

Abstract
The c-kit gene encodes a tyrosine kinase receptor (KIT) that is required in normal spermatogenesis and is expressed in seminomas and dysgerminomas, a subset of human germ cell tumors (GCTs). To determine whether activating mutations of the c-kit gene occur in GCTs, primary tissue samples of 33 testicular and ovarian tumors were examined for mutations in the juxtamembrane and phosphotransferase domains by polymerase chain reaction amplification and DNA sequencing. A novel missense mutation (D816H) was found in the phosphotransferase domain in tumors of seminoma/dysgerminoma differentiation. The c-kit alleles in nonneoplastic tissues from these patients were wild type, suggesting that the mutant alleles were acquired and selected for during malignant transformation. In cell transfection experiments, the D816H mutant protein was a constitutively activated kinase and was constitutively phosphorylated on tyrosine residues. This is the first description of an activating c-kit mutation in GCTs and is evidence that the KIT signal transduction pathway is important in the pathogenesis of neoplasms with seminoma differentiation.
The c-kit protooncogene encodes a type III transmembrane tyrosine kinase receptor (KIT). Upon binding of the ligand stem cell factor (SCF), KIT dimerizes, is phosphorylated, and initiates a signaling cascade that induces cell growth. 1 KIT is expressed in a number of cell types during development, as well as in a subset of malignant neoplasms. Gene mutations that cause constitutive activation of KIT have been found in human mast cell disease 2,3 and gastrointestinal stromal tumors, 4,5 and these mutant genes induce cell transformation in vivo. SCF/KIT-mediated signaling is known to play a crucial role in normal spermatogenesis, 6 and KIT has been shown to be expressed in some human germ cell tumors (GCTs). 7,8 GCTs are a heterogeneous group of neoplasms derived from primordial germ cells and are broadly divided into seminoma/dysgerminoma and nonseminoma/nondysgerminoma categories. It is the former category of tumors that is known to express the KIT receptor. To determine whether gain of function mutations is involved in the pathogenesis of these tumors, we screened the DNA sequences of c-kit from archival specimens of primary human GCTs and characterized the kinase activity and phosphorylation status of KIT proteins found to be mutated in these neoplasms.
Materials and Methods
Tissues
Hematoxylin and eosin-stained slides and formalin-fixed paraffin-embedded blocks were retrieved from the files of the division of Surgical Pathology at the University of Virginia Health Sciences Center. All cases were reviewed and categorized according to World Health Organization criteria for the classification of GCTs. 9
DNA Extraction
Histologic sections (7 μm) were stained with hematoxylin and eosin and rehydrated in a buffer solution containing 5% glycerol, as described previously. 10 Tumor and benign tissues were dissected separately, with a scalpel under direct microscopic visualization. Microdissected tumor samples were collected that contained as few nonneoplastic cells as possible (70–90% tumor cellularity). The cells were digested with proteinase K, treated with Chelex resin, and subjected to heat inactivation as described previously. 10
Polymerase Chain Reactions
Polymerase chain reaction (PCR) primers were designed to amplify exons 11 and 17 of the c-kit gene (GDB: 120117), which have been shown to harbor the vast majority of activating mutations in previous studies. 1 The PCR product lengths are 257 bp and 220 bp, respectively. The primer sequences for exon 11 anneal within flanking introns: 5′-ATTATTAAAAGGTGATCTATTTTTC-3′ (forward), 5′-ACTGTTATGTGTACCCAAAAAG-3′ (reverse).
The primer sequences for exon 17 anneal within flanking introns: 5′-TTCACTCTTTACAAGTTAAAATG-3′ (forward), 5′-GGACTGTCAAGCAGAGAATG-3′ (reverse).
PCR was carried out with the following conditions in a thermocycler (Touchdown; Hybaid, Ltd.): 50-μl total reaction volume (67 mM Tris-HCl (pH 8.8), 16 mM (NH4)2SO4, 10 mM β-mercaptoethanol, 0.1 mg/ml acetylated bovine serum albumin, 2 mM MgCl2, 0.4 mM deoxynucleoside triphosphates, 1 μM primers, 10% dimethyl sulfoxide). Fifty to one hundred equivalents of genomic DNA was used per reaction. Cycling conditions were as follows: 98°C for 2 minutes; hold temperature at 78°C, at which time 2.5 units Taq polymerase (Gibco BRL) was added; then 40 cycles at 95°C for 30 seconds, 55°C for 30 seconds, 72°C for 30 seconds, followed by 1 cycle at 72°C for 5 minutes. A negative control (no DNA) was included with each PCR reaction run to monitor for contamination. PCR products were visualized after electrophoresis in 2% agarose before sequence analysis.
DNA Sequencing
PCR products were prepared for cycle sequencing by the addition of 1 μl of 10 μ/μl Exonuclease I (USB/Amersham Life Sciences) at 37°C incubation for 15 minutes, followed by the addition of 5 μl of 1 μ/μl shrimp alkaline phosphatase (Boehringer Mannheim) and 37°C incubation for 30 minutes, followed by 80°C incubation for 15 minutes. The PCR products were then sequenced using a 32P-end-labeled primer and the EXCEL II cycle sequencing kit (Epicentre Technologies), by the protocol supplied by the manufacturer. The sequencing primers used were 5′-TGTGTACCCAAAAAGGTGACATGG-3′ (reverse intron sequence for exon 11) and 5′-ATGGTTTTCTTTTCTCCTCCAACCT-3′ (forward intron sequence for exon 17). Cycling conditions were as follows: 30 cycles of 30 seconds at 94°C, 30 seconds at 55°C, 1 minute at 70°C.
Verification of Mutations
All mutations were confirmed by a second independent round of tissue microdissection, PCR, and cycle sequencing. Samples of normal tissue were subjected to PCR and sequencing to determine whether changes in the DNA sequence detected in tumors were germline or somatic.
KIT Expression Vectors
The complete coding sequence of the human c-kit gene was excised as a BamH1-Xmn1 fragment (bases 1–3192), eliminating most of the 3′ untranslated region from a larger cDNA clone. 11 This fragment was ligated into BamH1/Sma1 restricted eukaryotic expression vector pJ3Ω (American Type Culture Collection no. 37719). The D816V and D816H mutations were introduced by site-directed mutagenesis using the QuikChange kit (Stratagene) and mutagenic oligonucleotides. The introduced mutations were confirmed by sequence analysis.
Cell Transfection and Protein Extract Preparation
COS cells were transfected with the KIT expression plasmids, using diethylaminoethyl dextran. After 72 hours of culture, transfected cells were solubilized with lysis buffer, per published protocols. 12
Immunoprecipitation and Western Blot Procedure
These procedures were carried out per published protocols. 12 KIT immunoprecipitation was performed with 2 μg polyclonal anti-KIT antibody directed against amino acid residues 958–976 at the carboxy terminal domain of human KIT (p145; Research Diagnostics) per 500-μl protein extract. Immunoprecipitated proteins were separated by 7.5% sodium dodecyl sulfate polyacrylamide gel electrophoresis, electroblotted onto nitrocellulose membranes, and probed with either the p145 polyclonal anti-KIT antibody (0.5 μg/ml) and protein A/horseradish peroxidase conjugate (Amersham) or with an anti-phosphotyrosine antibody/horseradish peroxidase conjugate (clone RC20; Transduction Laboratories; 1:2500 dilution of 250 μg/ml stock). Immunoblots were developed by chemiluminescence.
Kinase Assay
Protein extracts were incubated with 20 μCi [γ-32P]ATP and processed as previously described, 13 with the exception that protein gels were electroblotted onto nitrocellulose membranes, and the KOH wash was omitted.
Immunohistochemistry
Histological sections of GCTs were stained with the p145 polyclonal antibody raised against the C-terminus of KIT, by methods previously described. 5
Results
A Novel Human c-kit Mutation (D816H) Is Found in Germ Cell Tumors
Thirty-three human GCTs (22 testicular, nine ovarian, two extragonadal), which included 17 seminomas/dysgerminomas, 10 nonseminomas/nondysgerminomas, and six tumors with a mixture of these components, were evaluated for mutations in exons 11 and 17 of c-kit. Mutations in exon 17, corresponding to the phosphotransferase protein domain, were found in two GCTs (a seminoma and a mixed ovarian dysgerminoma/yolk sac tumor). Both tumors showed a G-to-C substitution at nucleotide 2467, causing a change from aspartic acid to histidine at amino acid position 816 (Figure 1) ▶ . In both tumors, the wild-type sequence was retained in proportions similar to those of the mutant sequence, consistent with retention of the other wild-type allele in the tumor cells. The ovarian mixed GCT had the mutation in each tumor component. Corresponding nonneoplastic tissues from these two patients demonstrated only wild-type c-kit sequences, consistent with somatic acquisition of the c-kit gene mutation during neoplastic transformation of precursor cells.
Figure 1.

Autoradiographs of DNA sequencing. Cycle sequencing of c-kit PT domain PCR amplified products from GCTs (designated as T) and from nonneoplastic tissue (designated as N) are shown. In nonneoplastic tissue only a wild-type sequence is present; in tumor samples mutant and wild-type sequences are coexistent, consistent with retention of one wild-type allele in the tumor cells. In all tumor samples there is a G-to-C substitution at position 2467 (solid arrows), resulting in an amino acid coding substitution (D816H). A: The two components of a mixed ovarian GCT, a yolk sac tumor component (designated as T1) and a dysgerminoma component (designated as T2), were sampled separately. Both tumor components contain the G-to-C substitution resulting in the D816H mutation (solid arrow). In addition, there is a C-to-T substitution at nucleotide position 2469 (open arrow), which does not result in a change in amino acid coding. B: Results from a seminoma showing the G-to-C substitution at position 2467 that results in the D816H amino acid missense mutation.
D816H KIT Is Constitutively Phosphorylated on Tyrosine Residues and Has Constitutive Kinase Activity
To characterize the biochemical properties of the mutated gene product in GCTs, the D816H mutation was introduced into a KIT expression vector and transiently transfected into COS cells. After cell lysis and immunoprecipitation with anti-KIT antibodies, an in vitro kinase assay showed that the D816H mutant gene product had at least the same level of constitutive activity as the well-characterized D816V mutant (Figure 2A) ▶ . In contrast, wild-type KIT showed no appreciable kinase activity in the absence of ligand stimulation.
Figure 2.
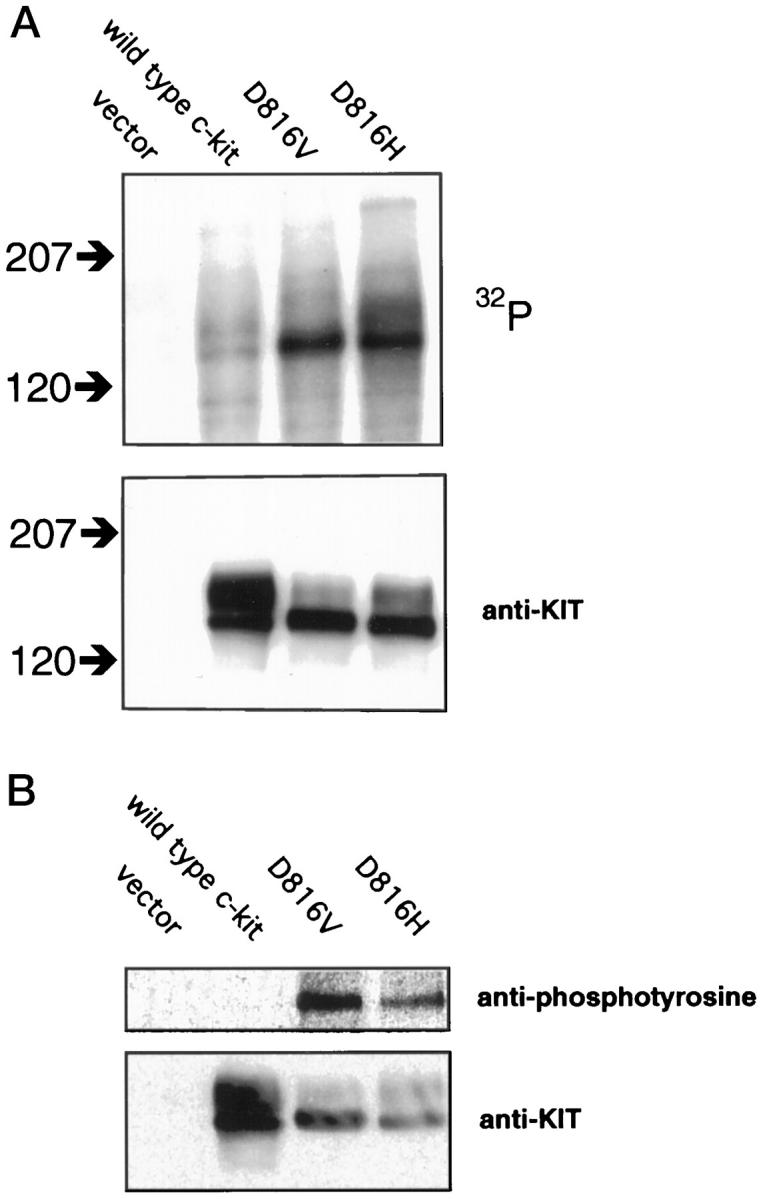
The D816H KIT gene product is a constitutively activated kinase. COS cells were transfected with eukaryotic expression vector constructs containing wild-type and mutant forms of c-kit, as well as the vector alone as a negative control. A: In vitro kinase assay. Protein extracts of transfectants were immunoprecipitated with anti-KIT antibodies and incubated with [γ-32P]ATP. The upper panel shows an autoradiograph of a blot of the kinase assays of equal amounts of protein extracts after separation by gel electrophoresis. The location of molecular mass standards are shown to the left in kd. The location of major phosphorylation targets are consistent with KIT protein. Both the well-characterized D816V mutant and the D816H mutant show a marked increase in kinase activity when compared to wild-type KIT. The same blot was probed with anti-KIT antibody to control for levels of KIT protein in the extracts (lower panel). Similar levels of D816V and D816H mutant KIT proteins were expressed by the transfected cells, with higher levels of expression of wild-type KIT. B: Anti-phosphotyrosine immunoblot. Protein extracts of transfectants were immunoprecipitated with anti-KIT antibodies, separated by gel electrophoresis, and blotted. The upper panel shows a blot probed with anti-phosphotyrosine antibody. A protein is detected at the appropriate molecular mass for KIT in extracts containing D816V and D816H KIT, but not wild-type KIT. The same blot was stripped and reprobed with anti-KIT antibody to control for the presence and quantity of KIT proteins (lower panel).
To determine if the D816H mutant was constitutively phosphorylated on a tyrosine residue in vivo (a state associated with constitutive kinase activity), protein extracts from COS cell transfectants were immunoprecipitated with anti-KIT antibodies and then analyzed by Western blots with anti-phosphotyrosine antibodies. In the absence of ligand stimulation, D816V and D816H KIT showed tyrosine phosphorylation, whereas wild-type KIT did not (Figure 2B) ▶ .
Immunohistochemical Detection of KIT in Germ Cell Tumors
The GCTs were evaluated for KIT protein expression by immunohistochemical staining. All seminomas/dysgerminomas, including the tumors with c-kit mutation, displayed cell membrane staining of KIT receptor (23/23, 100%). This included seminoma/dysgerminoma components of mixed GCT. In contrast, only two yolk sac tumors of 16 nonseminomas/nondysgerminomas showed KIT staining. In both cases the distribution of staining was focal “aberrant” intracytoplasmic staining rather than typical cell membrane staining (Figure 3) ▶ . Both of the focally KIT positive yolk sac tumors were components of mixed GCT, which also contained seminoma or dysgerminoma components. KIT staining was also observed in all intratubular germ cell neoplasia (ITGCN) present in examined histological sections (8/8, 100%).
Figure 3.
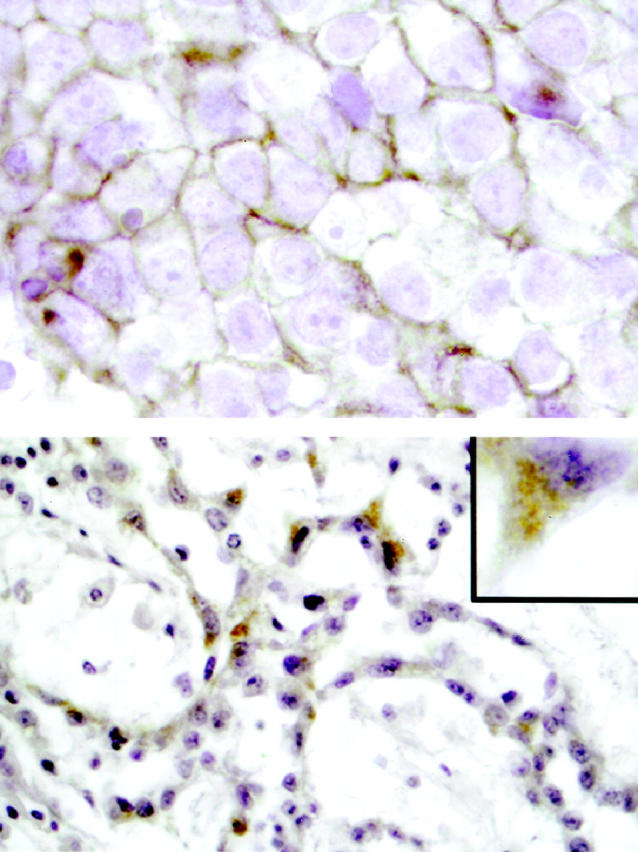
Example of KIT immunohistochemical staining in an ovarian GCT with dysgerminoma and yolk sac tumor components. Upper panel: The dysgerminoma component reveals uniform membrane staining (brown) of tumor cells. Original magnification ×1000. Lower panel: Focal intracytoplasmic staining of tumor cells in the yolk sac tumor component is demonstrated. The majority of the yolk sac tumor showed no KIT expression. Original magnification ×400. Inset: Detail of single yolk sac tumor cell, showing cytoplasmic, but not membrane staining. Original magnification ×1000.
Discussion
We found a gene mutation (D816H) in the c-kit phosphotransferase domain in tumors of the seminoma/dysgerminoma category and showed in cell transfection experiments that this mutation causes constitutive activation of KIT kinase activity. This is the first description of an activating c-kit mutation in GCTs. 14 The human KIT D816H mutant has not previously been reported, although a different missense mutation at the same residue, D816V, isolated from cases of mast cell disease, has been found to be constitutively activated. 15 The wild-type aspartic acid residue at this position is conserved in the mouse and rat c-kit genes, and missense mutations at positions analogous to the human 816 aspartic acid residue have been found in the murine mastocytoma cell line P-185 (D814Y) and the rat mast cell leukemia cell line RBL-2H3 (D817Y) that also constitutively activate KIT. 15 After the completion of our studies a murine c-kit mutation, D814H, analogous to the human D816H mutation, was reported in the ELM-I-1 erythroleukemia cell line. 16 The murine D814H KIT had constitutive kinase activity, appeared to signal through the mitogen-activated protein kinase (MAPK) pathway, and resulted in growth factor-independent cell proliferation.
Our findings of KIT expression predominantly in the seminoma/dysgerminoma subtype of GCT tumors are in agreement with previous studies. 7,8 The occurrence of mixed forms of GCT strongly suggests that there is a continuum of phenotypic differentiation in this class of neoplasms, and previous morphological studies have been consistent with seminoma/dysgerminoma occurring an early stage in tumor progression of GCT. 17 The finding of KIT expression in both intratubular germ cell neoplasia (ITGCN) and in seminomas is consistent with the theory that tumor progression may pass first through ITCGN, then seminoma, then to more aggressive forms of GCT (yolk sac tumor, embryonal carcinoma, etc.). Our finding of the D816H mutation in both forms of the mixed ovarian germ cell tumor is also consistent with this pattern of progression. The KIT gene products were not highly expressed in the yolk sac component of this tumor, nor were they normally situated in the plasma membrane, and hence presumably were not as active in signal transduction as in the dysgerminoma component of the tumor. Presumably the KIT mutation was first selected on a functional basis in the dysgerminoma component and later carried as a silent change in the clone of cells that developed the yolk sac tumor phenotype.
The results of immunohistochemical surveys of GCTs suggest that the effects of KIT-mediated signaling are associated with cellular differentiation to a seminoma/dysgerminoma phenotype. This phenotype recapitulates the early phase of spermatogenesis and is in agreement with studies that show that KIT-mediated signaling is crucial for germ cell development. 6 Conversely, the other subtypes of GCTs (yolk sac tumor, embryonal carcinoma, choriocarcinoma) do not simulate spermatogonia and do not appear to depend on KIT for their maintenance. In addition to the evidence of absence of KIT protein in nonseminomas/nondysgerminomas, mRNA levels of c-kit are absent or greatly diminished in these neoplasms, 7 and there is no detectable protein or mRNA expression of the KIT ligand SCF. 7,8
Our finding of constitutively activated mutant KIT in seminomas/dysgerminomas confirms an important role of this signaling pathway in this class of neoplasms. The fact that we found mutations in only a minority of tumors does not necessarily signify that genetic alteration affecting this pathway occurs in only a minority of these neoplasms. Because of the nature of formalin-fixed paraffin-embedded archival tumor specimens, our methodology was confined to assaying small portions of tumor genomes by PCR analysis. Prospective acquisition of frozen tumor specimens will allow more comprehensive surveys of the entire c-kit gene to determine whether areas outside of the JM and PT domains are targets of activation. In addition, because one study detected SCF production in seminomas, 8 it is possible that the KIT signaling pathway may be activated in nonmutated seminomas/dysgerminomas in an autocrine loop, as has been demonstrated in small cell carcinoma. 18 Our results do suggest that signaling pathways involving KIT are important in seminomas/dysgerminomas, although all of the components of this signal cascade in GCT are unknown. Elucidation of the signaling proteins and their interactions will yield other potential targets for study of the genetic alterations critical to the progression of these neoplasms.
Acknowledgments
We thank Mark S. Clem for help in histological sectioning and immunohistochemical staining of tissue. We also thank Michael Cox and Sarah Parsons for technical assistance and the gifts of reagents for the kinase and phosphotyrosine immunoblot assays.
Footnotes
Address reprint requests to Dr. Chris Moskaluk, Department of Pathology, Box 214, University of Virginia Health Sciences Center, Charlottesville, VA 22908. E-mail: cam5p@virginia.edu.
Supported by grant 5K08CA74431-02 from the National Cancer Institute. The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of the National Cancer Institute. G.W.K. is supported by a Merit Review Award from the Department of Veteran Affairs.
References
- 1.Vliagoftis H, Worobec AS, Metcalfe DD: The protooncogene c-kit and c-kit ligand in human disease. J Allergy Clin Immunol 1997, 100:435-440 [DOI] [PubMed] [Google Scholar]
- 2.Longley BJ, Tyrrell L, Lu SZ, Ma YS, Langley K, Ding TG, Duffy T, Jacobs P, Tang LH, Modlin I: Somatic c-KIT activating mutation in urticaria pigmentosa and aggressive mastocytosis: establishment of clonality in a human mast cell neoplasm. Nature Genet 1996, 12:312–314 [DOI] [PubMed]
- 3.Nagata H, Worobec AS, Oh CK, Chowdhury BA, Tannenbaum S, Suzuki Y, Metcalfe DD: Identification of a point mutation in the catalytic domain of the protooncogene c-kit in peripheral blood mononuclear cells of patients who have mastocytosis with an associated hematologic disorder. Proc Natl Acad Sci USA 1995, 92:10560-10564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hirota S, Isozaki K, Moriyama Y, Hashimoto K, Nishida T, Ishiguro S, Kawano K, Hanada M, Kurata A, Takeda M, Muhammad Tunio G, Matsuzawa Y, Kanakura Y, Shinomura Y, Kitamura Y: Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science 1998, 279:577-580 [DOI] [PubMed] [Google Scholar]
- 5.Moskaluk CA, Tian Q, Marshall CR, Rumpel CA, Franquemont DW, Frierson HF Jr.: Mutations in the c-kit JM domain are found in a minority of human gastrointestinal stromal tumors. Oncogene 1999, 18:1897–1902 [DOI] [PubMed]
- 6.Loveland KL, Schlatt S: Stem cell factor and c-kit in the mammalian testis: lessons originating from Mother Nature’s gene knockouts. J Endocrinol 1997, 153:337-344 [DOI] [PubMed] [Google Scholar]
- 7.Strohmeyer T, Reese D, Press M, Ackermann R, Hartmann M, Slamon D: Expression of the c-kit proto-oncogene and its ligand stem cell factor (SCF) in normal and malignant human testicular tissue. J Urol 1995, 153:511-515 [DOI] [PubMed] [Google Scholar]
- 8.Bokemeyer C, Kuczyk MA, Dunn T, Serth J, Hartmann K, Jonasson J, Pietsch T, Jonas U, Schmoll HJ: Expression of stem-cell factor and its receptor c-kit protein in normal testicular tissue and malignant germ-cell tumours. J Cancer Res Clin Oncol 1996, 122:301-306 [DOI] [PubMed] [Google Scholar]
- 9.Mostofi FK, Sobin LH: Histological typing of testis tumor. International Histological Classification of Tumors, 1977, vol 16. World Health Organization, Geneva
- 10.Moskaluk C, Kern S: Microdissection and PCR amplification of genomic DNA from histologic tissue sections. Am J Pathol 1997, 150:1547-1552 [PMC free article] [PubMed] [Google Scholar]
- 11.Hines SJ, Organ C, Kornstein MJ, Krystal GW: Coexpression of the c-kit and stem cell factor genes in breast carcinomas. Cell Growth Differ 1995, 6:769-779 [PubMed] [Google Scholar]
- 12.Harlow E, Lane D: Antibodies: A Laboratory Manual. 1988. Cold Spring Harbor Laboratory Cold Spring Harbor, NY
- 13.Tian Q, Taupin J, Elledge S, Robertson M, Anderson P: Fas-activated serine/threonine kinase (FAST) phosphorylates TIA-1 during Fas-mediated apoptosis. J Exp Med 1995, 182:865-874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Murty VVVS, Chaganti RSK: A genetic perspective of male germ cell tumors. Semin Oncol 1998, 25:133-144 [PubMed] [Google Scholar]
- 15.Tsujimura T, Kanakura Y, Kitamura Y: Mechanisms of constitutive activation of c-kit receptor tyrosine kinase. Leukemia 1997, 11:396-398 [PubMed] [Google Scholar]
- 16.Leslie NR, O’Prey J, Bartholomew C, Harrison PR: An activating mutation in the KIT receptor abolishes the stroma requirement for growth of ELM erythroleukemia cells, but does not prevent their differentiation to erythropoietin. Blood 1998, 92:4798-4807 [PubMed] [Google Scholar]
- 17.Ulbright TM, Roth LM: Testicular and paratesticular neoplasms. Sternberg SS eds. Diagnostic Surgical Pathology. 1994, :pp 1885-1947 Raven Press, New York [Google Scholar]
- 18.Krystal GW, Hines SJ, Organ CP: Autocrine growth of small cell lung cancer mediated by coexpression of c-kit and stem cell factor. Cancer Res 1996, 56:370-376 [PubMed] [Google Scholar]