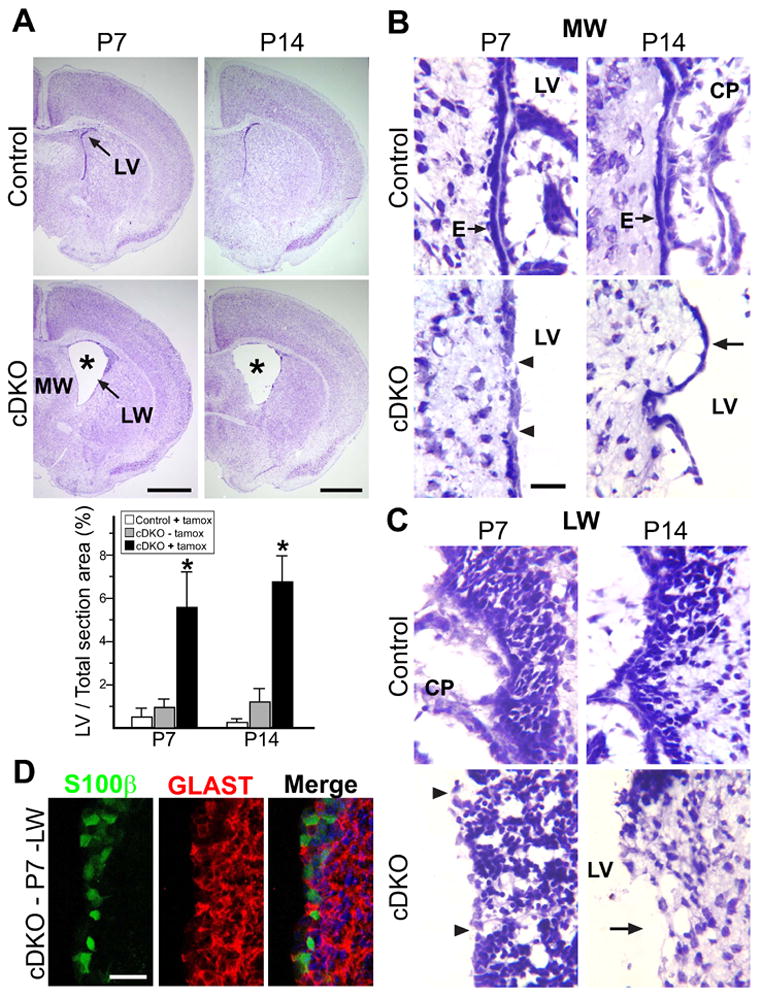
(A) Nissl staining of nestin-creERtm4, NumbF/F, Numbl+/- (Control) and nestin-creERtm4; NumbF/F, Numbl−/− (cDKO) brain sections with P0-tamoxifenind. Sections of P7 and P14 brains showing lateral ventricle (LV) at or just rostral to the anterior commissure. Note the increased LV size in mutant mice (asterisks). MW: medial wall; LW: lateral wall. Quantitative analysis of LV size, represented as % of total LV area in section just rostral to AC, over total brain section area (to control for variations in brain section sizes during processing). *: P < 0.01, Wilcoxon Two Sample Test; n = 5 in each group; mean ± standard deviation (SD). (B) Close-up views of Nissl-stained LV MW ependymal cells from P7 and P14 control and cDKO sections after P0 induction. Arrows point to ependymal cells (E), showing gaps (arrowheads) in P7 and detachment (arrow) in P14 cDKO samples. CP: choroid plexus. (C) Close-up views of LV LW neurogenic niche. Arrowheads point to gaps at P7, and arrow points to denuded area at P14 in cDKO samples. (D) Antibody staining of P7 mutant LW neurogenic niche. S100β+ ependymal cells were made postnatally in mutant mice. Nuclei labeled with DAPI (blue). Scale bars: (A) P7, P14 = 1 mm; (B, C, D) 20 μm.
Having found Numb expression in ependymal cells during the postnatal period (Figure S2C), we closely examined these areas in the induced cDKO mice. At P7, the ependymal layer lining the medial walls of the LVs was formed in the induced cDKO brain (Figure 2B). However, unlike controls which had a continuous ependymal lining, the induced P7 cDKO ependyma showed occasional gaps (Figure 2B, arrowheads), which progressed to ependymal detachment by P14 (Figure 2B, arrow). Unlike the medial wall, where ependymal cells are specified before birth, the lateral walls of the LVs gradually transitions from radial glial-formed neuroepithelium to the mature ependymal-lined epithelium shortly after birth (Spassky et al., 2005). In the induced cDKO P7 brains, we saw occasional defects in lateral wall integrity when compared to control brains (Figure 2C, arrowheads). But by P14, the defects became severe, as many areas lost both the ependyma and the underlying SVZ niche (Figure 2C, arrow). Staining of induced cDKO P7 LV walls revealed abundant S100β+ ependymal cells comparable to control brains, which showed that the structural defects were not caused by a lack of ependymal cells (Figure 2D and data not shown). We examined serial sections to analyze the ventricular system in the induced cDKO brains, but did not find obvious restrictions or obstructions (data not shown). These data suggest that compromises in ventricular wall integrity lead to the LV enlargement seen in the tamoxifen induced cDKO mice. Since the nestin-creERtm4 transgene had negligible background Cre activity (Figure 1), and we did not observe any significant differences between the uninduced cDKO mice and induced control littermates (Figure 2, Figure S3 and data not shown), for consistency we continued with our analysis comparing mutant mice to their littermates (nestin-creERtm4; NumbF/F; Numbl+/−) injected with tamoxifen at the same time.
Numb/Numbl are required for postnatal ependymal adhesion and SVZ neuroblast survival
We used EM to further characterize the ventricular wall defects in the induced cDKO mice. Semi-thin brain sections showed that P7 mutant ependymal cells extended cilia from their apical membrane like control cells (Figure 3A, arrowheads), thus retaining basic epithelial apical-basal polarity. However, instead of a flat epithelial sheath, the P7 mutant mice showed poorly-formed ependyma with pseudo-columnar cells (Figure 3A, arrows). By P14, the normal ependyma developed complex, interdigitating junctions between neighboring cells (Figure 3A, yellow arrowheads), but in the induced cDKO mice, while some ependymal cells detached, the remaining cells formed short, defective borders that showed separation between cells (Figure 3A, yellow arrows/asterisk). The mutant ependymal cells were TUNEL negative and did not show apoptotic nuclei (Figure 3A and data not shown), suggesting that these defects were not due to cell death.
Figure 3. Ependymal adhesion and SVZ neuroblast survival defects in Numb/Numbl mutant mice.

(A) EM images of control and cDKO mutant ependymal cells (P0-tamoxifenind). Toluene blue staining of P7 semi-thin sections showing the ependymal layer (E, arrows) and apical cilia (arrowheads). EM images of P14 ependymal cell borders showing interdigitation in control cells (yellow arrowheads), but a lack of interdigitation (yellow arrows) and cell separation (asterisk) between mutant ependymal cells. (B) Antibody staining of P7 Control and cDKO mutant lateral ventricle (LV) medial wall (MW) ependymal cells (P0-tamoxifenind). Note the lack of apical Phalloidin staining in mutant cells (arrowheads), low level of border E-cadherin staining in mutant cells except for an occasional Numb-expressing cell (arrows), and co-localization of E-cadherin and Numb staining in control cells (arrowheads). (C) Antibody staining of control P0 and P14 MW ependymal cells, showing upregulation of cell-border E-cadherin during maturation. (D) Co-immunoprecipitation (IP) of E-cadherin and FLAG-Numb. Protein extract from postnatal ependymal cells electroporated with FLAG-Numb expression construct was subjected to IP with beads and antibodies as indicated. Grin1 was used as positive control for Numb binding and negative control for non-specific E-cadherin interaction. The upper and lower panels were immunoblotted (IB) with anti-FLAG and anti-E-cadherin (E-cad.) antibodies, respectively. Note that E-cadherin was part of the FLAG-Numb IP complex, and vise versa. (E) Nissl, Doublecortin (DCX), and TUNEL staining of P7 Control and cDKO brain sections (P0-tamoxifenind). Note the greatly increased striato-cortical junction (SCJ) cellularity in the cDKO sample. DCX and TUNEL co-staining on P7 Control and cDKO brain sections shows greatly increased numbers of apoptotic neuroblasts in the mutant SCJ. (F) Nissl, DCX, and TUNEL staining of P14 CALSL-NICD; nestin-creERtm4 mice either with (+) or without (-) P0-tamoxifenind. Note the greatly increased SCJ cellularity, as well as apoptotic DCX+ neuroblasts after tamoxifenind. Nuclei labeled with DAPI (blue). Analysis of SCJ area and TUNEL+ cells: SCJ from single coronal section just rostral to anterior commissure; n = 5 mice in each group. For all quantitative analyses: *: P < 0.01, Wilcoxon Two Sample Test; mean ± SD. Scale bars: (A) Semi-thin = 10 μm; P14 = 500 nm; (B, C) 20 μm; (E, F) SCJ Nissl = 100 μm, brain sections = 1 mm, DCX/TUNEL = 50 μm.
Since EM revealed substantial ependymal border abnormalities by P14, we stained P7 brain sections with antibodies against cell border and adhesion junction molecules. While there were no apparent differences in tight junction marker ZO-1 and desmosomal protein Desmoglein staining between control and mutant ependymal cells (data not shown), anti-Phalloidin antibody against F-actin showed that the strong apical border staining in P7 control ependyma was completely abolished in Numb/Numbl mutant ependyma (Figure 3B, arrowheads). Given that polymerized actin is linked to epithelial adhesion and adherens junction formation (Vaezi et al., 2002), we next examined the expression pattern of the adherens junction molecule E-cadherin. In control P7 mice, E-cadherin staining showed ependymal cell border localization, as well as co-localization with Numb in many places (Figure 3B, arrowheads). However, in P7 mutant mice we saw low and diffuse E-cadherin staining in ependymal cells (Figure 3B). When we did see E-cadherin levels equivalent to controls at ependymal cell borders in the mutant mice, these cells retained Numb protein and likely escaped tamoxifenind (Figure 3B, arrows). The presence of E-cadherin in escaper ependymal cells served as an internal control for E-cadherin levels in mutant cells.
These results, together with the fact that E-cadherin was up-regulated during postnatal ependymal maturation similarly to Numb (Figure 3C), suggested to us that Numb may have a cell-autonomous function in regulating E-cadherin localization to ependymal cell borders. To test whether Numb physically interacts with E-cadherin in ependymal cells, we injected and electroporated a FLAG-tagged Numb-GFP expression construct, pFLAG-Numb65 (Roncarati et al., 2002), into the LV of P4 mice, and micro-dissected GFP+ ependymal cells at P7 for co-immunoprecipitation (IP) analysis. We used Grin1 as a positive IP control as it interacts with Numb (Nishimura et al., 2006) and is expressed in epithelial cells including skin (Morhenn et al., 2004), neuroepithelial cells (Sharp et al., 2003), and ependymal cells (data not shown). IPs with the Grin antibody, as well as the E-cadherin antibody were able to pull down ependymal interacting proteins that contained FLAG-tagged Numb when immuno-blotted (IB) with the FLAG antibody (Figure 3D). In the reverse experiment, using Grin1 antibody as a negative control, FLAG IP antibody pulled down Numb interacting proteins that included E-cadherin (Figure 3D). These results show that Numb physically interacts with E-cadherin, and plays an unexpected post-mitotic role in regulating ependymal integrity.
In addition to ependymal abnormalities, we observed significant postnatal SVZ neurogenesis defects in the induced cDKO mice. At P7 and prior to ependymal detachment, the mutant mice showed significantly increased cellularity at the striato-cortical junction (SCJ) of the LV, which marks the beginning of the rostral neuroblast migratory path (Figure 3E). These SCJ cells expressed the immature neuroblast marker Doublecortin (DCX), and many of them were apoptotic as shown by both EM and TUNEL assays (Figure 3E and data not shown). This accumulation of SCJ neuroblasts was not caused by over-proliferation, as transient BrdU-pulse experiments did not reveal increased cell proliferation in either the SVZ or the SCJ of P7 mutant mice (Figure S4A). Since ependymal cells have been implicated in directing SVZ neuroblast migration towards the OB (Sawamoto et al., 2006), it was not surprising that ependymal defects together with SCJ neuroblast apoptosis led to P14 mutant mice having smaller OBs containing fewer interneurons than control mice with comparable brain size (Figure S4B, arrows and data not shown). In particular, the RMS entering the mutant OBs contained far fewer neuroblasts than control samples (Figure S4B, dashed-lines).
The SVZ neurogenesis defects could be secondary to ependymal abnormalities, so to determine if Numb/Numbl played cell-intrinsic roles in regulating SVZ neurogenesis, we used an adeno-Cre virus to generate subpopulations of Numb/Numbl mutant radial glia (Merkle et al., 2004). This technique targets SVZ progenitor cells that later give rise to migrating neuroblasts while sparing LV ependymal cells. In P14 Numb(F/F); Numbl(+/−); r26r control mice injected with adeno-Cre at P0, we saw β-gal+ cells in the SVZ as well as in the OBs, which increased in numbers by 5 weeks of age, indicating that recombined SVZ progenitors were giving rise to neuroblasts that migrated into the OB (Figure S4C). Although we also detected β-gal+ cells in the SVZ of P14 Numb(F/F); Numbl(−/ −); r26r mutant mice, we saw few, if any β-gal+ cells in the corresponding OBs, both at P14 and at 5 weeks of age (Figure S4C). The ability of Numb/Numbl mutant SVZ progenitors to proliferate and differentiate was confirmed by harvesting postnatal SVZ progenitors from uninduced cDKO mice and then inducing Numb deletion in culture via tamoxifenind (data not shown). These results show that Numb/Numbl are required for multiple aspects of postnatal SVZ homeostasis, including ependymal wall integrity and neuroblast survival.
Since Numb antagonizes Notch during fly neural precursor division (reviewed in Roegiers and Jan, 2004), and aberrant Notch activity can inhibit SVZ neuroblast maturation and survival (Chambers et al., 2001), we wondered if increased Notch activation in mutant neuroblasts could have caused the survival defects. Because spatial and temporal regulation of Notch activity is difficult to detect in vivo, we took a genetic approach to over-express activated intracellular Notch (NICD) (Yang et al., 2004) in the postnatal SVZ using the nestin-creERtm4 transgene. This resulted in greatly increased DCX+ neuroblast cellularity and apoptosis at the SCJ (Figure 3F), and phenocopied the neuroblast defects in Numb/Numbl mutant mice, which is consistent with reported functions for Numb to inhibit Notch activity. Interestingly, we did not observe any LV enlargement, ependymal adhesion, or E-cadherin expression defects in the induced NICD mice (Figure 3F and data not shown), supporting the idea that Numb/Numbl’s regulation of ependymal integrity is through a Notch-independent mechanism.
Ventricular wall remodeling in adult cDKO mutant mice
With the massive LV enlargement seen at P14 (Figure 2A), we expected these animals to physically deteriorate over time. However, none of the induced cDKO mutant mice died prematurely either during the postnatal period or as they became adults. To investigate the progression of LV wall defects, we examined 6-week old cDKO mutant brains after P0-tamoxifenind. Much to our surprise, the LV enlargement we observed at P14 was significantly reduced by 6 weeks of age (Figure 4A). The brain ventricular walls were again lined by cell layers, but most of these cells had a morphology different from that found in control littermates (Figure 4A, arrows). Whereas the control ependyma was a single-cell layer epithelium, in the cDKO mutant mice we saw ventricular lining composed of multi-layered stratified epithelium (Figure 4A, arrows).
Figure 4. Ventricular wall repair/remodeling in adult Numb/Numbl mutant mice.
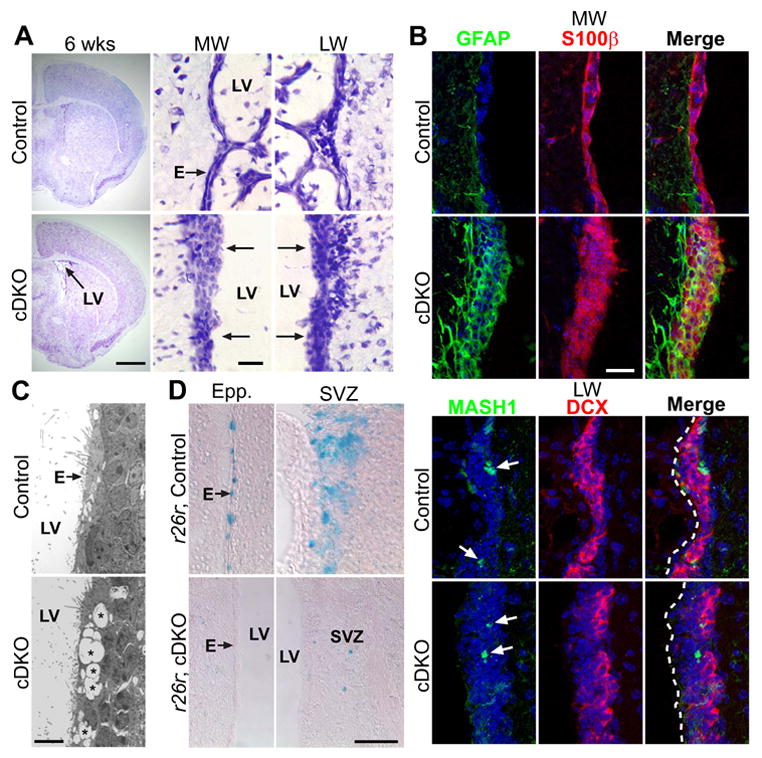
(A) Nissl staining of 6 wk old adult Control and cDKO brain sections (P0-tamoxifenind). Note the decreased cDKO lateral ventricle (LV) size when compared to cDKO in 2A. Also note the abnormal stratified medial (MW) and lateral (LW) ventricular walls. (B) IHC staining of 6 wk old adult Control and cDKO LV MW and LW. Control MW ependymal cells are GFAP- and S100β+, but some remodeled mutant MW highly expressed both GFAP and S100β. Both Control and cDKO LW SVZ showed MASH1+ transient amplifying C cells (arrows) and Doublecortin (DCX+) neuroblasts, but the mutant neurogenic niche is separated from the ventricular surface (dashed lines) via a much thicker wall. Nuclei labeled with DAPI (blue). (C) EM images of remodeled 6 wk LW. Instead of normal ependyma (E), the cDKO mutant mice showed abnormal free space (*) in the remodeled wall adjacent to the LV. (D) β-gal staining of 6 wk old rosa26r−/ −, Control and rosa26r+/−, cDKO mice injected with tamoxifen at P0. Control mice showed significant numbers of β-gal+ cells in the ependymal layer (E) and the SVZ, but the cDKO mice had very few β-gal+ cells along the remodeled walls. Scale bars: (A) brain section = 1 mm, close-up = 20 μm; (B) 20 μm; (C) 5 μm; (D) 50 μm.
To further characterize this remodeled ventricular wall, we stained the 6 week LV wall with markers for both ependymal cells (S100β) and astrocytes (GFAP). In control mice, as expected the medial wall was covered by ependymal cells expressing S100β but not GFAP (Figure 4B). However, in 6 week old P0-induced mutant mice, the stratified wall sections stained positive for both S100β and GFAP, suggesting that a mixture of astrocytes and ependymal cells now form this epithelial barrier (Figure 4B). Some of these cells highly expressed both S100β and GFAP, and may represent transformed astrocytic cells during the repair process. Although the remodeled mutant lateral wall showed similar histological appearance and immunoreactivity to the medial wall, its subventricular composition was different. In transient cell proliferation assays we found BrdU+ GFAP-expressing cells in the 6 week mutant SVZ (data not shown). Closer examination of this proliferative area demonstrated the presence of MASH1+ transient amplifying C cells as well as DCX+ neuroblasts, similar to control mice, showing that postnatal neurogenesis had been reestablished adjacent to the repaired ventricular wall (Figure 4B). Consistent with this, we also saw a recovery in DCX+ neuroblasts migrating via the RMS into the OBs of 6 week old mutant mice (Figure S5A). Structurally, the remodeled ventricular wall showed very few ependymal cells, intermixed with scar tissue that exhibited abnormal hollow space (Figure 4C, asterisks), as well as robust E-cadherin expression unlike the mutant wall during the postnatal period (Figure S5B). These observations, together with the fact that we did not see such ventricular wall remodeling in control mice (Figure 4 and data not shown), suggested that remodeling of the LV wall was triggered by earlier structural defects in the mutant mice.
How did this remodeling come about? We considered the possibility that Numb/Numbl mutant cells changed their developmental fate and contributed to the generation of this new ventricular lining, and tested this by crossing rosa26-reporter onto the cDKO background. We reasoned that if Numb/Numbl mutant cells could repair the damaged ventricular wall, then these structures should be β-gal+ from Cre-mediated recombination. For the mutant nestin-creERtm4; NumbF/F; Numbl−/ −; r26r+/− mice and control littermates, again we induced with tamoxifen at P0 and studied the ventricular wall at 6 weeks of age. However, unlike control littermates which showed populations of β-gal+ cells in both the SVZ and in the ependyma, in the induced mutant animals we could not detect significant contributions of β-gal+ cells in the remodeled ventricular wall (Figure 4D). This indicates that Numb/Numbl mutant cells do not contribute significantly to the repair of damaged ventricular walls.
Numb-expressing SVZ progenitors participate in local repair and remodeling
It is also possible that existing cells in the CNS can line the damaged walls, or that Numb+ SVZ progenitor cells escaping initial P0-tamoxifenind can participate in the repair. To see if there are indeed neuroprogenitors that escaped P0 tamoxifen-mediated Numb deletion, we looked for Numb expression in the damaged SVZ of P14 mutant mice, which revealed that the remaining cell clusters along the damaged lateral wall were Numb+ (Figure 5A, arrowheads), and expressed high level of GLAST on the ventricular surface, which meant they were not mature ependymal cells (Figure 5A). GFAP and one hour transient BrdU double labeling showed that these Numb+ clusters not only contained GFAP+ progenitors with proliferative capacity, but in the absence of ependymal cells, these cells divided directly on the ventricular wall (Figure 5A, arrows). Ultra-structural analysis of the ventricular wall showed that unlike controls, which were covered by ciliated mature ependymal cells (Figure 5B, arrowheads), the P14 mutant samples contained numerous cells with a globular morphology similar to Type C transient amplifying SVZ cells sitting directly adjacent to the cerebrospinal fluid (Figure 5B, arrows).
Figure 5. Numb-expressing SVZ progenitor cells can mediate lateral ventricle wall remodeling in Numb/Numbl mutant mice.
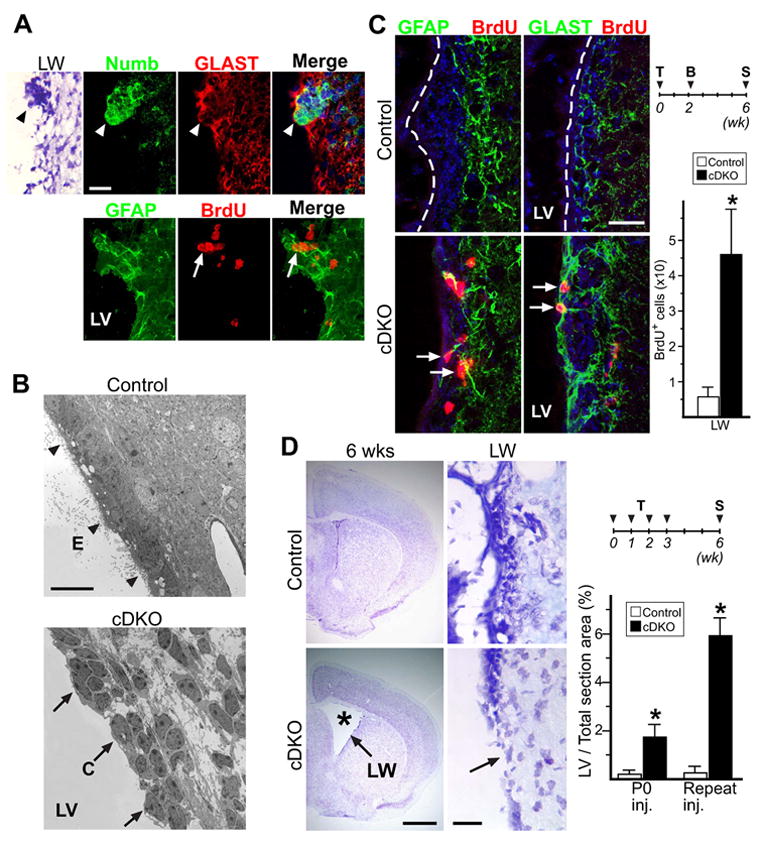
(A) Nissl and antibody staining of P14 cDKO lateral ventricle (LV) lateral wall (LW). Nissl staining showing cellular aggregate on damaged LW (arrowhead). These aggregates stain positive for Numb and GLAST, representing cells that escaped CreER-mediated recombination (arrowheads). They also express GFAP, and are highly proliferative as shown by transient 1 hr BrdU pulse (arrows). (B) EM images of P14 Control and cDKO LW lining. Unlike Control LW, lined with ciliated ependyma (E, arrowheads), mutant LW showed clumps of transient amplifying C cells bordering LV lumen (C, arrows). (C) IHC staining of Control and cDKO mice injected with tamoxifen (T) at P0, pulsed with BrdU (B) at P16, then sacrificed (S) at 6 wks of age. In Control LW, the ependymal wall (dash line) is largely GFAP- and GLAST-, and the SVZ showed very sparse BrdU incorporation throughout. In the remodeled cDKO LW, there are many BrdU+ cells that are also GFAP+ or GLAST+ (arrows). Quantitative analysis of LW BrdU+ cells: from single coronal section at or just rostral to anterior commissure. (D) Nissl stain of 6 wk brain sections from Control and cDKO mice injected repeatedly with tamoxifen. Note the ventricular enlargement (asterisk) in cDKO mice, due to poor ventricular wall structural integrity (arrow). Quantitative analysis of 6 wk old LV size, represented as % of total LV area in coronal section just rostral to anterior commissure, over total brain section area. For all quantitative analysis: *: P < 0.01, Wilcoxon Two Sample Test; n = 5 mice in each group; mean ± SD. Scale bars: (A, C) 20 μm; (B) 10 μm; (D) brain section = 1 mm, closeup = 20 μm.
Since the Numb-expressing clusters along the damaged P14 mutant ventricular wall were highly proliferative, we wondered if these cells contributed to the remodeling of the ventricular wall and SVZ niche. To answer this question, we used BrdU birth-dating to track the cells participating in the repair process. We reasoned that as the SVZ progenitors divide and proliferate during the postnatal period, transient BrdU exposure will be diluted out weeks later after proliferating BrdU-labeled progenitors produced their migrating progeny. However, if the Numb+ SVZ progenitors in the P14 mutant mice were able to give rise to terminally differentiated cells that repaired the damaged ventricular wall, we should be able to detect their local BrdU incorporation. For both control and cDKO mice we again tamoxifenind at P0, gave the animals 3 days of twice per day BrdU injections at P16, and analyzed their brains at 6 weeks of age (Figure 5C). Consistent with previous observations, we did not detect any BrdU incorporation in the control ependymal wall (Spassky et al., 2005), and very few BrdU retaining cells within the control SVZ (Figure 5C). In contrast, we observed large numbers of BrdU+ cells along the ventricular wall in the induced cDKO mice (Figure 5C, arrows). These BrdU+ cells appeared at all levels within the remodeled wall, and many showed co-labeling with GFAP and GLAST, 2 markers not normally expressed by mature ependymal cells (Figure 5C, arrows). These results showed that SVZ cells born during the repair process participated in generating the remodeled ventricular wall.
Results from these experiments are consistent with the notion that Numb-expressing SVZ progenitors escaping initial tamoxifenind can participate in the subsequent structural repair. To further test this, we reasoned that repeated tamoxifen injections during the postnatal period to delete the floxed-Numb gene in more SVZ progenitors should delay or abolish this repair. In 6-week old cDKO mice injected with tamoxifen serially at P0, P7, P14, and P21, we saw dramatic ventricular enlargement in contrast to normal morphology of littermate controls injected with the same dosing schedule, as well as reduction in the numbers of neuroblasts reaching the OB (Figure 5D, asterisk and data not shown). Closer examination of the LV walls showed that many areas in the repeatedly-injected cDKO mice still lacked epithelial covering (Figure 5D, arrow). These data showed a self-repair mechanism along the LV of mammalian brain involving Numb-expressing SVZ progenitors.
DISCUSSION
Using a tamoxifen-inducible Cre transgenic line we showed that Numb/Numbl are required to maintain postnatal SVZ neurogenesis, and revealed a post-mitotic function for Numb/Numbl in regulating ependymal wall integrity. The severely damaged brain ventricular wall resulting from Numb/Numbl deletion allowed us to examine how the SVZ stem cell niche responds to environmental change. These results showed surprisingly that postnatal ventricular wall damage can be self-repaired, and that the SVZ stem cell niche has considerable plasticity for local repair and remodeling, results that will likely have implications in other areas of stem cell biology.
Numb/Numbl functions in SVZ niche homeostasis
Postnatally, Numb-expression is induced during ependymal maturation and is required for ependymal wall integrity. Based on our current knowledge of Numb’s function during asymmetric cell division, these ependymal results are unexpected. Our data showing that a single Numb-expressing ependymal cell, in an otherwise mutant environment, can localize E-cadherin strongly suggests that the loss of cell border E-cadherin expression in mutant Numb/Numbl mice is a primary, cell-autonomous defect. Given that Numb and E-cadherin are concurrently upregulated in ependymal cells during the postnatal period, co-localized to cell borders, and associate with each other in vivo, it is possible that Numb is involved in cell border E-cadherin localization during ependymal maturation. The inability of NICD over-expression to mimic the ependymal phenotype of Numb/Numbl mutant mice is also consistent with the notion that Numb regulates the ependymal integrity via a Notch-independent mechanism. In Drosophila, Numb is known to inhibit Notch signaling through the endocytotic machinery (Berdnik et al., 2002), so it would be of interest to know whether Numb’s interaction with E-cadherin also involves similar mechanisms. Taken together, our results suggest that Numb/Numbl maintain the homeostasis of the SVZ neurogenic niche by regulating ependymal wall integrity through E-cadherin, and by ensuring neuroblast survival through Notch inhibition. During cortical neurogenesis, radial glia function both as neuroprogenitors and as neuroepithelial cells lining the ventricular wall, thus our findings here that Numb can participate in different pathways may help to explain the apparently contradictory Numb/Numbl embryonic knockout phenotypes (Petersen et al., 2002; Li et al., 2003; Petersen et al., 2004).
SVZ plasticity and the ability to remodel after injury
In deleting Numb/Numbl postnatally, we found that brain ventricular wall damage can be self-repaired. Part of this repair was likely accomplished by astrocytic invasion, in a process similar to scar tissue formation. But it was surprising that Numb-expressing SVZ progenitors can mediate this re-lining of the ventricular wall, and can subsequently maintain neurogenesis in this highly modified environment. As with inducible gene-deletion techniques, it is often not possible to target all cells. Since postnatal SVZ progenitors expressed Numb, it was difficult to accurately estimate our targeting efficiency in these cells prior to ependymal detachment due to residual Numb protein after gene deletion. The ependymal cells only up-regulated Numb expression postnatally, and revealed that we can target 80 to 90% of these cells after single P0-tamoxifenind. We were able to observe the ventricular wall repair because the nestin-creERtm4 transgene did not target SVZ progenitors with 100% efficiency.
There are several possible explanations for how this niche remodeling occurs in the Numb/Numbl mutant mice. It is possible that Numb-expressing SVZ progenitors simply responded to the ependymal detachment by making differentiated progeny that provided similar niche functions to ependymal cells, and continued to support postnatal SVZ neurogenesis. It is also possible that the SVZ progenitors initially responding to ependymal detachment recreated a functional niche through differentiation, thus allowing other SVZ progenitors to populate this niche and continue with postnatal neurogenesis. The cells within the remodeled lateral wall of the LV, some of which expressed S100β, GFAP, and GLAST, do not resemble any known cell types within the normal SVZ neurogenic niche. To understand the behavior of these escapee Numb-expressing progenitor cells during the ventricular wall remodeling phase, we will need new tools to positively identify this cell population and their progeny.
These results demonstrating a self-repair mechanism in the brain may have implications for niche remodeling in other areas of postnatal/adult stem cell biology. It has been reported that SVZ neuroblasts can migrate to distant sites caused by brain injury such as stroke (reviewed in Lindvall and Kokaia, 2004; Carmichael, 2006). Although the exact mechanisms are still being work on, these observations raise the possibility that postnatal neurogenesis can one day be used to repair brain damage. Our results here show that self-repair and local remodeling can indeed happen along the brain LV wall, and further insights into this process should shed light on whether SVZ neural stem cells participate in stroke/trauma-induced brain remodeling and postnatal/adult brain tumor formation. Furthermore, understanding how the SVZ cells participate in local repair should help bring us closer to the goal of using neural stem cells as therapeutic agents in neurodegenerative diseases.
EXPERIMENTAL PROCEDURES
Animals
All mouse experiments were performed according to an approved protocol by the Institutional Animal Care and Use Committee at UCSF. Mice were backcrossed 3 generations onto the C57BL/6 background.
Generation of nestin-creERtm mice
Nestin-CreERtm DNA construct was made by replacing Cre coding sequence in pNesCre (Tronche et al., 1999) with CreERtm from pBS-CreERtm (Hayashi and McMahon, 2002). The resulting construct was subjected to pronuclear injection by standard methods. The following PCR primers: NesPr1 (CGCTTCCGCTGGGTCACTGTCG) and Cre3 (TCGTTGCATCGACCGGTAATGCAGGC) resulted in a roughly 250 base-pair product.
Tamoxifen, BrdU administration
Tamoxifen (Sigma) and 4OH-tamoxifen (Sigma) stocks, and embryonic CreERtm inductions were as described (Hayashi and McMahon, 2002). For postnatal induction, subcutaneous injection of tamoxifen (20 mg/ml) at 8 mg/40gm of body weight was administered at the times indicated. For proliferation assays, BrdU (Sigma) (10 mg/ml, dissolved in 0.9% saline) (50 μg/gm of body weight) was either injected once per 12 hrs for 3 continuous days, or just once 1 hr before sacrifice for transient assays.
In vivo electroporation and immunoprecipitation
For gene transfer into postnatal ependymal cells, P4 pups were injected with 3-4 μl of pFLAG-Numb65 (Roncarati et al., 2002) (concentrated at 4μg/l) into LV followed by electroporation (two 50 ms pulses of 150 V with 950 ms interval). P7 brains were sectioned (350 μm), and ependyma dissected under dissecting loupe (Zeiss Stemi SV6), and subjected to co-immunoprecipitation assays. Details are available in Supplemental Data.
In vivo viral injection
In vivo adeno-cre viral injection was performed as described (Merkle et al., 2004).
EM, β-galactosidase, immunohistochemistry
Processing for electron microscopy was performed as described (Herrera et al., 1999).β-galactosidase staining was performed as described: http://axon.med.harvard.edu/~cepko/protocol/xgalplap-stain.htm. Preparation of brain tissue for immunohistochemistry (IHC) and BrdU labeling was as described (Spassky et al., 2005). IHC was performed on 12 μm cryostat samples. Comparisons between control and mutant samples were imaged using identical confocal settings. Details are available in Supplemental Data.
Supplementary Material
Acknowledgments
We thank R. Klein for the Nestin promoter / 2nd intron enhancer construct; A. McMahon for CreERtm construct; Dev. Studies Hybridoma Bank for RC2 antibody; T. Cheng for technical assistance; J. Rubenstein, S. Pleasure, N. Shah, D. Castaneda, J. Wildonger, P. Soba, M. Berger, and A. Buckley for helpful comments. This work was supported by National Institutes of Health Grant 5 R01 NS047200. C.T.K. is a Calif. Inst. of Regenerative Medicine postdoctoral scholar. Y.N.J. and L.Y.J. are Howard Hughes Medical Institute investigators.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahn S, Joyner AL. In vivo analysis of quiescent adult neural stem cells responding to Sonic hedgehog. Nature. 2005;437:894–897. doi: 10.1038/nature03994. [DOI] [PubMed] [Google Scholar]
- Alvarez-Buylla A, Lim DA. For the long run: maintaining germinal niches in the adult brain. Neuron. 2004;41:683–686. doi: 10.1016/s0896-6273(04)00111-4. [DOI] [PubMed] [Google Scholar]
- Berdnik D, Torok T, Gonzalez-Gaitan M, Knoblich JA. The endocytic protein alpha-Adaptin is required for numb-mediated asymmetric cell division in Drosophila. Dev Cell. 2002;3:221–231. doi: 10.1016/s1534-5807(02)00215-0. [DOI] [PubMed] [Google Scholar]
- Bertrand N, Castro DS, Guillemot F. Proneural genes and the specification of neural cell types. Nat Rev Neurosci. 2002;3:517–530. doi: 10.1038/nrn874. [DOI] [PubMed] [Google Scholar]
- Campbell K. Cortical neuron specification: it has its time and place. Neuron. 2005;46:373–376. doi: 10.1016/j.neuron.2005.04.014. [DOI] [PubMed] [Google Scholar]
- Carmichael ST. Cellular and molecular mechanisms of neural repair after stroke: making waves. Ann Neurol. 2006;59:735–742. doi: 10.1002/ana.20845. [DOI] [PubMed] [Google Scholar]
- Chambers CB, Peng Y, Nguyen H, Gaiano N, Fishell G, Nye JS. Spatiotemporal selectivity of response to Notch1 signals in mammalian forebrain precursors. Development. 2001;128:689–702. doi: 10.1242/dev.128.5.689. [DOI] [PubMed] [Google Scholar]
- Conover JC, Doetsch F, Garcia-Verdugo JM, Gale NW, Yancopoulos GD, Alvarez-Buylla A. Disruption of Eph/ephrin signaling affects migration and proliferation in the adult subventricular zone. Nat Neurosci. 2000;3:1091–1097. doi: 10.1038/80606. [DOI] [PubMed] [Google Scholar]
- Doetsch F, Garcia-Verdugo JM, Alvarez-Buylla A. Regeneration of a germinal layer in the adult mammalian brain. Proc Natl Acad Sci USA. 1999;96:11619–11624. doi: 10.1073/pnas.96.20.11619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuccillo M, Joyner AL, Fishell G. Morphogen to mitogen: the multiple roles of hedgehog signalling in vertebrate neural development. Nat Rev Neurosci. 2006;7:772–783. doi: 10.1038/nrn1990. [DOI] [PubMed] [Google Scholar]
- Gotz M, Barde YA. Radial glial cells defined and major intermediates between embryonic stem cells and CNS neurons. Neuron. 2005;46:369–372. doi: 10.1016/j.neuron.2005.04.012. [DOI] [PubMed] [Google Scholar]
- Hayashi S, McMahon AP. Efficient recombination in diverse tissues by a tamoxifen-inducible form of Cre: a tool for temporally regulated gene activation/inactivation in the mouse. Dev Biol. 2002;244:305–318. doi: 10.1006/dbio.2002.0597. [DOI] [PubMed] [Google Scholar]
- Herrera DG, Garcia-Verdugo JM, Alvarez-Buylla A. Adult-derived neural precursors transplanted into multiple regions in the adult brain. Ann Neurol. 1999;46:867–877. doi: 10.1002/1531-8249(199912)46:6<867::aid-ana9>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- Kawaguchi A, Miyata T, Sawamoto K, Takashita N, Murayama A, Akamatsu W, Ogawa M, Okabe M, Tano Y, Goldman SA, Okano H. Nestin-EGFP transgenic mice: visualization of the self-renewal and multipotency of CNS stem cells. Mol Cell Neurosci. 2001;17:259–273. doi: 10.1006/mcne.2000.0925. [DOI] [PubMed] [Google Scholar]
- Kriegstein AR, Noctor SC. Patterns of neuronal migration in the embryonic cortex. Trends Neurosci. 2004;27:392–399. doi: 10.1016/j.tins.2004.05.001. [DOI] [PubMed] [Google Scholar]
- Li HS, Wang D, Shen Q, Schonemann MD, Gorski JA, Jones KR, Temple S, Jan LY, Jan YN. Inactivation of Numb and Numblike in embryonic dorsal forebrain impairs neurogenesis and disrupts cortical morphogenesis. Neuron. 2003;40:1105–1118. doi: 10.1016/s0896-6273(03)00755-4. [DOI] [PubMed] [Google Scholar]
- Lim DA, Tramontin AD, Trevejo JM, Herrera DG, Garcia-Verdugo JM, Alvarez-Buylla A. Noggin antagonizes BMP signaling to create a niche for adult neurogenesis. Neuron. 2000;28:713–726. doi: 10.1016/s0896-6273(00)00148-3. [DOI] [PubMed] [Google Scholar]
- Lindvall O, Kokaia Z. Recovery and rehabilitation in stroke: stem cells. Stroke. 2004;35:2691–2694. doi: 10.1161/01.STR.0000143323.84008.f4. [DOI] [PubMed] [Google Scholar]
- Machold R, Hayashi S, Rutlin M, Muzumdar MD, Nery S, Corbin JG, Gritli-Linde A, Dellovade T, Porter JA, Rubin LL, et al. Sonic hedgehog is required for progenitor cell maintenance in telencephalic stem cell niches. Neuron. 2003;39:937–950. doi: 10.1016/s0896-6273(03)00561-0. [DOI] [PubMed] [Google Scholar]
- Merkle FT, Tramontin AD, Garcia-Verdugo JM, Alvarez-Buylla A. Radial glia give rise to adult neural stem cells in the subventricular zone. Proc Natl Acad Sci USA. 2004;101:17528–17532. doi: 10.1073/pnas.0407893101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morhenn VB, Murakami M, O'Grady T, Nordberg J, Gallo RL. Characterization of the expression and function of N-methyl-D-aspartate receptor in keratinocytes. Exp Dermatol. 2004;13:505–511. doi: 10.1111/j.0906-6705.2004.00186.x. [DOI] [PubMed] [Google Scholar]
- Nishimura T, Yamaguchi T, Tokunaga A, Hara A, Hamaguchi T, Kato K, Iwamatsu A, Okano H, Kaibuchi K. Role of numb in dendritic spine development with a Cdc42 GEF intersectin and EphB2. Mol Biol Cell. 2006;17:1273–1285. doi: 10.1091/mbc.E05-07-0700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen PH, Zou K, Hwang JK, Jan YN, Zhong W. Progenitor cell maintenance requires numb and numblike during mouse neurogenesis. Nature. 2002;419:929–934. doi: 10.1038/nature01124. [DOI] [PubMed] [Google Scholar]
- Petersen PH, Zou K, Krauss S, Zhong W. Continuing role for mouse Numb and Numbl in maintaining progenitor cells during cortical neurogenesis. Nat Neurosci. 2004;7:803–811. doi: 10.1038/nn1289. [DOI] [PubMed] [Google Scholar]
- Rakic P. Developmental and evolutionary adaptations of cortical radial glia. Cereb Cortex. 2003;13:541–549. doi: 10.1093/cercor/13.6.541. [DOI] [PubMed] [Google Scholar]
- Roegiers F, Jan YN. Asymmetric cell division. Curr Opin Cell Biol. 2004;16:195–205. doi: 10.1016/j.ceb.2004.02.010. [DOI] [PubMed] [Google Scholar]
- Roncarati R, Sestan N, Scheinfeld MH, Berechid BE, Lopez PA, Meucci O, McGlade JC, Rakic P, D'Adamio L. The gamma-secretase-generated intracellular domain of beta-amyloid precursor protein binds Numb and inhibits Notch signaling. Proc Natl Acad Sci U S A. 2002;99:7102–7107. doi: 10.1073/pnas.102192599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawamoto K, Wichterle H, Gonzalez-Perez O, Cholfin JA, Yamada M, Spassky N, Murcia NS, Garcia-Verdugo JM, Marin O, Rubenstein JL, et al. New neurons follow the flow of cerebrospinal fluid in the adult brain. Science. 2006;311:629–632. doi: 10.1126/science.1119133. [DOI] [PubMed] [Google Scholar]
- Sharp CD, Fowler M, Jackson THt, Houghton J, Warren A, Nanda A, Chandler I, Cappell B, Long A, Minagar A, Alexander JS. Human neuroepithelial cells express NMDA receptors. BMC Neurosci. 2003;4:28. doi: 10.1186/1471-2202-4-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Q, Zhong W, Jan YN, Temple S. Asymmetric Numb distribution is critical for asymmetric cell division of mouse cerebral cortical stem cells and neuroblasts. Development. 2002;129:4843–4853. doi: 10.1242/dev.129.20.4843. [DOI] [PubMed] [Google Scholar]
- Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- Spassky N, Merkle FT, Flames N, Tramontin AD, Garcia-Verdugo JM, Alvarez-Buylla A. Adult ependymal cells are postmitotic and are derived from radial glial cells during embryogenesis. J Neurosci. 2005;25:10–18. doi: 10.1523/JNEUROSCI.1108-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sur M, Rubenstein JL. Patterning and plasticity of the cerebral cortex. Science. 2005;310:805–810. doi: 10.1126/science.1112070. [DOI] [PubMed] [Google Scholar]
- Temple S. The development of neural stem cells. Nature. 2001;414:112–117. doi: 10.1038/35102174. [DOI] [PubMed] [Google Scholar]
- Tramontin AD, Garcia-Verdugo JM, Lim DA, Alvarez-Buylla A. Postnatal development of radial glia and the ventricular zone (VZ): a continuum of the neural stem cell compartment. Cereb Cortex. 2003;13:580–587. doi: 10.1093/cercor/13.6.580. [DOI] [PubMed] [Google Scholar]
- Tronche F, Kellendonk C, Kretz O, Gass P, Anlag K, Orban PC, Bock R, Klein R, Schutz G. Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat Genet. 1999;23:99–103. doi: 10.1038/12703. [DOI] [PubMed] [Google Scholar]
- Tropepe V, Craig CG, Morshead CM, van der Kooy D. Transforming growth factor-alpha null and senescent mice show decreased neural progenitor cell proliferation in the forebrain subependyma. J Neurosci. 1997;17:7850–7859. doi: 10.1523/JNEUROSCI.17-20-07850.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai LH, Gleeson JG. Nucleokinesis in neuronal migration. Neuron. 2005;46:383–388. doi: 10.1016/j.neuron.2005.04.013. [DOI] [PubMed] [Google Scholar]
- Vaezi A, Bauer C, Vasioukhin V, Fuchs E. Actin cable dynamics and Rho/Rock orchestrate a polarized cytoskeletal architecture in the early steps of assembling a stratified epithelium. Dev Cell. 2002;3:367–381. doi: 10.1016/s1534-5807(02)00259-9. [DOI] [PubMed] [Google Scholar]
- Yang X, Klein R, Tian X, Cheng HT, Kopan R, Shen J. Notch activation induces apoptosis in neural progenitor cells through a p53-dependent pathway. Dev Biol. 2004;269:81–94. doi: 10.1016/j.ydbio.2004.01.014. [DOI] [PubMed] [Google Scholar]
- Zhong W, Feder JN, Jiang MM, Jan LY, Jan YN. Asymmetric localization of a mammalian numb homolog during mouse cortical neurogenesis. Neuron. 1996;17:43–53. doi: 10.1016/s0896-6273(00)80279-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.