

Abstract
Chemoprevention involves the use of natural or synthetic substances to reduce the risk of developing cancer. Strategies for protecting cells from initiation events include decreasing metabolic enzymes responsible for generating reactive species (phase I enzymes) while increasing phase II enzymes that can deactivate radicals and electrophiles known to intercede in normal cellular processes. Reduction of electrophilic quinones by quinone reductase is an important detoxification pathway. Following evaluation of approximately 3000 plant and marine organism extracts, the number characterized as “active” was established in the range of 12% of the total, and over 60 active compounds have been isolated as quinone reductase inducers. One of them, isoliquiritigenin (1), isolated from tonka bean, was shown to be a monofunctional inducer by having similar quinone reductase inducing ability in wild-type Hepa 1c1c7 cells and two mutant cell lines. To further investigate the mechanism of induction, HepG2 human hepatoma cells stably transfected with ARE-luciferase plasmid were used. Isoliquiritigenin (1) significantly induced the luciferase activity in a dose-dependent manner. On the basis of these results, a full-term cancer chemoprevention study was conducted with 7,12-dimethylbenz[a]anthracene (DMBA)-treated female Sprague-Dawley rats. Dietary administration of 1 increased tumor latency. Based on these promising preliminary results, additional mechanistic studies are underway, as well as full-term carcinogenesis studies with chronic administration schedules.
Introduction
Cancer chemoprevention involves prevention, delay, or reversal of the process of carcinogenesis through ingestion of dietary or pharmaceutical agents.1,2 Recent advances that have defined the cellular and molecular events associated with carcinogenesis, along with a growing body of experimental, epidemiological, and clinical trial data, provide a foundation for relatively new strategies of cancer prevention.3,4 One such strategy involves suppression of carcinogen metabolic activation or blocking the formation of ultimate carcinogens.5 In particular, the induction of phase II enzymes can offer protection against toxic and reactive chemical species.6 Many recent studies have shown that elevation of phase II enzymes, such as NAD(P)H:quinone reductase (QR) and GST, correlates with protection against chemical-induced carcinogenesis in animal models,7,8 in the stage of promotion9 as well as initiation.7
In our program for the procurement of novel plant-derived chemotherapeutic/chemopreventive agents, induction of QR has been used as one marker of activity.10 This led to the identification of isoliquiritigenin (1) as an inducer of quinone reductase.11 The evaluation of 1 in a mouse mammary organ culture (MMOC) assay, which is used as a secondary discriminator, also exhibited a significant response.11 Here, the effect of 1 was evaluated in cultured Hepa 1c1c7 murine hepatoma cells and two mutants thereof.12 Furthermore, we have analyzed the mechanism of the observed enzyme induction in HepG2 cells stably transfected with ARE. Finally, the chemopreventative potential was investigated in a full-term carcinogenesis study.
Quinone Reductase Induction and Cancer Chemoprevention
Carcinogenesis is a complex and protracted multistage process, yet the entire course can be initiated by a single event wherein a cellular macromolecule is damaged by an endogenous or exogenous agent. Strategies for protecting cells from these initiating events include decreasing metabolic enzymes responsible for generating reactive species (phase I enzymes) while increasing phase II enzymes that can deactivate radicals and electrophiles known to intercede in normal cellular processes. Reduction of electrophilic quinones by QR is an important detoxification pathway, which converts quinones to hydroquinones and reduces oxidative cycling.13,14 Enzyme inducers are of two types: monofunctional and bifunctional.15 Bifunctional inducers increase phase II enzymes as well as phase I enzymes, such as aryl hydrocarbon hydroxylase, and bind with high affinity to the aryl hydrocarbon (Ah) receptor-xenobiotic response element (XRE).16,17 Monofunctional inducers induce phase II enzymes selectively and will activate the antioxidant response element (ARE) through Keap1 and Nrf2.18,19 Since phase I enzymes can activate procarcinogens to their ultimate reactive species, monofunctional agents that induce phase II enzymes selectively would theoretically appear to be more desirable candidates for cancer chemoprevention.20 In addition, selective phase II enzyme inducers would be anticipated to serve as anticarcinogens early in the process of carcinogenesis, but it has been established that inhibition of carcinogenesis at later stages is also possible.9
QR elevation with in vitro and in vivo systems has been shown to correlate with induction of other protective phase II enzymes and provides a reasonable biomarker for the potential chemoprotective effect of test agents against cancer initiation.21 The murine hepatoma cell line Hepa 1c1c7 contains easily measurable inducible QR that provides a reliable, high-throughput system for detecting inducers of phase II enzymes.22 This assay can also be used to determine if an agent is monofunctional or bifunctional. This is accomplished by comparing the induction capability of a compound in wild-type Hepa 1c1c7 cells with that observed in two mutant cell lines designated TAOc1BPrc1 and BPrc1, which are defective in a functional Ah receptor or unable to translocate the receptor–ligand complex to the nucleus, respectively.23 Compounds that have similar inducing ability in the wild-type and mutant Hepa lines are considered monofunctional inducers.
Screening of Plants and Marine Organisms
We have procured plant and marine organism materials from throughout the world for investigation of natural inhibitors of carcinogenesis. The overall experimental approach for obtaining natural product anticarcinogens from these plants and marine organisms has been described in detail.24–29 Crude nonpolar and polar extracts, prepared from each plant or marine organism obtained, are evaluated for their potential chemopreventive activity using a battery of short-term in vitro bioassays developed to monitor inhibition of tumorigenesis at the various stages.30 On the basis of the results of these bioassays, selected extracts are further evaluated in a MMOC model. In this assay, test materials are evaluated for their ability to inhibit 7,12-dimethylbenz(a)anthracene (DMBA)-induced preneoplastic lesions.31 One of the assays used to study the initiation stage is the induction of QR activity.7 In the next stage, extracts showing potency in the in vitro bioassays are selected for bioassay-guided fractionation to uncover their active principles. Pure active compounds are then evaluated in the QR assay, and selected compounds are further processed for evaluation in the MMOC model. Finally, the in vivo cancer chemopreventive activity of highly promising pure plant or marine organism constituents is evaluated in animal full-term tumorigenesis models, including the two-stage mouse skin model using DMBA as an initiator and 12-O-tetradecanoylphorbol-13-acetate (TPA) as a promoter and the rat mammary carcinogenesis model with N-methyl-N-nitrosourea (MNU) or DMBA as a carcinogen.32,33 Additional in vivo models are used as required.
To date, we have evaluated 2675 plant extracts and 528 marine organism extracts in the QR induction assay system by using cultured Hepa 1c1c7 cells. Of these, 191 plant extracts (7.1%) and 98 marine organism extracts (18.6%) showed QR induction activity. When concentration to double activity (CD) values of extracts are below 10 μg/mL, they are considered as active leads. Many have been discussed in various review articles.10,24 Numerous epidemiological studies, together with data from in vivo and in vitro experiments, have shown that vegetables, especially cruciferous vegetables, have an important role in protection against various cancers.34–36 Many cruciferous vegetables such as cabbage, broccoli, Brussels sprouts, watercress, and cauliflower induce phase II enzymes.37 Active compounds isolated from these vegetables include glucosinolates, sulforaphane, indole-3-carbinol, and brassinin.38–41 In garlic and onion, the organosulfur compounds induce phase II detoxification enzymes and seem to be responsible for the chemoprotective action.42 Recently, curcumin, a yellow pigment of turmeric, was reported to induce phase II detoxification enzymes, while inhibiting procarcinogen activating phase I enzymes, such as cytochrome P4501A1.43 In tomatillo, an ingredient used in Latin America for salsas, over 30 withanolides were isolated and showed potent QR induction.44 Some medicinal plants such as Tephrosia purpurea and species of the genus Renealmia were also found to be active.45,46 In total, over 60 active compounds have been isolated as QR inducers, and substantial molecular diversity has been observed. These active compounds comprise ceramides, terpenoids, withanolides, flavonoids, chalcones, alkaloids, and diarylheptanoids. In addition to the active phytochemicals, semi-synthetic or synthetic derivatives of bioactive compounds can provide more promising lead compounds. For example, the flavonoid 4′-bromoflavone was found to be an extremely potent inducer of QR and an effective cancer chemopreventive agent.7
Cancer Chemopreventive Potential of Isoliquiritigenin (1)
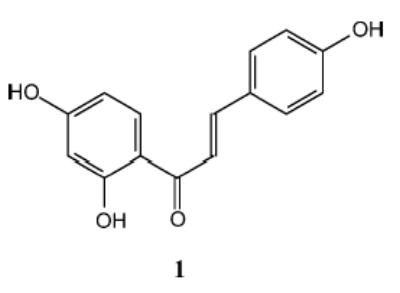
In our search for novel cancer chemopreventive agents, 1, a compound isolated from Dipteryx odorata (Aubl.) Willd. (tonka been), but also present in licorice and shallots, was found to significantly induce QR activity in Hepa 1c1c7 cells (CD: 2 μM) and exhibited a significant response in a carcinogen-treated MMOC assay (76% inhibition at 10 μg/mL).11 In addition, 1 inhibited azoxymethane (AOM)-induced murine colon carcinogenesis and AOM-induced murine colon aberrant crypt focus formation.47 This compound has also been found to suppress metastasis in a pulmonary metastasis model of mouse renal cell carcinoma and to prevent severe 5-fluorouracil-induced leukocytopenia in this model.48
In this study, we tested the potential of 1 to induce QR activity in Hepa 1c1c7 cells and two mutant cell lines using the method described previously.10 Compound 1 induced QR activity in a dose-dependent manner in the concentration range of 2–30 μM with a maximum of 7-fold induction at the highest concentration tested. As summarized in Table 1, CD values obtained with 1 were similar between the wild-type and the mutant cell lines, indicative of a monofunctional induction pattern. Compound 1 is thus lacking phase I enzyme-inducing properties and is devoid of cytochrome P450-activating properties. Induction profiles of two well-known inducers, sulforaphane, a monofunctional inducer, and 4′-bromoflavone (4′BF), a bifunctional inducer, are also shown.
Table 1.
Effect of Isoliquiritigenin (1) on QR Activity in Hepa 1c1c7 Hepatoma Cells and Hepa 1c1c7 Mutants
| isoliquiritigenin (1)
|
4′-bromoflavone
|
sulforaphane
|
|||||||
|---|---|---|---|---|---|---|---|---|---|
| cell line | CDa (μM) | IC50b (μM) | CIc | CDa (μM) | IC50b (μM) | CIc | CDa (μM) | IC50b (μM) | CIc |
| Hepa 1c1c7 | 1.80 ± 0.44 | 19.6 ± 2.5 | 11 | 0.023 ± 0.010 | >62.5 | >2700 | 0.30 ± 0.04 | 6.3 ± 0.9 | 21 |
| BPrc1 | 9.91 ± 0.87 | 16.2 ± 3.8 | 2 | >250 | >62.5 | NAd | 0.49 ± 0.13 | 7.6 ± 0.3 | 16 |
| TAOc1BPrc1 | 2.22 ± 0.36 | 22.0 ± 3.0 | 10 | >250 | >62.5 | NAd | 0.33 ± 0.07 | 5.9 ± 0.0 | 18 |
Mean value of the concentration required to double the specific activity of QR ± SD (n = 2).
Mean value of the half-maximal inhibitory concentration of cell viability ± SD (n = 2).
Chemoprevention index: ratio between IC50 and CD.
NA, not applicable.
To further investigate the mechanism of induction, HepG2 human hepatoma cells stably transfected with ARE-luciferase plasmid were used.27 As shown in Figure 1, treatment with 1 (7.5–30 μM) significantly induced luciferase expression via interaction with ARE in a dose-dependent manner and did not show any cytotoxicity. The positive controls 4′BF (10 μM) and sulforaphane (10 μM) also induced the luciferase expression.
Figure 1.
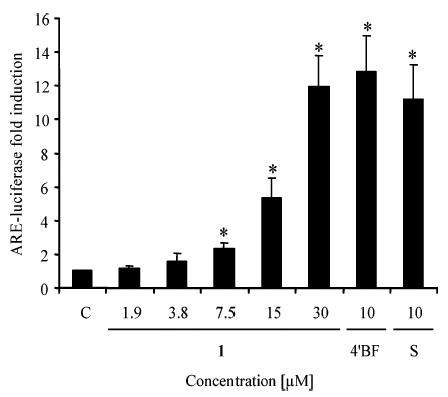
Isoliquiritigenin (1) induces the luciferase activity in HepG2 cells stably transfected with an ARE-luciferase plasmid. Transfected cells were treated with 1.9–30 μM isoliquiritigenin (1), 10 μM 4′-bromoflavone (4′BF), 10 μM sulforaphane (S), or DMSO (0.5% final concentration) as control (C) and then analyzed for chemiluminescence using the luciferase assay system from Promega. Results are shown as a fold induction relative to the level observed in the control. Results are the means of three determinations ± SD. *Significantly different from control values, determined by Student’s t-test (p < 0.05).
Finally, 1 was evaluated in the DMBA-induced rat mammary tumorigenesis model as described previously.7 To determine the doses to use, a MTD was performed using 1000, 2000, 3000, 4000, 5000, and 20 000 mg/kg diet, and no toxicity was observed, based on body weight change and necropsy. As shown in Figure 2A, administration of 1 (5000 mg/kg diet) increased tumor latency in Sprague-Dawley rats, but had little effect on the incidence of mammary tumors after 120 days (Figure 2B). There were no significant differences in body weight (Figure 2C) between the different groups. On the basis of these preliminary results, additional mechanistic studies are underway, as well as full-term carcinogenesis studies with chronic administration schedules.
Figure 2.

Effect of dietary isoliquiritigenin (1) on percent incidence of observable mammary tumors (A), number of tumors (B), and body weight (C). Female Sprague-Dawley rats were given a single i.g. dose of 7,12-dimethylbenz(a)anthracene (DMBA) on day 0. The rat treatment groups were (○) DMBA in sesame oil; (■) DMBA and 2500 mg/kg diet of isoliquiritigenin; (▲) DMBA and 5000 mg/kg diet of isoliquiritigenin; and (×) 5000 mg/kg diet of isoliquiritigenin. Isoliquiritigenin was included in the diet during the period of 7 days prior to DMBA administration (−7) to 7 days post-DMBA administration (+7). During the remainder of the experimental period, unsupplemented diet was given to the animals (20 rats/group).
Conclusions
Induction of the phase II detoxification enzymes such as QR is a useful strategy for cancer chemoprevention. Many edible plants have been found to contain cancer chemopreventive agents capable of inducing phase II enzymes. Notably, since some of the lead compounds are found in vegetables, administration of cancer chemopreventive agents through the diet may be viewed as a convenient and effective strategy in cancer prevention.
Acknowledgments
This work was supported by program project P01 CA48112 funded by the National Cancer Institute, NIH, Bethesda, MD.
Footnotes
Dedicated to Dr. Norman R. Farnsworth of the University of Illinois at Chicago for his pioneering work on bioactive natural products.
References and Notes
- 1.Sporn MB, Newton DL. Fed Proc. 1979;38:2528–2534. [PubMed] [Google Scholar]
- 2.Hong WK, Sporn MB. Science. 1997;278:1073–1077. doi: 10.1126/science.278.5340.1073. [DOI] [PubMed] [Google Scholar]
- 3.Kelloff GJ, Sigman CC, Greenwald P. Eur J Cancer. 1999;35:1755–1762. doi: 10.1016/s0959-8049(99)00164-1. [DOI] [PubMed] [Google Scholar]
- 4.Kensler TW. Environ Health Perspect. 1997;105(Suppl 4):965–970. doi: 10.1289/ehp.97105s4965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wattenberg LW. Cancer Res. 1985;45:1–8. [PubMed] [Google Scholar]
- 6.Begleiter A, Leith MK, Curphey TJ, Doherty GP. Oncol Res. 1997;9:371–382. [PubMed] [Google Scholar]
- 7.Song LL, Kosmeder JW, II, Lee SK, Gerhäuser C, Lantvit D, Moon RC, Moriarty RM, Pezzuto JM. Cancer Res. 1999;59:578–585. [PubMed] [Google Scholar]
- 8.Boone CW, Steele VE, Kelloff GJ. Mutat Res. 1992;267:251–255. doi: 10.1016/0027-5107(92)90069-e. [DOI] [PubMed] [Google Scholar]
- 9.Gills JJ, Jeffery EH, Matusheski NV, Moon RC, Lantvit DD, Pezzuto JM. Cancer Lett. 2005 doi: 10.1016/j.canlet.2005.05.007. in press. [DOI] [PubMed] [Google Scholar]
- 10.Kang YH, Pezzuto JM. Methods Enzymol. 2004;382:380–414. doi: 10.1016/S0076-6879(04)82021-4. [DOI] [PubMed] [Google Scholar]
- 11.Jang DS, Park EJ, Hawthorne ME, Vigo JS, Graham JG, Cabieses F, Santarsiero BD, Mesecar AD, Fong HH, Mehta RG, Pezzuto JM, Kinghorn AD. J Nat Prod. 2003;66:583–587. doi: 10.1021/np020522n. [DOI] [PubMed] [Google Scholar]
- 12.De Long MJ, Santamaria AB, Talalay P. Carcinogenesis. 1987;8:1549–1553. doi: 10.1093/carcin/8.10.1549. [DOI] [PubMed] [Google Scholar]
- 13.Ross D, Kepa JK, Winski SL, Beall HD, Anwar A, Siegel D. Chem Biol Interact. 2000;129:77–97. doi: 10.1016/s0009-2797(00)00199-x. [DOI] [PubMed] [Google Scholar]
- 14.Talalay P, De Long MJ, Prochaska HJ. Proc Natl Acad Sci USA. 1988;85:8261–8265. doi: 10.1073/pnas.85.21.8261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Prochaska HJ, Talalay P. Cancer Res. 1988;48:4776–4782. [PubMed] [Google Scholar]
- 16.Sogawa K, Fujii-Kuriyama Y. J Biochem (Tokyo) 1997;122:1075–1079. doi: 10.1093/oxfordjournals.jbchem.a021864. [DOI] [PubMed] [Google Scholar]
- 17.Yang CS, Smith TJ, Hong JY. Cancer Res. 1994;54:1982s–1986s. [PubMed] [Google Scholar]
- 18.Dinkova-Kostova AT, Holtzclaw WD, Cole RN, Itoh K, Wakabayashi N, Katoh Y, Yamamoto M, Talalay P. Proc Natl Acad Sci USA. 2002;99:11908–11913. doi: 10.1073/pnas.172398899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eggler AL, Liu G, Pezzuto JM, van Breemen RB, Mesecar AD. Proc Natl Acad Sci USA. 2005;102:10070–10075. doi: 10.1073/pnas.0502402102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Talalay P. Biofactors. 2000;12:5–11. doi: 10.1002/biof.5520120102. [DOI] [PubMed] [Google Scholar]
- 21.Pezzuto JM. In: In Recent Advances in Phytochemistry, Phytochemistry of Medicinal Plants. Arnason J, Mata R, Romeo J, editors. Vol. 29. Plenum Press; New York: 1995. pp. 19–44. [Google Scholar]
- 22.Prochaska HJ, Santamaria AB. Anal Biochem. 1988;169:328–336. doi: 10.1016/0003-2697(88)90292-8. [DOI] [PubMed] [Google Scholar]
- 23.Gerhäuser C, Lee SK, Kosmeder JW, Moriarty RM, Hamel E, Mehta RG, Moon RC, Pezzuto JM. Cancer Res. 1997;57:3429–3435. [PubMed] [Google Scholar]
- 24.Kinghorn AD, Su BN, Jang DS, Chang LC, Lee D, Gu JQ, Carcache-Blanco EJ, Pawlus AD, Lee SK, Park EJ, Cuendet M, Gills JJ, Bhat K, Park HS, Mata-Greenwood E, Song LL, Jang M, Pezzuto JM. Planta Med. 2004;70:691–705. doi: 10.1055/s-2004-827198. [DOI] [PubMed] [Google Scholar]
- 25.Mehta RG, Pezzuto JM. Curr Oncol Rep. 2002;4:478–486. doi: 10.1007/s11912-002-0059-2. [DOI] [PubMed] [Google Scholar]
- 26.Pezzuto JM. Biochem Pharmacol. 1997;53:121–133. doi: 10.1016/s0006-2952(96)00654-5. [DOI] [PubMed] [Google Scholar]
- 27.Pezzuto JM, Kosmeder JW, Park EJ, Lee SK, Cuendet M, Gills JJ, Bhat K, Grubjesic S, Park HS, Mata-Greenwood E, Tan YM, Yu R, Lantvit DD, Kinghorn AD. In: In Cancer Chemoprevention, Volume 2: Strategies for Cancer Chemoprevention. Kelloff GJ, Hawk ET, Sigman CC, editors. Vol. 2. Humana Press Inc; Totowa, NJ: 2005. pp. 3–37. [Google Scholar]
- 28.Jensen P, Fenical W. Annu Rev Microbiol. 1994;48:559–584. doi: 10.1146/annurev.mi.48.100194.003015. [DOI] [PubMed] [Google Scholar]
- 29.Mincer TJ, Jensen P, Kauffman CA, Fenical W. Appl Envir Microbiol. 2002;68:5005–5011. doi: 10.1128/AEM.68.10.5005-5011.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pezzuto JM, Angerhofer CK, Mehdi H. Stud Nat Prod Chem. 1998;20:507–560. [Google Scholar]
- 31.Mehta RG, Hawthorne ME, Steele VE. Methods Cell Sci. 1997;19:19–24. [Google Scholar]
- 32.Jang M, Cai L, Udeani GO, Slowing KV, Thomas CF, Beecher CW, Fong HH, Farnsworth NR, Kinghorn AD, Mehta RG, Moon RC, Pezzuto JM. Science. 1997;275:218–220. doi: 10.1126/science.275.5297.218. [DOI] [PubMed] [Google Scholar]
- 33.Udeani GO, Gerhäuser C, Thomas CF, Moon RC, Kosmeder JW, Kinghorn AD, Moriarty RM, Pezzuto JM. Cancer Res. 1997;57:3424–3428. [PubMed] [Google Scholar]
- 34.Go VL, Wong DA, Resnick MS, Heber D. J Nutr. 2001;131:179S–180S. doi: 10.1093/jn/131.1.179S. [DOI] [PubMed] [Google Scholar]
- 35.Park EJ, Pezzuto JM. Cancer Metastasis Rev. 2002;21:231–255. doi: 10.1023/a:1021254725842. [DOI] [PubMed] [Google Scholar]
- 36.Van Duyn MA, Pivonka E. J Am Diet Assoc. 2000;100:1511–1521. doi: 10.1016/S0002-8223(00)00420-X. [DOI] [PubMed] [Google Scholar]
- 37.Talalay P, Zhang Y. Biochem Soc Trans. 1996;24:806–810. doi: 10.1042/bst0240806. [DOI] [PubMed] [Google Scholar]
- 38.Nho CW, Jeffery E. Toxicol Appl Pharmacol. 2001;174:146–152. doi: 10.1006/taap.2001.9207. [DOI] [PubMed] [Google Scholar]
- 39.Zhang Y, Talalay P, Cho CG, Posner GH. Proc Natl Acad Sci USA. 1992;89:2399–2403. doi: 10.1073/pnas.89.6.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Keck AS, Staack R, Jeffery EH. Nutr Cancer. 2002;42:233–240. doi: 10.1207/S15327914NC422_13. [DOI] [PubMed] [Google Scholar]
- 41.Rushmore TH, Kong AN. Curr Drug Metab. 2002;3:481–490. doi: 10.2174/1389200023337171. [DOI] [PubMed] [Google Scholar]
- 42.Singh SV, Pan SS, Srivastava SK, Xia H, Hu X, Zaren HA, Orchard JL. Biochem Biophys Res Commun. 1998;244:917–920. doi: 10.1006/bbrc.1998.8352. [DOI] [PubMed] [Google Scholar]
- 43.Khafif A, Schantz SP, Chou TC, Edelstein D, Sacks PG. Carcinogenesis. 1998;19:419–424. doi: 10.1093/carcin/19.3.419. [DOI] [PubMed] [Google Scholar]
- 44.Misico RI, Song LL, Veleiro AS, Cirigliano AM, Tettamanzi MC, Burton G, Bonetto GM, Nicotra VE, Silva GL, Gil RR, Oberti JC, Kinghorn AD, Pezzuto JM. J Nat Prod. 2002;65:677–680. doi: 10.1021/np0106337. [DOI] [PubMed] [Google Scholar]
- 45.Chang LC, Gerhauser C, Song L, Farnsworth NR, Pezzuto JM, Kinghorn AD. J Nat Prod. 1997;60:869–873. doi: 10.1021/np970236p. [DOI] [PubMed] [Google Scholar]
- 46.Gu JQ, Park EJ, Vigo JS, Graham JG, Fong HH, Pezzuto JM, Kinghorn AD. J Nat Prod. 2002;65:1616–1620. doi: 10.1021/np020249p. [DOI] [PubMed] [Google Scholar]
- 47.Baba M, Asano R, Takigami I, Takahashi T, Ohmura M, Okada Y, Sugimoto H, Arika T, Nishino H, Okuyama T. Biol Pharm Bull. 2002;25:247–250. doi: 10.1248/bpb.25.247. [DOI] [PubMed] [Google Scholar]
- 48.Yamazaki S, Morita T, Endo H, Hamamoto T, Baba M, Joichi Y, Kaneko S, Okada Y, Okuyama T, Nishino H, Tokue A. Cancer Lett. 2002;183:23–30. doi: 10.1016/s0304-3835(02)00113-1. [DOI] [PubMed] [Google Scholar]