

Abstract
A new methodology for quantitative analysis of proteins is described, applying stable isotope labeling by small organic molecules combined with 1- or 2-dimensional electrophoresis and MALDI-TOF-MS, also allowing concurrent protein identification by peptide mass fingerprinting. Our method eliminates fundamental problems in other existing isotope-tagging methods requiring liquid chromatography and MS/MS, such as isotope-effects, fragmentation, and solubility. It is also anticipated to be more practical and accessible than those LC-dependent methods.
Keywords: Quantitative analysis, Proteomics, Stable-isotope labeling, Cysteine modifier
Introduction
Proteomics is rapidly becoming an important research area for comprehensive study of protein expression patterns under a specific set of conditions. A great deal of effort has been focused on developing methods of measuring global changes in relative protein abundance between two distinct cell systems, such as control vs. treated cells. Thus, it is essential to develop efficient methodologies for quantitative analysis of proteins within complex mixtures expressed under certain physiological conditions.
Conventional methods for quantitative analysis of proteins include two-dimensional densitometry of gels1 and radioisotope labeling.2 While these methods are still useful for certain purposes, more recently, application of stable-isotope labeling followed by mass spectrometry analysis has been emerging as a powerful technology for quantification and concurrent identification of proteins for proteomics research.3
Some of the earliest examples are metabolic labeling methods in which cells are cultured in isotope-enriched or normal media and the relative abundance of specific proteins is quantitatively analyzed from peak intensities from each sample in the mass spectra.4,5 The disadvantage of metabolic labeling methods in general is that, although they are applicable to cultured cells or small organisms, it is rather difficult to label larger organisms.
Instead, chemical modifications by covalent labeling on specific amino acid residues using isotope-labeled reagents followed by mass spectrometry analysis are expected to significantly expand the scope of isotope-labeling methods, because in theory, such methods are applicable to any protein samples.
Some of the most pioneering work has been reported by Aebersold et al., applying deuterium-labeled isotope-coded affinity tags (ICATs).6 The first generation of ICAT reagents is a set of reagents that consists of a biotin conjugate of a well-known cysteine-modifying reagent, iodoacetamide, and its deuterated version, whose molecular weight is 8 Da heavier.
In this method, proteins are modified with the cysteine-specific reactive group (iodo group) and the biotin tag in the ICAT reagent allows the specific isolation of the modified Cys-containing peptides by immobilized avidin. Changes in the relative abundance of peptides from distinct proteome samples are accomplished by the use of isotopically labeled and unlabeled ICAT reagents, which show chemically identical behavior. After the derivatized proteomes are pooled and digested with trypsin, the tryptic peptide mixtures isolated by avidin affinity chromatography can be quantitatively analyzed by liquid chromatography/electrospray ionization-tandem mass spectrometry (LC/ESI-MS/MS). Subsequently many other researchers reported other LC-dependent methods inspired by this ICAT method.7 Coupled with recent development of multidimensional liquid chromatography, these LC-based methods have been playing important roles in proteomics.
However, several fundamental problems with this ICAT method have been reported, such as primary isotope effects especially for d-labeling, causing differential elution during microcapillary reversed-phase liquid chromatography (μLC), and fragmentation of the labels during collision-induced dissociation (CID) conditions, complicating the interpretation of tandem mass spectra. These problems are likely derived from the use of large hydrophobic organic molecules, which have decreased solubility in the aqueous media. Although several improvements have been made for the later versions of ICAT reagents, such as acid- or photo-cleavable versions,8 and other different types of labeling reagents have also been reported,7 these LC-dependent methods essentially require a large amount of aqueous media for purification of the derivatized peptides. For this reason, it is imperative to develop fundamentally different approaches that use small-molecule labeling reagents and that eliminate the liquid chromatography step.
Therefore, we have been developing our own methodology for quantitative analysis of proteins by a combination of isotope-labeled and unlabeled chemical modification of specific amino acid residues by small organic molecules followed by 2D electrophoresis and matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF-MS). The concept is outlined in Scheme 1.
Scheme 1.
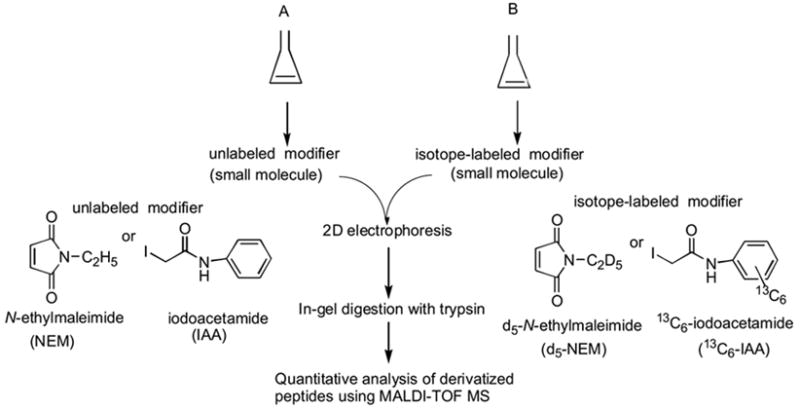
Our strategy for quantitative analysis of proteins
Since our method uses 2D electrophoresis instead of LC for the purification of protein, the procedure is more convenient and more economical, and the small organic molecules are also expected to be more easily accessible as well as to have better solubility in the sample mixture. The use of small molecules and a soft ionization mass spectrometry, MALDI-MS, also eliminates the fragmentation problems. While the choice of mass spectrometry is not critical and either MALDI or electrospray ionization (ESI) is possible, we chose MALDI rather than ESI, since more laboratories tend to have MALDI and the operation and maintenance are easier, making our method more practical.
The 2D polyacrylamide gel electrophoresis (2D PAGE), which displays all separated proteins on a single gel, has been the standard tool for protein expression in proteomics. Two dimensional gel separation using ICAT reagents has been reported.9 The 2D gel electrophoresis also provides information about the molecular weights and pI values of proteins. Therefore our method allows us to identify each protein through peptide mass fingerprinting prior to quantitative analysis without a need for the MS/MS mode for identification of proteins, which will not only eliminate the fragmentation problems, but also make it simpler and more economical and therefore accessible to more researchers.
We previously synthesized sulfhydryl (-SH)-group specific modifiers that are small molecules for quantitative analysis of proteins, since a sulfhydryl group is one of the most nucleophilic functional groups within peptide sequences. The combinations we have synthesized are N-ethylmaleimide (NEM) and its d5-deuterated versions (d5-NEM)10 as well as iodoacetanilide (IAA) and 13C6-labeled iodoacetoanilide (13C6-IAA).11 Iodoacetanilide (IAA) is a derivative of a well-known sulfhydryl group modifier, iodoacetamide, and both NEM and IAA are known to specifically react with -SH groups of cysteine residues (Scheme 2). The unlabeled N-ethylmaleimide is commercially available.
Scheme 2.
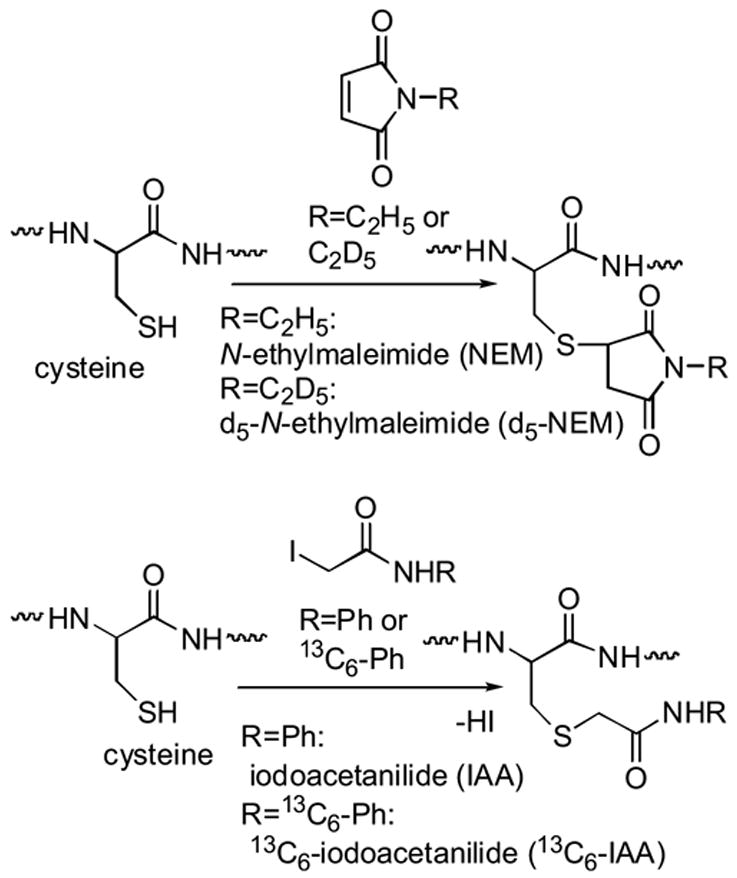
Reactions of N-ethylmaleimide(NEM) or iodoacetanilide (IAA) with cysteine
As both modifiers react at different pHs, these modifiers may be used according to the sample environment. The iodoacetanilides are expected not to interfere with phosphine-based reducing reagents,12 and are anticipated to be more reactive than N-ethylmaleimides due to the existence of a reactive leaving group, an iodo group. The mass difference between IAA and 13C6-IAA is 6 Da rather than 5 Da as in NEM and d5-NEM; therefore the combination of IAA and13C6-IAA is expected to provide sharper separation of the IAA and 13C6-IAA-modified peptide peaks in the MALDI spectra rather than the NEM and d5-NEM-modified ones, and hence more accurate quantification, while both combinations are quite visible in the mass spectra separated by 1D and 2D electrophoresis.
We have shown that both combinations are successful for quantitative analysis of various peptides.10,11 Using known amounts of various peptides, we confirmed that ionization efficiencies for isotope-labeled and –unlabeled peptides are identical within experimental errors. Here we show their application to proteomic experiments, quantitative analysis of proteins.
Experimental Methods
1(a)Preparation of standard protein solutions for 1D SDS-PAGE (without modifiers)
Each of the following four commercial proteins (1mg each, purchased from Sigma), bovine serum albumin (66 kDa), egg albumin (45 kDa), carbonic anhydrase (29 kDa), or α-lactalbumin (14.2 kDa), was dissolved in a sample buffer (1mL) containing Trizma® Pre-Set Crystals (purchased from Sigma), ethylenediaminetetraacetic acid (EDTA), sodium dodecyl sulfate (SDS), and β-mercaptoethanol. The pHs and the concentrations of Trizma®, EDTA, SDS, and β-mercaptoethanol were as follows. For the Trizma® Pre-Set Crystals the initial pH was 7.0 and the final concentration was 78 mM. For the EDTA, the final concentration was 6.3 mM. For the SDS, the final concentration was 2.5% (w/v). For the β-mercaptoethanol, the final concentration (v/v) was 1.25%. The mixed protein solution was left for 1 hour at room temperature.
1(b) Preparation of standard protein solutions for 1D SDS-PAGE (with NEM/d5-NEM)
Each of the following four commercial proteins (1mg each, purchased from Sigma), bovine serum albumin (66 kDa), egg albumin (45 kDa), carbonic anhydrase (29 kDa), or α-lactalbumin (14.2 kDa), was dissolved in a sample buffer (1mL) containing Trizma® Pre-Set Crystals (Sigma), EDTA, SDS, and tributylphosphine (TBP). The pHs and the concentrations of the Trizma®, EDTA, SDS, and TBP were as follows. For the Trizma® Pre-Set Crystals, the initial pH and the final concentration were 7.8 and 84 mM. For the EDTA, the final concentration was 6.8 mM. For the SDS, the final concentration was 2.7% (w/v). For the tributylphosphine (TBP), the final concentration was 20mM. The mixed protein solution was left for 1 hour at room temperature for sufficient reduction with TBP.
Each of the above protein solutions (10 μL) with different molar ratios was incubated with 1 μL of 1 M NEM or d5-NEM DMSO solution, and these two solutions were mixed and incubated for 2 hours at 37 °C. In this way, the following five molar ratio pairs of protein solutions were examined: 9:1, 6:1, 3:1, 1:1, and 1:3 (NEM-modified:d5-NEM-modified).
Preparation of standard protein solutions for 1D SDS-PAGE (with IAA/13C6-IAA)
Each of the following four commercial proteins (1mg each, purchased from Sigma), bovine serum albumin (66 kDa), egg albumin (45 kDa), carbonic anhydrase (29 kDa), or α-lactalbumin (14.2 kDa), was dissolved in a sample buffer (1mL) containing Trizma® Pre-Set Crystals (Sigma), EDTA, SDS, and tributylphosphine (TBP). The pHs and the concentrations of the Trizma®, EDTA, SDS, and TBP were as follows. For the Trizma® Pre-Set Crystals, the initial pH was 9.0 and the final concentration was 84 mM. For the EDTA, the final concentration was 6.8 mM. For the SDS, the final concentration was 2.7% (w/v). For the tributylphosphine (TBP), the final concentration of was 20mM. The mixed protein solution was left for 1 hour at room temperature for sufficient reduction with TBP.
Each of the above protein solutions (10 μL) with different molar ratios was incubated with 1μL of 200 mM IAA or 13C6-IAA DMSO solution, and the two solutions were mixed and incubated for 2 hours at 37 °C. In this way, the following five molar ratio pairs of protein solutions were examined: 9:1, 6:1, 3:1, 1:1, and 1:3 (NEM-modified:d5-NEM-modified or IAA-modified:13C6-IAA-modified).
Separation of proteins in 1D SDS-PAGE
The following SDS-PAGE gels were used: 0.8 mm thick gels (17 cm wide) that consist of 5% acrylamide in stacking gels (2 cm long) and 11% acrylamide in separating gels (13.5 cm long). Right before loading the samples, 20 μL of the protein mixture solutions were mixed with 5 μL of a solution consisting of 20% (v/v) glycerol and 0.1% (w/v) bromophenol blue, and the solution was subjected to SDS-PAGE. The electrophoresis conditions were as follows: 10 mA for about 1 hour and 30 minutes, until the bromophenol blue reached the end of the stacking gel, followed by 25 mA for about 2 hours and 30 minutes until the bromophenol blue reached the end of the separating gel.
Preparation of protein extracts from Drosophila heads for 2D IEF/SDS-PAGE (without modifiers)
A total of 100 precisely dissected Drosophila heads, excluding eyes, were homogenized using 120 μL of a lysis buffer consisting of 8.5 M urea, 2% 3-[3-(cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS), 0.5% Bio-Lyte 3/10 ampholyte (Bio-Rad), and 5% β-mercaptoethanol. After one hour, the supernatant was collected by centrifugation, and 100μL was loaded onto the IEF.
Preparation of protein extracts from Drosophila heads for 2D IEF/SDS-PAGE (with NEM/d5-NEM)
A total of 100 precisely dissected Drosophila heads, excluding eyes, were homogenized using 80–120 μL of a lysis buffer consisting of 47.25 mM Trizma® Pre-Set Crystals pH 7.0, including 8.5 M urea, 2% CHAPS, 0.5% Bio-Lyte 3/10 ampholyte (Bio-Rad), and 20 mM TBP. After one hour, the supernatant collected by centrifugation was divided into the volume ratios of 3:1, 1:1 or 1:3, and then the above lysis buffer was added to the smaller amount of supernatant in each pair to make the volumes equal. Each solution in each pair was incubated with 2μL of 1M NEM or d5NEM DMSO solution for 2 hours at 37 °C, and subsequently 47.5μL from each solution were mixed. After 5μL of β-mercaptoethanol was added to the mixture and incubated for 10 minutes at room temperature to stop the reaction with NEMs, the mixture (a total of 100 μL) was loaded onto the IEF.
Preparation of protein extracts from Drosophila heads for 2D IEF/SDS-PAGE(with IAA/13C6IAA)
A total of 100 precisely dissected Drosophila heads, excluding eyes, were homogenized using 80–120 μL of a lysis buffer consisting of 47.25 mM Trizma® Pre-Set Crystals pH 9.0 for incubation with IAA or 13C6-IAA, including 8.5 M urea, 2% CHAPS, 0.5% Bio-Lyte 3/10 ampholyte (Bio-Rad), and 20 mM TBP. After one hour, the supernatant collected by centrifugation was divided into the volume ratios of 3:1, 1:1 or 1:3, and then the above lysis buffer was added to the smaller amount of supernatant to make the volumes equal. Each solution in each pair was incubated with 2μL of 200 mM IAA or 13C6-IAA DMSO solution for 2 hours at 37 °C, and subsequently 47.5μL from each solution were mixed. After 5μL of β-mercaptoethanol was added to the mixture and incubated for 10 minutes at room temperature to stop the reaction with IAAs, the mixture (a total of 100 μL) was loaded onto the IEF.
Separation of proteins in 2D IEF/SDS-PAGE
For IEF analysis, the ImmobilineTM DryStrips (pH 3–10, 13 cm long, 3 mm wide, and 0.5 mm thickness after rehydration, Amersham Biosciences) were rehydrated overnight with the lysis buffer including 5% β-mercaptoethanol (v/v) and 0.1% bromophenol blue aqueous solution. Each 90 μL of four sets of samples was loaded into a sample cup and focused on IPGphor (Amersham Pharmacia Biotech), and then IEF was performed. The voltage was linearly increased from 0 to 500 V for 3 minutes, then increased up to 4000 V over 1 hour and 30 minutes and held constant at 4000 V for an additional 30 minutes, and for 4 hours at 8000 V with a maximum current per strip of 70 μA. A total Vh product rather than 17 kVh was used to focus the strip. The electrophoresis conditions for the second SDS-PAGE were as follows: 15 mA for about 1 hour until the bromophenol blue reached the end of the stacking gel, followed by 25 mA for about 3 hours, until the bromophenol blue reached the end of the separating gel.
In-gel digestion
The protein bands (from 1D SDS-PAGE) or spots (from 2D IEF/SDS-PAGE) stained with Coomassie® Brilliant Blue G-250 (Bio-Rad) or Brilliant Blue R-250 (Sigma) were excised and destained with 50% acetonitrile/100 mM NH4HCO3 (pH 8.0). After they were dried briefly, 2 μL (1 μL at a time, twice) of N-p-tosyl-L-phenylalanine chloromethyl ketone (TPCK)-treated trypsin (Promega) solution (0.2 μg/μL in 50 mM acetic acid) was applied to each gel piece, followed by incubation in 50 μL of 2.5 mM NH4HCO3 overnight at 30°C. After that, the incubated solution was collected and refrigerated. For extraction of the tryptic peptides, the gel piece was immersed in 60% acetonitrile/0.1% trifluoroacetic acid (TFA) and shaken for 1 hour. The above refrigerated incubated solution and the extracted peptide solutions were mixed and dried. The dried tryptic peptides were dissolved in 5 μL milli-Q® water.
MALDI-TOF-MS
The extracted peptide solution was mixed with a matrix (α-cyano-4-hydroxycinnamic acid, CHCA) solution (prepared by dissolving 10 mg of CHCA in 1 mL of 50% acetonitrile/0.1% TFA) and then subjected to MALDI-TOF-MS analysis. The MALDI spectra were obtained by a Voyager Elite BioSpectrometry Research Station, equipped with a delayed extraction option (Applied Biosystems) operated at a 20 kV accelerating voltage, 75% grid voltage, 0.05% guide wire voltage, 150ns pulse delay time, 100mV vertical scale, and 3% vertical offset. A pulsed nitrogen laser operating at 337 nm was used as a desorption/ionization source. Mass spectrometry was performed in a reflector with positive ion detection. The ion signal was recorded using a 500 MHz transient digitizer. The data were analyzed using GRAMS/386 (Galactic Industries Corporation).
Database search
The monoisotopic m/z values of tryptic peptide peaks were entered into the MS-Fit in the ProteinProspector (UCSF: http://prospector.ucsf.edu/) for a database search for characterization of proteins.
Peak area calculation
The areas of the monoisotopic peak and the following four isotopic peaks were added together for each modified peptide using GRAMS/386. The areas of the small 6- or 7-10th isotopic peaks of NEM- or IAA-modified peptide were subtracted from the monoisotopic peaks and the following isotopic peaks of d5-NEM- or13C6-IAA-modified peptide respectively. The observed relative ratios of unlabeled and isotope-labeled peptides in the five sets of 9:1, 6:1, 3:1, 1:1, and 1:3 for 1D SDS-PAGE, or three sets of 3:1, 1:1, and 1:3 for 2D IEF/SDS-PAGE were plotted against their theoretical ratios as shown in the graphs in the following section.
Results and Discussion
Quantitative analysis of standard proteins for 1-D SDS-PAGE
In order to test the applicability of our method to proteins, first we applied it to quantitative analysis of four commercially available proteins: α-lactalbumin, bovine serum albumin (BSA), carbonic anhydrase, and ovalbumin, each of which was purified by 1D electrophoresis. These proteins were individually reacted with NEM, d5-NEM, IAA, or13C6-IAAs in the presence of a reducing reagent, tributylphosphine, which reduces intramolecular S-S bonds to sulfhydryl groups. Figure 1a shows the MALDI spectrum of the mixture of α-lactalbumin modified with NEM or d5-NEM (NEM-modified: d5-NEM-modified=1:1) loaded onto 1-D SDS-PAGE after tryptic digestion.
Figure 1(a), (b), and (c).
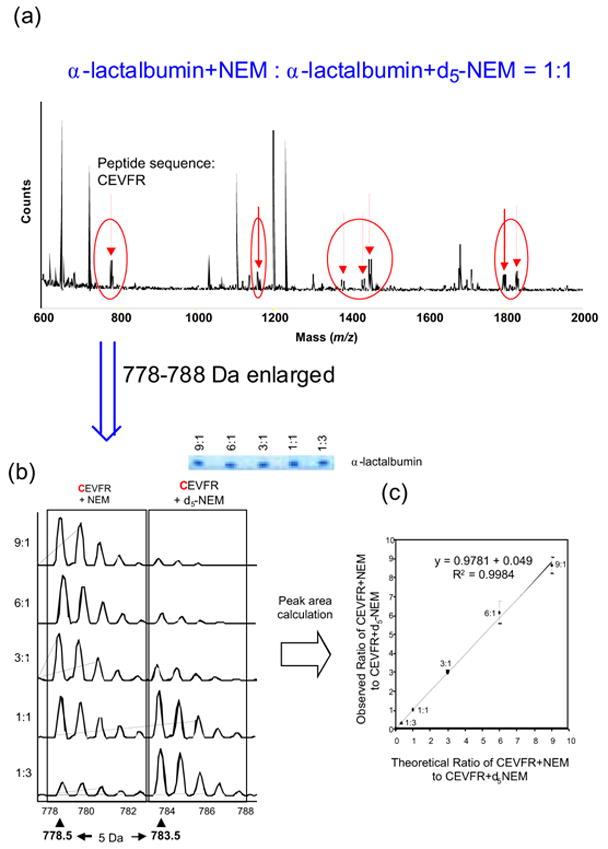
MALDI Spectrum of α-Lactalbumin Modified with NEM or d5-NEM
Seven pairs of tryptic peptides modified with NEM or d5-NEM that were 5Da apart with nearly equal intensities were detected, which are indicated by the arrows in Figure 1a. These seven pairs of peptides reacted with NEM or d5-NEM include peptides with unknown amino acid sequences and peptides formed by cleavage of other longer peptides (data not shown). Figure 1b shows the enlarged peak area of the NEM- or d5-NEM-modified peptides of α-lactalbumin consisting of residues 6–10, CEVFR, with five molar ratios, 9:1, 6:1, 3:1, 1:1, and 1:3. The correlation parameters and inclinations between the observed ratio and the theoretical ratio for the NEM- and d5-NEM-CEVFR were R2 = 0.9984 and inclination = 0.9781, as shown in Figure 1c and Table 1. Some peptides with unknown amino acid sequences may not contain any cysteines, but these data indicate that these peptides still showed reasonable correlation between the observed ratios and the theoretical ratios.
Table 1.
Results of Quantitative Analysis of Commercial Proteins with NEM and d5-NEM after 1D SDS-PAGE
| Protein | Number of Cys | R2 | Inclination |
|---|---|---|---|
| bovine serum albumin | 35 | 0.9998 | 1.1036 |
| ovalbumin | 6 | 0.9928 | 1.2822 |
| carbonic anhydrase | 0 | ND1) | ND1) |
| α-lactalbumin | 8 | 0.9984 | 0.9781 |
ND; not detectable
The same experiments were conducted for three other commercial proteins, and the correlation parameters and inclinations for these proteins modified with NEM or d5-NEM are summarized in Table 1 along with α-lactalbumin. The three proteins other than carbonic anhydrase show the reasonable correlation parameters and inclinations. No NEM- and d5-NEM-modified peptide peaks were detected from carbonic anhydrase because this protein has no cysteine residues in its sequence.
Figure 2a shows the expanded MALDI MS spectra of tryptic peptides digested from bovine serum albumin (BSA) unmodified (upper) and modified with IAA or 13C6-IAA (IAA-modified:13C6-IAA-modified=1:1) (lower).
Figure 2(a )and (b).
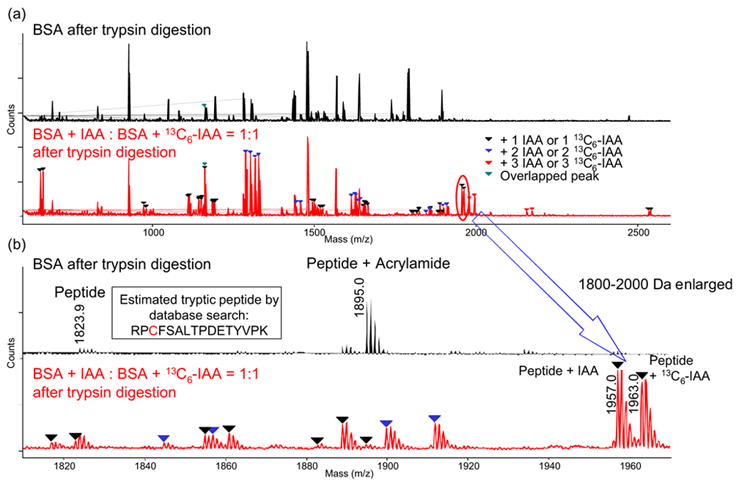
MALDI Spectrum of Bovie Serum Albumin(BSA) Modified with IAA or 13C6-IAA (IAA-modified:13C6-IAA-modified=1:1)
In a comparison of the two spectra, many modified peaks were observed as pairs having the same intensities. Some tryptic peptides reacted with more than one equivalent of IAAs or 13C6-IAA, but these IAA-modified and 13C6-IAA-modified peptides still showed the same intensities. Around 90% of cysteine residues (31 out of 35) in BSA were found to be modified with IAA or 13C6-IAA, and they are listed in Table 2.
Table 2.
. Tryptic Peptide Peaks of BSA modified with IAA or 13C6-IAA (BSA-IAA: BSA-13C6-IAA=1:1)
| Number of Peptides | Observed m/z Value of IAA Modified Peptide | Observed m/z Value of 13C6IAA Modified Peptide | Theoretical1)m/z Value of Unmodified Peptide | Identified Peptide from Database | Number of Cysteines | Number of Adding IAAs (13C6IAAs) | |
|---|---|---|---|---|---|---|---|
| Position on Protein | Sequence | ||||||
| 1 | 1655.3 | 1661.3 | 11 522.3 | unknown | unknown | unknown | 1 |
| 2 | 1974.5 | 1980.5 | 11 841.5 | 483–489 | LCVLHEK | 1 | 1 |
| 3 | 1110.5 | 1116.5 | 1 977.5 | 123–130 | NECFLSHK | 1 | 1 |
| 4 | 1144.5 | 1150.5 | 1011.4 (low)2) | 413–420 | QNCDQFEK | 1 | 1 |
| 5 | 1148.6 | 1154.5 | 11 1015.5 | 310–318 | SHCIAEVEK | 1 | 1 |
| 6 | 1157.6 | 1163.6 | 11 1024.5 | 499–507 | CCTESLVNR | 2 | 1 |
| 7 | 1290.6 | 1302.7 | 11 1024.5 | 499–507 | CCTESLVNR | 2 | 2 |
| 8 | 1183.7 | 1189.6 | 1050.5 (low) | 588–597 | EACFAVEGPK | 1 | 1 |
| 9 | 1185.6 | 1191.7 | 1052.5 (low) | 460–468 | CCTKPESER | 2 | 1 |
| 10 | 1318.7 | 1330.6 | 1052.5 (low) | 460–468 | CCTKPESER | 2 | 2 |
| 11 | 1443.8 | 1455.8 | 1177.6 (ND)3) | 300–309 | ECCDKPLLEK | 2 | 2 |
| 12 | 1615.8 | 1627.8 | 1349.5 (ND) | 76–88 | TCVADESHAG CEK | 2 | 2 |
| 13 | 1495.9 | 1501.7 | 1362.7 (ND) | 89–100 | SLHTLFGDE CK | 1 | 1 |
| 14 | 1630.8 | 1642.7 | 1364.5 (ND) | 106–117 | ETYGDMAD CCEK | 2 | 2 |
| 15 | 1519.8 | 1525.7 | 1386.6 (ND) | 286–297 | YICDNQDTISSK | 1 | 1 |
| 16 | 1654.8 | 1666.9 | 1388.6 (ND) | 375–386 | EYEATLE CCAK | 2 | 2 |
| 17 | 1630.8 | 1636.7 | 1497.6 (ND) | 387–399 | DDPHACYSTVFDK | 1 | 1 |
| 18 | 1652.9 | 1658.8 | 1519.7 (ND) | 139–151 | LKPDPNTLC DEFK | 1 | 1 |
| 19 | 1844.9 | 1856.8 | 1578.6 (ND) | 267–280 | ECCHGDLL CADDR | 3 | 2 |
| 20 | 1977.8 | 1995.8 | 1578.6 (ND) | 267–280 | ECCHGDLL CADDR | 3 | 3 |
| 21 | 1899.8 | 1911.8 | 1633.7 (ND) | 184–197 | YNGVFQ CC QAEDK | 2 | 2 |
| 22 | 1801.0 | 1806.9 | 1667.8 (ND) | 469–482 | MPCTEDYLSLILN | 1 | 1 |
| 23 | 1817.0 | 1823.0 | 1683.8 (ND) | 469–482 1Met-Ox4) | MPCTEDYLSLILN | 1 | 1 |
| 24 | 1854.9 | 1860.9 | unknown | unknown | unknown | 1 | |
| 25 | 1882.9 | 1889.0 | 1749.9 (ND) | unknown | unknown | unknown | 1 |
| 26 | 1889.0 | 1894.8 | 1756.0 (ND) | unknown | unknown | unknown | 1 |
| 27 | 2155.9 | 2174.0 | 1756.7 (ND) | 581–597 | CCAADDKEA CFAVEGPK | 3 | 3 |
| 28 | 1957.0 | 1963.0 | 111823.9 | 508–523 | RPCFSALTPDETYVPK | 1 | 1 |
| 29 | 2534.3 | 2540.3 | 2401.2 (ND) | 319–340 | DAIPENLPPLTADFAEDKDV CK | 1 | 1 |
Theoretical m/z Value of Unmodified Peptide: calculated from the peptide sequence or the observed m/z value of IAA/13C6-IAA modified peptides.
low: low intensity.
ND: not detectable.
Met-Ox; methionine is oxidized.
Figure 2b shows the enlarged peak area around m/z 1800 to 2000 in Figure 2a. The peaks of peptides consisting of residues 508–523, RPCFSALTPDETYVPK, that were modified with IAA or 13C6-IAA are clearly seen at m/z 1957.0 or 1963.0, respectively, and many other modified peaks were clearly detected with high intensities. As one example, Figure 3(a) shows expansion of the peak areas around 1950–1970 Da, in which the IAA- and 13C6-IAA-modified peptide residues RPCFSALTPDETYVPK appear in various ratios, and Figure 3(b) shows the correlation between the theoretical and observed ratios calculated from these results. As in this scheme, we also observed that the modified peak intensity ratios between these pair peaks well represent the five molar ratios of the loaded BSAs modified with IAA or 13C6-IAA (IAA-modified:13C6-IAA-modified=9:1, 6:1, 3:1, 1:1, and 1:3). The correlation parameters and inclinations between the observed ratio and the theoretical ratio for this peptide RPCFSALTPDETYVPK were R2 = 0.9996 and inclination = 0.9678 as shown in Figures 3a, 3b, and Table 3.
Figure 3(a )and (b).
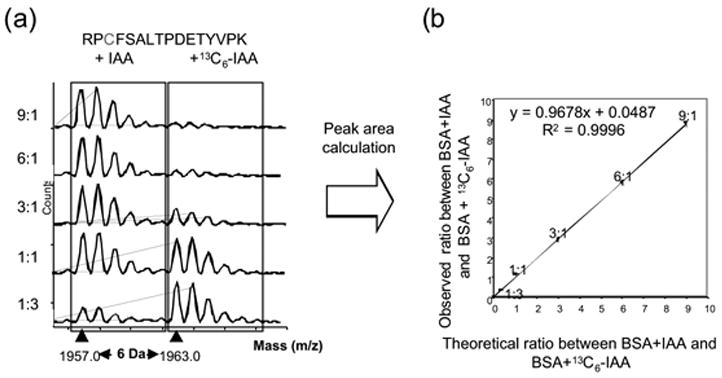
Quantitative Analysis of Bovine Serum Albumin(BSA) by 1D-SDS-PAGE
Table 3.
Results of Quantitative Analysis of Commercial Proteins with IAA and 13C6-IAA after 1D SDS-PAGE
| Protein | Number of Cys | R2 | Inclination |
|---|---|---|---|
| bovine serum albumin | 35 | 0.9996 | 0.9678 |
| ovalbumin | 6 | 0.9999 | 1.0044 |
| carbonic anhydrase | 0 | ND1) | ND1) |
| α-lactalbumin | 8 | 0.9997 | 0.9931 |
ND: not detectable
The correlation parameters and inclinations for the other commercial proteins modified with IAA or 13C6-IAA are summarized in Table 3 as well. The three proteins other than carbonic anhydrase show the good correlation parameters and inclinations. As in the case of NEM and d5-NEM, no IAA- and 13C6-IAA-modified peptide peaks were detected from carbonic anhydrase due to nonexistence of a cysteine residue.
As in the peptide at m/z 1895.0 in Figure 2(b) (above), sometimes acrylamide-reacted peptides are detected when SDS-PAGE is performed, however, such peaks were rarely observed in these NEM, d5-NEM, IAA, or13C6-IAA-modified proteins. While acrylamide is occasionally used as a modifier,13 this observation may mean that IAA and 13C6-IAA are more reactive than acrylamide toward cysteine residues and hence more suitable cysteine modifiers.
From these results we conclude that both characterization and quantification of commercially available proteins are possible by the use of NEM and d5-NEM, or IAA and13C6-IAA combined with 1-D SDS-PAGE and MALDIMS. It should also be noted that Corthals et al. have reported successful application of a combination of IAA and d5-IAA to quantitative analysis of commercially available proteins, bovine serum albumin (BSA) and lactoperoxidase, separated by 1D SDS PAGE.14
Quantitative analysis of protein extracts from Drosophila heads for 2-D IEF/SDS-PAGE
Next, we applied our method to quantitative analysis of the above four proteins separated by 2D electrophoresis, and confirmed that our method is also applicable to these commercial proteins as in the experiments by 1D electrophoresis described above (data not shown). Therefore we applied our method to quantitative analysis of proteins from the extract obtained from Drosophila heads that were separated by 2D electrophoresis. The protein extracts from 100 Drosophila heads were split into a set of two solutions in the ratios of 3:1, 1:1, and 1:3, and were reacted with an excess of NEM, d5-NEM, IAA, or 13C6-IAA in the presence of the reducing agent, tributylphosphine, as in the experiment with 1D electrophoresis above. We then performed quantitative analysis of many proteins in these protein samples developed on 2-D IEF/SDS-PAGE described in the experimental section above. We also identified these proteins by peptide mass fingerprinting concurrently with or prior to the quantitative analysis. All the protein samples were developed under the same conditions. Although occasionally poor reproducibility has been pointed out for 2D electrophoresis, all the 2D gel images were highly superimposable throughout our experiments. Although recently thiourea is frequently used for performing 2D electrophoresis, we used urea instead in order to avoid potential side reactions between thiourea and these tagging reagents.
For example, Figure 4 is the 2-D IEF/SDS-PAGE gel of the mixture of the above protein samples in the ratio of 3:1 that were reacted with an excess of IAA or 13C6-IAA.
Figure 4.
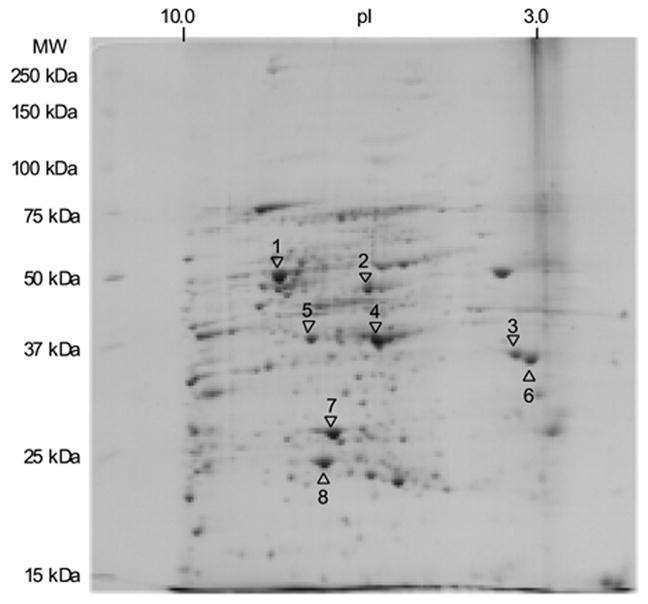
2-D IEF-SDS-PAGE of Drosophila Head Proteins:Mixture of IAA-Modified and 13C6-IAA-Modified Proteins in the Proportion of 3:1
The ratio reflects the amount of the sample protein reacted with IAA and 13C6-IAA, namely, IAA-reacted protein:13C6-IAA-reacted protein=3:1 in this case. Figures 5a and 5b show the MALDI spectra of the tryptic peptides digested from the protein labeled No. 3 (tropomyosin 1) in the above protein sample prepared by mixing the NEM-reacted protein extract and the d5-NEM-reacted protein extract in the ratio of 3:1 (NEM-reacted: d5-NEM-reacted) (Figure 5a) and that prepared by mixing the IAA-reacted protein extract and the 13C6-IAA-reacted protein extract in the ratio of 3:1 (IAA-reacted: 13C6-IAA-reacted) (Figure 5b), respectively. Mass (m/z)
Figure 5(a) and(b).
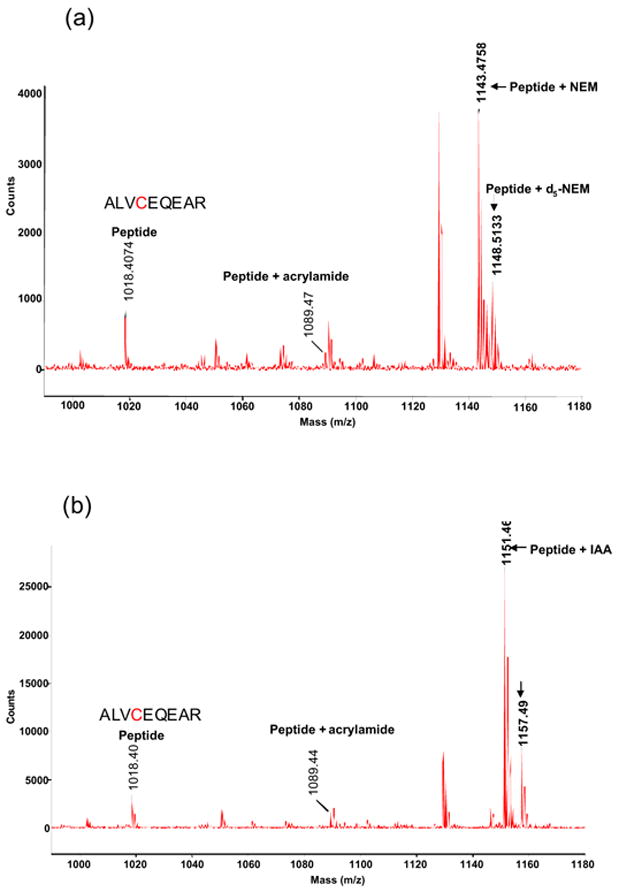
MALDI Spectrum of Spot 3 (tropomyosin 1)
(a): NEM-modified:d5-NEM-modified=3:1
(b): IAA-modified:13C6-IAA-modified=3:1
These spectra indicate that the proteins extracted from living organisms react with NEM, d5-NEM, IAA, or 13C6-IAA successfully in the presence of tributylphosphine, while some differences in reactivity between the proteins extracted from Drosophila heads and the commercial proteins can be pointed out. Among the proteins developed on 2-D IEF/SDS-PAGE, eight proteins identified through peptide mass fingerprinting and their quantitative analysis using IAA and 13C6-IAA are summarized in Table 4.
Table 4.
Results of Quantitative Analysis of Proteins from Drosophila Heads separated by 2D IEF-SDS-PAGE
| Spot No. | Protein Candidate | Number of Cys | R2 | Inclination |
|---|---|---|---|---|
| 1 | CG3612-PA | 4 | 0.9997 | 1.0647 |
| 2 | enolase | 4 | 1.0 | 0.9467 |
| 3 | tropomyosin 1 | 1 | 0.9989 | 1.092 |
| 4 | arginine kinase | 5 | 0.9995 | 1.0457 |
| 5 | aldolase | 4 | 1.0 | 1.0143 |
| 6 | tropomyosin 2 | 3 | 0.998 | 0.9542 |
| 7 | mitochondrial porin | 0 | ND1) | ND1) |
| 8 | alcohol dehydrogenase | 2 | 0.9953 | 1.017 |
ND: not detectable
In a comparison of the observed and the theoretical ratios between IAA and 13C6-IAA-modified tryptic peptides, each analytical graph at each ratio shows reasonable linearity and inclination values. Among the eight proteins in Table 4, mitochondrial porin has no cysteine residues. Therefore as in the above results in 1D electrophoresis with commercially available proteins, no IAA- or 13C6-IAA-modified peptide peaks were detected from this protein (data not shown). As in the case of the commercial proteins purified by 1D electrophoresis, acrylamide-reacted peptides were rarely observed after this 2D electrophoresis.15
Although NEM and IAA were both sufficiently reactive and hence equally efficient modifiers for quantitative analysis of model proteins in the above experiments, we observed substantial differences in performance between these two reagents when they were applied to the 2D electrophoresis experiments on biological samples. We were able to obtain reasonable quantitative results only from tropomyosin 1 when using NEM or d5-NEM (data not shown). Although we tried to increase the reaction time of the proteins with NEM or d5-NEM to 24 hours and 48 hours, and also to raise the reaction temperature up to 37°C, these results did not change significantly. While the reason is unclear, one plausible reason is that NEM and d5-NEM are less reactive than IAA and13C6-IAA to cysteine residues to begin with. It may be pointed out that these N-alkylmaleimides are in general known to interfere with reducing reagents more than iodoacetamide derivatives.12 In other words, iodoacetamide derivatives tend to react with sulfhydryl groups more specifically. The pH conditions that are known to be suitable for reactions of cysteines and iodoacetamide are around 8–9 while those suitable for reactions of cysteines with N-alkylmaleimide are around 7. This slightly basic pH may also contribute to enhancing the reactivity of sulfhydryl group as a nucleophile, and hence explain the higher reactivity of the iodoacetamide derivatives, IAA or 13C6-IAA, than of the N-alkylmaleimide derivatives, NEM or d5-NEM. Some studies also indicate that the optimal pH for reduction of S-S bonds appears to be around 8.5–8.9,16 which would also account for the enhanced reactivity of the cysteines with IAA or 13C6-IAA. However, it is reasonable to exclude the conditions of 2D electrophoresis as a cause, because this method was successful with either combination, NEM/d5-NEM or IAA/13C6-IAA, for the commercial proteins separated by 2D electrophoresis. Another explanation may be attributed to the buffer solutions. Since some components of the lysis buffer to prepare the protein extract from Drosophila heads and the buffer to prepare commercial protein solutions are different, it is possible that the components in the buffer for the protein extracts from Drosophila heads may have interfered with the less specific N-alkylmaleimides under our experimental conditions before the N-alkylmaleimides reacted with sulfhydryl groups. It is also possible that since commercial proteins are relatively pure the protein concentration was higher than that of the proteins from Drosophila heads, while the protein extracts from Drosophila heads have many ingredients, which may also have interfered with the less specific N-alkylamides. We are examining the possible application of our method to other protein extract samples to obtain further information about its applicability.
From these results we conclude that both characterization and quantification of most proteins are possible by the use of IAA and 13C6-IAA combined with 2-D IEF/SDS-PAGE and MALDI MS.
Summary and Conclusions
We have demonstrated that the combination of isotope-labeled and unlabeled small organic molecules, NEM and d5-NEM as well as IAA and13C6-IAA, and MALDI MS with 1D or 2D electrophoresis is applicable to quantitative analysis of commercial proteins as well as proteins extracted from Drosophila heads. Ionization efficiencies of the peptides do not appear to be significantly influenced by modifications with these reagents, even though some of the peptides show slightly decreased ionization efficiency, possibly caused by their modification. Both combinations have proven to exhibit differences in the molecular mass (5Da and 6Da) sufficient to overcome overlapping problems caused by the monoisotopic peaks from the isotope-labeled compounds and the isotopic peaks from unlabeled compounds, as we had shown in similar experiments with peptides.10,11 Earlier, Sechi,17 Hamdan et al.18 and Schrattenholz et al.19 independently reported their methods for quantitative analysis of proteins separated by 1D or 2D electrophoresis applying d0- and d3-acrylamides as cysteine modifiers. Another set of small molecule cysteine modifiers, d0- and d4-vinylpyridine20 is also anticipated to be equally useful in combination with 2D electrophoresis. However, from our previous study using d3-methylmaleimide,10 we conclude that at least 5Da is necessary to sufficiently overcome this overlapping problem.21 Consisting of only three carbons, acrylamide limits possibilities for further synthetic modification to yield a greater difference in the molecular mass. Our simple synthetic manipulation offers an attractive approach for further modifying various types of modifiers.
Although recently a number of proteomic methods for quantitative analysis of proteins have been reported, most methods are based on liquid chromatography for separation, purification and observation of protein expression profiles, and proteomic methods based on the use of 2D electrophoresis as a means to obtain protein expression profiles are rather limited.22 The major reasons for the preference are the ease with which LC-based methods adjust to high-throughput systems as well as the ability of these methods to deal with a wider range of proteins than 2D electrophoresis does. However, 2D PAGE has been the most mature technique for obtaining protein expression patterns, separating thousands of proteins. As there is no single proteomic procedure useful to comprehend all the necessary data, many different technologies are required for analysis of various features of proteins for proteomics.
Our method revealed several advantageous points over LC-dependent methods. First, we did not observe any isotope effects, although they have been common and fundamental problems in LC-dependent methods such as ICAT. We conclude that the reactivities of these isotope-labeled and unlabeled modifiers are identical. Therefore, even if several cysteine residues are rather poorly reactive, they do not affect the quantitative analysis of proteins because the reactivity toward the isotope-labeled or unlabeled modifier is identical. In fact, no particular discrepancies in the ratios were observed from different cysteine residues within the same protein. We think that this outcome is due to the use of small organic molecules which are less hydrophobic and therefore more soluble in SDS-PAGE conditions. When there is more than one cysteine residue, these residues provide multiple data points for quantitative analysis, ensuring accuracy. Furthermore, we identify each protein through peptide mass fingerprinting prior to quantitative analysis, and therefore even when non-specific binding is detected between non-sulfhydryl groups and these labeling reagents, such non-specific binding can simply be ignored. We have demonstrated that even in the presence of potential non-specific binding, the results of the quantitative analysis still indicated reasonable correlation between the theoretical and observed ratios in the above experiments. This is a significant advantage over the ICAT method in which protein identification is conducted only at the end of the entire process by tandem mass spectrometry, while in our method tandem mass spectrometry is optional for confirmation of the proteins identified through peptide mass fingerprinting.
Recent comparative studies of commonly used proteomic quantitative methods indicate significant discrepancies among the quantitative data obtained by the DIGE methods and two LC-dependent methods that use cleavable ICAT or iTRAQ.23 Other studies on the shotgun approach in protein identification by microcapillary liquid chromatography-tandem mass spectrometry (μCL-MS/MS) indicate that the efficacy of digestion heavily depends on the protocols followed in this approach.24 These results also suggest the importance of an alternative approach by electrophoresis and small molecule isotope-labeling.
Therefore, depending on the types of the sample proteins that contain cysteines and the purpose of the projects, our method is anticipated to complement other existing LC-dependent methods and to be applicable to quantitative proteomics in many types of projects, its greater applicability thus making it more readily accessible to diverse laboratories. We are currently synthesizing other modifiers that are small organic molecules of which the differences of molecular mass between isotope-labeled and unlabeled versions are greater than 6Da, and investigating the possibility of expanding our method toward these reagents as well as those that modify non-cysteine-containing proteins, and further studies will be reported in due course.
Acknowledgments
We thank Dr. Nobuaki Takemori and Mr. Hisao Haniu for kindly helping with the electrophoresis, and Mr. Anil Singh for providing excellent technical assistance throughout these studies. This work is supported by grants from the National Institute of Health (EY13877, EY12190, and RR17703). S.N. thanks Texas Tech University for start-up funds.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.For example, Matsumoto H, Kurien B, Takagi Y, Kahn ES, Kinumi T, Komori N, Yamada T, Hayashi F, Isono K, Pak WL, Jackson KW, Tobin SL. Phosrestin I undergoes the earliest light-induced phosphorylation by a calcium/calmodulin-dependent protein kinase in Drosophila photoreceptors. Neuron. 1994;12:997–1010. doi: 10.1016/0896-6273(94)90309-3.Fach EM, Garulacan LA, Gao J, Xiao Q, Storm SM, Dubaquie YP, Hefta SA, Opiteck GJ. In vitro biomarker discovery for atherosclerosis by proteomics. Mol Cell Proteomics. 2004;12:120–1210. doi: 10.1074/mcp.M400160-MCP200.Bernardini G, Renzone G, Comanducci M, Mini R, Arena S, D'Ambrosio C, Bambini S, Trabalzini L, Grandi G, Martelli P, Achtman M, Scaloni A, Ratti G, Santucci A. Proteome analysis of Neisseria meningitidis serogroup A. Proteomics. 2004;10:2893–2926. doi: 10.1002/pmic.200400946.Lamanda A, Zahn A, Roder D, Langen H. Improved ruthenium II Tris (bathophenantroline disulfonate) Staining and Destaining Protocol for a better signal-to-background ratio and improved baseline resolution. Proteomics. 2004;3:599–608. doi: 10.1002/pmic.200300587.
- 2.For example, Gygi SP, Rochon Y, Franza BR, Aebersold R. Correlation between protein and mRNA abundance in yeast. Mol Cell Biol. 1999;19:1720–1730. doi: 10.1128/mcb.19.3.1720.Vuong GL, Weiss SM, Kammer W, Priemer M, Vingron M, Nordheim A, Cahill MA. Improved sensitivity proteomics by postharvest alkylation and radioactive labelling of proteins. Electrophoresis. 2000;21:2594–2605. doi: 10.1002/1522-2683(20000701)21:13<2594::AID-ELPS2594>3.0.CO;2-K.
- 3.For example, Goshe MB, Smith RD. Stable isotope-coded proteomics mass spectrometry. Curr Opin Biotechnol. 2003;14:101–109. doi: 10.1016/s0958-1669(02)00014-9.Tao WA, Aebersold R. Advances in quantitative proteomics via stable isotope tagging and mass spectrometry. Curr Opin Biotechnol. 2003;14:110–118. doi: 10.1016/s0958-1669(02)00018-6.Flory MR, Griffin TJ, Martin D, Aebersold R. Advances in quantitative proteomics using stable isotope tags. Trends Biotechnol. 2002;20:S23–S29. doi: 10.1016/s1471-1931(02)00203-3.Patterson SD. Proteomics: the industrialization of protein chemistry. Curr Opin Biotechnol. 2000;11:413–418. doi: 10.1016/s0958-1669(00)00119-1.Griffin TJ, Goodlett DR, Aebersold R. Advances in proteome analysis by mass spectrometry. Curr Opin Biotechnol. 2001;12:607–612. doi: 10.1016/s0958-1669(01)00268-3.Aebersold R, Goodlett DR. Mass spectrometry in proteomics. Chem Rev. 2001;101:269–295. doi: 10.1021/cr990076h.Hamdan M, Righetti PG. Modern strategies for protein quantification in proteome analysis: advantages and limitations. Mass Spectrom Rev. 2002;21:287–302. doi: 10.1002/mas.10032.Aebersold R, Mann M. Mass spectrometry-based proteomics. Nature. 2003;422:198–207. doi: 10.1038/nature01511.Julka S, Regnier F. Quantification in proteomics through stable isotope coding: A Review. J Proteome Res. 2004;3:350–363. doi: 10.1021/pr0340734.Broenstrup M. Absolute quantification strategies in proteomics based on mass spectrometry. Expert Review of Proteomics. 2004;1:503–512. doi: 10.1586/14789450.1.4.503.Righetti PG, Campostrini N, Pascali J, Hamdan M, Astner H. Quantitative proteomics: a review of different methodologies. Eur J Mass Spectrom. 2004;10:335–348. doi: 10.1255/ejms.600.
- 4.For example, Pasa-Tolic L, Jensen PK, Anderson GA, Lipton MS, Peden KK, Martinovic S, Tolic N, Bruce JE, Smith RD. High throughput proteome-wide measurements of protein expression using mass spectrometry. J Am Chem Soc. 1999;121:7949–7950.Oda Y, Huang K, Cross FR, Cowburn D, Chait BT. Accurate quantitation of protein expression and site-specific phosphorylation. Proc Natl Acad Sci U S A. 1999;96:6591–6596. doi: 10.1073/pnas.96.12.6591.Veenstra TV, Martinovic S, Anderson G, Pasa-Tolic L, Smith R. Proteome analysis using selective incorporation of isotopically labeled amino acids. J Am Soc Mass Spectrom. 2000;11:78–82. doi: 10.1016/S1044-0305(99)00120-8.Chen X, Smith LM, Bradbury EM. Site-specific mass tagging with stable isotope isotopes in proteins for accurate and efficient protein identification. Anal Chem. 2000;72:1134–1143. doi: 10.1021/ac9911600.Conrads TP, Alving K, Veenstra TD, Belov ME, Anderson GA, Anderson DJ, Lipton MS, Pasa-Tolic L, Udseth HR, Chrisler WB, Thrall BD, Smith RD. Quantitative analysis of bacterial and mammalian proteomes using a combination of cysteine affinity tags and 15N-metabolic labeling. Anal Chem. 2001;73:2132–2139. doi: 10.1021/ac001487x.Zhu H, Pan S, Gu S, Bradbury EM, Chen X. Amino acid residue specific stable isotope labeling for quantitative proteomics. Rapid Commun Mass Spectrom. 2002;16:2115–2123. doi: 10.1002/rcm.831.Berger SJ, Lee SW, Anderson GA, Pasa-Tolic L, Tolic N, Shen Y, Zhao R, Smith RD. High-throughput global peptide proteomic analysis by combining stable isotope amino acid labeling and data-dependent multiplexed-MS/MS. Anal Chem. 2002;74:4994–5000. doi: 10.1021/ac020105f.Gu S, Pan S, Bradbury EM, Chen X. Precise peptide sequencing and protein quantification in the human proteome through in vivo lysin-specific mass tagging. J Am Soc Mass Spectrom. 2003;14:1–7. doi: 10.1016/S1044-0305(02)00799-7.Stump MJ, Jones JJ, Fleming RC, Lay JO, Jr, Wilkins CL. Use of double-depleted 13C and 15N culture media for analysis of whole cell bacteria by MALDI time-of-flight and Fourier transform mass spectrometry. J Am Soc Mass Spectrom. 2003;14:1306–1314. doi: 10.1016/S1044-0305(03)00577-4.Wu CC, MacCoss MJ, Howell KE, Matthews DE, Yates JR., III Metabolic labeling of mammalian organisms with stable isotope for quantitative proteomics analysis. Anal Chem. 2004;76:4951–4959. doi: 10.1021/ac049208j.
- 5.Recently, Mann et al. reported a method called “stable isotope labeling by amino acid in cell culture (SILAC),” in which isotope-labeled essential amino acids are quantitatively incorporated into cell lines. Ong S-E, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, Mann M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics. 2002;1:376–386. doi: 10.1074/mcp.m200025-mcp200.
- 6.(a) Gygi SP, Rist B, Gerber SA, Turecek F, Gelb MH, Aebersold R. Quantitative analysis of complex protein mixtures, using isotope-coded affinity tags. Nat Biotechnol. 1999;17:994–999. doi: 10.1038/13690. [DOI] [PubMed] [Google Scholar]; (b) Han DK, Eng J, Zhou H, Aebersold R. Quantitative profiling of differentiation-induced microsomal proteins using isotope-coded affinity tags and mass spectrometry. Nat Biotechnol. 2001;19:946–951. doi: 10.1038/nbt1001-946. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Griffin TJ, Aebersold R. Advances in proteome analysis by mass spectrometry. J Biol Chem. 2001;276:45497–45500. doi: 10.1074/jbc.R100014200. [DOI] [PubMed] [Google Scholar]; (d) Griffin TJ, Gygi SP, Rist B, Aebersold R, Loboda A, Jilkine A, Ens W, Standing KG. Quantitative proteomic analysis using a MALDI quadrupole time-of-flight mass spectrometry. Anal Chem. 2001;73:978–986. doi: 10.1021/ac001169y. [DOI] [PubMed] [Google Scholar]; (e) Griffin TJ, Han DKM, Gygi SP, Rist B, Lee H, Aebersold R, Parker KC. Toward a high-throughput approach to quantitative proteomic analysis: expression-dependent protein identification by mass spectrometry. J Am Soc Mass Spectrom. 2001;12:1238–1246. doi: 10.1016/S1044-0305(01)00316-6. [DOI] [PubMed] [Google Scholar]; (f) Gygi SP, Rist B, Griffin TJ, Eng J, Aebersold R. Proteome analysis of low-abundance proteins using multidimensional chromatography and isotope-coded affinity tags. J Proteome Res. 2002;1:47–54. doi: 10.1021/pr015509n. [DOI] [PubMed] [Google Scholar]; (g) Ranish JA, Yi EC, Leslie DM, Purvine SO, Goodlett DR, Eng J, Aebersold R. The study of macromolecular complexes by quantitative proteomics. Nat Genet. 2003;33:349–355. doi: 10.1038/ng1101. [DOI] [PubMed] [Google Scholar]
- 7.For example, Che FY, Fricker LD. Quantitation of neuropeptides in Cpe(fat)/Cpe(fat) mice using differential isotopic tags and mass spectrometry. Anal Chem. 2002;74:3190–3198. doi: 10.1021/ac015681a.Ji J, Chakraborty A, Geng M, Zhang X, Amini A, Bina M, Regnier F. Strategy for qualitative and quantitative analysis in proteoics based on signature peptides. J Chromatogr B Biomed Sci Appl. 2000;745:197–210. doi: 10.1016/s0378-4347(00)00192-4.Geng M, Ji J, Regnier FE. Signature-peptide approach to detecting proteins in complex mixtures. J Chromatogr A. 2000;870:295–313. doi: 10.1016/s0021-9673(99)00951-6.Munchbach M, Quadroni M, Miotto G, James P. Quantitation and facilitated de novo sequencing of proteins by isotopic N-terminal labeling of peptides with a fragmentation-directing Moiety. Anal Chem. 2000;72:4047–4057. doi: 10.1021/ac000265w.Turecek F. Mass spectrometry in coupling with affinity capture-release and isotope-coded affinity tags for quantitative protein analysis. J Mass Spectrom. 2002;37:1–14. doi: 10.1002/jms.275.Thompson A, Schäfer J, Kuhn K, Kienle S, Schwarz J, Schmidt G, Neumann T, Hamon C. Tandem mass tags: a novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal Chem. 2003;75:1895–1904. doi: 10.1021/ac0262560.Hsu JL, Huang SY, Chow NH, Chen SH. Stable-isotope dimethyl labeling for quantitative proteomics. Anal Chem. 2003;75:6843–6852. doi: 10.1021/ac0348625.Shi Y, Xiang R, Crawford JK, Colangelo CM, Horvath C, Wilkins JA. A simple solid phase mass tagging approach for quantitative proteomics. J Proteome Res. 2004;3:104–111. doi: 10.1021/pr034081k.Ross PL, Huang YN, Marchese JN, Williamson B, Parker K, Hattan S, Khainovski N, Pillai S, Dey S, Daniel S, Purkayastha S, Juhasz P, Martin S, Bartlet-Jones M, He F, Jacobson A, Pappin DJ. Multiplexed protein quantitation in saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol Cell Proteomics. 2004;3:1154–1169. doi: 10.1074/mcp.M400129-MCP200.Kuyama H, Watanabe M, Toda C, Ando E, Tanaka K, Nishimura O. An approach to quantitative proteome analysis by labeling tryptophan residues. Rapid Commun Mass Spectrom. 2003;17:1642–1650. doi: 10.1002/rcm.1100.Wang S, Zhang X, Regnier FE. Quantitative proteomics strategy involving the selection of peptides containing both cysteine and histidine from tryptic digests of cell lysates. J Chromatogr A. 2002;949:153–162. doi: 10.1016/s0021-9673(01)01509-6.Weckwerth W, Willmitzer L, Fiehn O. Comparative quantification and identification of phosphoproteins using stable isotope labeling and liquid chromatography/mass spectrometry. Rapid Commun Mass Spectrom. 2000;14:1677–1681. doi: 10.1002/1097-0231(20000930)14:18<1677::AID-RCM84>3.0.CO;2-N.
- 8.For example, Zhou H, Ranish JA, Watts JD, Aebersold R. Qualitative proteome analysis by solid-phase isotope tagging and mass spectrometry. Nat Biotechnol. 2002;20:512–515. doi: 10.1038/nbt0502-512.Goshe MB, Conrads TP, Panisko EA, Angell NH, Veenstra TD, Smith RD. Phosphoprotein isotope-coded affinity tag approach for isolating and quantitating phosphopeptides in proteome-wide analyses. Anal Chem. 2001;73:2578–2586. doi: 10.1021/ac010081x.Qiu Y, Sousa EA, Hewick RM, Wang JH. Acid-labile isotope-coded extractants: a class of reagents for quantitative mass spectrometric analysis of complex protein mixtures. Anal Chem. 2002;74:4969–4979. doi: 10.1021/ac0256437.
- 9.Smolka M, Zhou H, Aebersold R. Quantitative protein profiling using two-dimensional gel electrophoresis, isotope-coded affinity tag labeling, and mass spectrometry. Mol Cell Proteomics. 2002;1:19–29. doi: 10.1074/mcp.m100013-mcp200. [DOI] [PubMed] [Google Scholar]
- 10.Niwayama S, Kurono S, Matsumoto H. Synthesis of d-labeled N-alkylmaleimides and application to quantitative peptide analysis by isotope differential mass spectrometry. Bioorg Med Chem Lett. 2001;11:2257–2261. doi: 10.1016/s0960-894x(01)00452-8. [DOI] [PubMed] [Google Scholar]
- 11.Niwayama S, Kurono S, Matsumoto H. Synthesis of 13C-labeled iodoacetanilide and application to quantitative peptide analysis by isotope differential mass spectrometry. Bioorg Med Chem Lett. 2003;13:2913–2916. doi: 10.1016/s0960-894x(03)00503-1. [DOI] [PubMed] [Google Scholar]
- 12.Getz EB, Xiao M, Chakrabarty T, Cooke R, Selvin PR. A comparison between the sulfhydryl reductants tris(2-carboxyethyl)phosphine and dithiothreitol for use in protein biochemistry. Anal Biochem. 1999;273:73–80. doi: 10.1006/abio.1999.4203. [DOI] [PubMed] [Google Scholar]
- 13.For example, Hamdan M, Bordini E, Galvani M, Righetti PG. Protein alkylation by acrylamide, its N-substituted derivatives and cross-linkers and its relevance to proteomics: a matrix assisted laser desorption/ionization-time of flight-mass spectrometry study. Electrophoresis. 2001;22:1633–44. doi: 10.1002/1522-2683(200105)22:9<1633::AID-ELPS1633>3.0.CO;2-C.Brueggemeier SB, Kron SJ, Palecek SP. Use of protein-acrylamide copolymer hydrogels for measuring protein concentration and activity. Anal Biochem. 2004;329:180–189. doi: 10.1016/j.ab.2004.02.012.Barber DS, LoPachin RM. Proteomic analysis of acrylamide-protein adduct formation in rat brain synaptosomes. Toxicol Appl Pharmacol. 2004;201:120–136. doi: 10.1016/j.taap.2004.05.008.Mineki R, Taka H, Fujimura T, Kikkawa M, Shindo N, Murayama K. In situ alkylation with acrylamimde for identification of cysteinyl residues in proteins during one- and two-dimensional sodium dodecyl sulphate-polyacrylamide gel electrophoresis. Proteomics. 2002;2:1672–1681. doi: 10.1002/1615-9861(200212)2:12<1672::AID-PROT1672>3.0.CO;2-#.
- 14.Pasquarello C, Sanchez JC, Hochstrasser DF, Corthals GF. N-t-Butyliodoacetamide and iodoacetanilide: two new cysteine alkylating reagents for relative quantitation of proteins. Rapid Commun Mass Spectrom. 2004;18:117–127. doi: 10.1002/rcm.1286. [DOI] [PubMed] [Google Scholar]
- 15.In rare cases we found cystein-containing proteins that do not react with IAA or 13C6-IAA.
- 16.Herbert B, Galvani M, Hamdan M, Olivieri E, MacCarthy J, Pedersen S, Righetti PG. Reduction and alkylation of proteins in preparation of two-dimensional map analysis: why, when, and how? Electrophoresis. 2001;22:2046–2057. doi: 10.1002/1522-2683(200106)22:10<2046::AID-ELPS2046>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 17.Sechi S. Method to identify and simultaneously determine the relative quantities of proteins isolated by gel electrophoresis. Rapid Commun Mass Spectrom. 2002;16:1416–1424. doi: 10.1002/rcm.734. [DOI] [PubMed] [Google Scholar]
- 18.Gehanne S, Cecconi D, Carboni L, Righetti PG, Domenici E, Hamdan M. Quantitative analysis of two-dimensional gel-separated proteins using isotopically marked alkylating agents and matrix-assisted laser desorption/ionization mass spectrometry. Rapid Commun Mass Spectrom. 2002;16:1692–1698. doi: 10.1002/rcm.773. [DOI] [PubMed] [Google Scholar]
- 19.Cahill MA, Wozny W, Schwall G, Schroer K, Hölzer K, Poznanovic S, Hunzinger C, Vogt JA, Stegmann W, Matthies H, Schrattenholz A. Analysis of relative isotopologue abundances for quantitative profiling of complex protein mixtures labelled with the acrylamide/D3-acrylamide alkylation tag system. Rapid Commun Mass Spectrom. 2003;17:1283–1290. doi: 10.1002/rcm.1046. [DOI] [PubMed] [Google Scholar]
- 20.Sebastiano R, Citterio A, Lapadula M, Righetti PG. A new deuterated alkylating agent for quantitative proteomics. Rapid Commun Mass Spectrom. 2003;17:2380–2386. doi: 10.1002/rcm.1206. [DOI] [PubMed] [Google Scholar]
- 21.While proteolytic O18 labeling using H2O18 is reported as a simple quantitation method, this method creates only a 2 to 4 Da difference in the molecular weight. For example, Mirgorodskaya OA, Kozmin YP, Titov MI, Körner R, Sönksen CP, Roepstorff P. Quantitation of peptides and proteins by matrix-assisted laser desorption/ionization mass spectrometry using 18O-labeled internal standards. Rapid Commun Mass Spectrom. 2000;14:1226–1232. doi: 10.1002/1097-0231(20000730)14:14<1226::AID-RCM14>3.0.CO;2-V.Yao X, Freas A, Ramirez J, Demirev PA, Fenselau C. Proteolytic 18O labeling for comparative proteomics: model studies with two serotypes of adenovirus. Anal Chem. 2001;73:2836–2842. doi: 10.1021/ac001404c.Sakai J, Kojima S, Yanagi K, Kanaoka M. 18O-Labeling quantitative proteomics using an ion trap mass spectrometer. Proteomics. 2005;5:16–23. doi: 10.1002/pmic.200300885.Stewart II, Thomson T, Figeys D. 18O Labeling: a tool for proteomics. Rapid Commun Mass Spectrom. 2001;15:2456–2465. doi: 10.1002/rcm.525.Hood BL, Lucas DA, Kim G, Chan KC, Blonder J, Issaq HJ, Veenstra TD, Conrads TP, Pollet I, Karsan A. Quantitative analysis of the low molecular weight serum proteome using 18O stable isotope labeling in a lung tumor xenograft mouse model. J Am Soc Mass Spectrom. 2005;16:1221–1230. doi: 10.1016/j.jasms.2005.02.005.
- 22.Other examples utilizing 2D electrophoresis and metabolic labeling, see Turko I, Murad F. Quantitative protein profiling in heart mitochondoria from diabetic rats. J Biol Chem. 2003;278:35844–35849. doi: 10.1074/jbc.M303139200.Vogt JA, Schroer K, Hölzer K, Hunzinger C, Klemm M, Biefang-Arndt K, Schillo S, Cahill MA, Schrattenholz A, Matthies H, Stegmann W. Protein abundance quantification in embryonic stem cells using incomplete metabolic labeling with 15N amino scids, matrix-assisted laser desorption/ionisation time-of-flight mass spectrometry, and analysis of relative isotopologue abundances of peptides. Rapid Commun Mass Spectrom. 2003;17:1273–1282. doi: 10.1002/rcm.1045.
- 23.Wu WW, Wang G, Baek SJ, Shen RF. Comparative study of three proteomic quantitative methods, DIGE, cICAT, and iTRAQ, using 2D gel-or LC-MALDI TOF/TOF. J Proteome Res. 2006;5:651–658. doi: 10.1021/pr050405o. [DOI] [PubMed] [Google Scholar]
- 24.Klammer AA, MacCoss MJ. Effects of modified digestion schemes on the identification of proteins from complex mixtures. J Proteome Res. 2006;5:695–700. doi: 10.1021/pr050315j. [DOI] [PMC free article] [PubMed] [Google Scholar]