

Abstract
We investigated the effect of tissue kallikrein infusion on cardiac protection at acute and sub-acute phases after myocardial infarction (MI). Immediately after MI, rats were infused with purified tissue kallikrein, with or without icatibant (a kinin B2 receptor antagonist). Intramyocardial injection of kallikrein reduced myocardial infarct size and inhibited cardiomyocyte apoptosis at 1 day after MI associated with increased nitric oxide levels, Akt and glycogen synthase kinase-3β phosphorylation and decreased caspase-3 activation. Kallikrein infusion for 7 days improved cardiac function, normalized left ventricular wall thickness and decreased monocyte/macrophage infiltration in the infarct heart. Kallikrein treatment reduced NADH oxidase expression and activity, superoxide formation and malondialdehyde levels, and reduced MAPK and Iκ-Bα phosphorylation, NF-κB activation and MCP-1 and VCAM-1 expression. Kallikrein’s effects were all blocked by icatibant. These results indicate that kallikrein through kinin B2 receptor activation prevents apoptosis, inflammation and ventricular remodeling by increased nitric oxide formation and suppression of oxidative stress-mediated signaling pathways.
Keywords: kallikrein, kinin B2 receptor, myocardial infarction, apoptosis, inflammation, oxidative stress, NF-κB
1. Introduction
Evidence from both animal and human studies suggests that oxidative stress may play an important role during the inflammatory phase of MI [1,2]. The lack of oxygen supply during ischemia disrupts the mitochondrial electron transport chain in cardiomyocytes, resulting in the generation and accumulation of reactive oxygen species (ROS) and thus cell death [3]. In addition, activated inflammatory cells infiltrate into cardiac muscle after MI and release ROS via NADH/NADPH oxidase [4]. Moreover, reduced function of cardiac anti-oxidant enzymes (such as glutathione peroxidase, catalase, and superoxide dismutase isoforms) has been observed in animal models with cardiac disease, thus causing an imbalance between anti-oxidant and oxidative enzymes [5]. Increased oxidative stress in ischemic cardiovascular diseases has been shown to play a key role in activating nuclear factor-κB (NF-κB) [6]. NF-κB is a ubiquitous rapid response transcription factor that modulates the expression of various cytokine and adhesion molecules. Activation of NF-κB up-regulates the expression of pro-inflammatory genes, such as vascular cell adhesion molecule 1 (VCAM-1) and monocyte chemoattractant protein 1 (MCP-1), that play pivotal roles in inflammation after ischemia [7,8]. These findings suggest that intervention with exogenous anti-oxidants may attenuate myocardial damage by inhibiting cardiomyocyte apoptosis and blunting the inflammatory response.
All components of the tissue kallikrein-kinin system have been identified in the cardiovascular system [9]. Tissue kallikrein is a serine proteinase that cleaves kininogen to release the vasoactive kinin peptide. Intact kinins bind to the kinin B2 receptor and activate second messengers such as nitric oxide (NO), cGMP, prostacyclin and cAMP, to produce a broad spectrum of biological effects [10]. A study using kallikrein- or kinin B2 receptor-deficient mice showed that both tissue kallikrein and the B2 receptor play an important role in cardioprotection during ischemic injury [11]. Moreover, a recent report demonstrated that expression of tissue kallikrein in transgenic mice reduces intramyocardial inflammation and oxidative stress in experimental diabetic cardiomyopathy [12]. In the present study, we examined the potential roles of tissue kallikrein/kinin in myocardial infarction injury by infusion of purified tissue kallikrein into rats after permanent coronary artery ligation. The objective was to investigate the protective role of tissue kallikrein in preventing cardiomyocyte apoptosis, inflammation and oxidative stress after acute myocardial infarction.
2. Materials and methods
2.1. Animals and treatments
Wistar rats (male, 250 to 280 g body weight, Harlan) were subjected to ligation of the left coronary artery as previously described [13]. This study complied with the Guides for the Care and Use of Laboratory Animals (Institute of Laboratory Resources, National Academy of Sciences).
Study 1
Animals were randomly divided into four groups (n=7 in each group). Rat tissue kallikrein was purified as previously described [14]. In two control groups, rats were subjected to either sham surgery or left anterior descending (LAD) coronary ligation followed by saline infusion. In the third group, tissue kallikrein (TK, 25 μg in 150 μl saline) was injected at 7 different sites into the border area of the left ventricle, immediately after LAD ligation. The fourth group received TK together with co-injection of the kinin B2 receptor antagonist (icatibant, obtained from Hoechst Marion Roussel, 15 μg/rat). One day after coronary artery ligation, hemodynamic parameters were analyzed, animals were euthanized, and heart tissues were harvested for morphological and biochemical analyses.
Study 2
Animals were randomly divided into four groups (n=7 in each group). In two control groups, rats were subjected to either sham surgery or LAD ligation followed by saline infusion. The third group received TK infusion by subcutaneous implantion of osmotic minipumps (ALZET) immediately after LAD ligation at an infusion rate of 1μg/hr. The fourth group received TK together with co-administration of icatibant at an infusion rate of 2 μg/hr. At 7 days after LAD ligation, hemodynamic parameters were analyzed, animals were euthanized, and heart tissues were harvested for morphological and biochemical analyses.
2.2. Hemodynamic parameters
At the end of the experiment, rats were anesthetized and cardiac function was measured. The common carotid artery was cannulated using a 2.5 French micromanometer (Millar Instrument) by advancement into the left ventricle. Mean arterial pressure (MAP), heart rate (HR) cardiac contractility (dP/dt maximum and dP/dt minimum), and left ventricle end diastolic pressure (LVEDP) were analyzed with a model 7E polygraph (Grass Instrument).
2.3. Myocardial infarct size determination
To measure infarct size at 24 hours after myocardial infarction, the middle part of the heart (1 mm) was sectioned transversely and incubated with 1.5% 2,3,5-triphenyltetrazolium chloride (TTC) (Sigma) for 5 minutes at 37°C. Infarct area, which approximately equals the area at risk in this setting, was then determined. The infarct area was distinguished by TTC staining using computer-assisted planimetry (NIH Image 1.57).
2.4. Histological analysis
For histological analyses, the left ventricle was cut into 3 transversal slices on basal, middle and apex levels. After fixation with 4% paraformaldehyde, the cardiac tissues were dehydrated and embedded. Four-micron (4 μm) sections were obtained for morphological analyses. Sections were subjected to Masson’s trichrome staining or immunohistochemical analysis using a staining kit (Universal Elite ABC, Vector) according to the manufacturer’s instructions. Primary antibody against ED-1 (Chemicon, 1:200) was used for immunostaining of monocytes/macrophages. The number of ED-1-positive cells was counted in a double-blind fashion from 20 different fields of each section (n=6 or 7) at 400 x magnification.
2.5. Western Blot Analysis
The infarct area was separated from the non-infarct region at 1-day after MI according to tissue color change due to lack of oxygen supply in the myocardium. Cardiac tissue from infarct area was homogenized and the cytosolic fraction was collected. Protein concentrations were measured by Bio-Rad Protein Assay kit (Bio-Rad). Western blot analysis was performed using cytosolic fractions to detect the total and phosphorylated forms of Akt, GSK-3β, p38MAPK, JNK, ERK1/2, IκB-α (Cell Signaling), as well as caspase-3, VCAM-1 (Santa Cruz) and GAPDH (Advanced Immunochemical). Membranes were incubated with secondary antibody conjugated to LumiGLO chemiluminescent reagent. Chemiluminescence was detected using an ECL-Plus kit (Perkin Elmer Life Science) and visualized by Kodak X-ray film. The bands were quantified by densitometry.
2.6. Nitrate/Nitrite, NADH oxidase and superoxide assays
Nitrate/nitrite (NOx) levels, an indicator of NO production, were measured by a fluorometric assay as previously described [15]. NADH oxidase activity was measured by chemiluminescent detection of superoxide using a luminometer (Turner Designs) [16]. Superoxide levels were measured by a spectrophotometric assay based on rapid reduction of ferricytochrome c to ferrocytochrome c. Non-superoxide-dependent reduction of cytochrome c was corrected for by deducting the activity not inhibited by superoxide dismutase [17].
2.7. Measurement of malondialdehyde levels
Lipid peroxidation, an indicator of oxidative stress, was determined by measurement of malondialdehyde (MDA) levels. Cytosolic proteins (500 μg) were mixed with 2% butylated hydroxytoluene and quintanilla reagent and boiled for 15 min. The reaction mixture was centrifuged at 3000 x g for 10 min. The soluble phase was measured with a spectrophotometer at 535 nm using MDA standards (0 to 30 μmol/mL) (Sigma).
2.8. Electrophoretic mobility shift assay (EMSA) for NF-κB activation
Nuclear proteins were isolated from cardiac extracts as previously described [18]. NF-κB DNA binding site oligonucleotides (AGTTGAGGGGACTTTCCCAGGC, Integrated DNA Technologies) were labeled using the Biotin 3' End DNA Labeling kit (Pierce) according to the manufacturer’s protocol. The gel shift assay was performed using the Light Shift Chemiluminescent electrophoretic mobility shift analysis kit (Pierce) following the manufacturer’s instructions. The relative nuclear NF-κB DNA binding activities were quantified by scanning densitometry.
2.9. Quantitative real-Time PCR
Total RNA was extracted from myocardium using Trizol reagent (Invitrogen). cDNA was transcribed using a cDNA Archive Kit (Applied Biosystems). The quantitative real-time PCR reaction was carried out using the Gene Expression Assay Rn01456716_gl for MCP-1, Rn00570921_ml for VCAM-1 and Rn00577357_ml for NADH/NADPH p22phox subunit (Applied Biosystems) running on a 7300 real time PCR system (Applied Biosystems). Transcription of the housekeeping gene GAPDH was determined by specific primer/probe mix (Applied Biosystems). The final quantification was determined by Relative Quantification software (Applied Biosystems).
2.10. Statistical analysis
Data were compared among experimental groups using ANOVA followed by Fisher’s PLSD. Data are expressed as mean ± SEM. Differences were considered statistically significant at a value of P<0.05.
3. Results
3.1. Kallikrein reduces infarct size and cardiomyocyte apoptosis
Intramyocardial injection of tissue kallikrein for 1 day significantly reduced infarct size in the left ventricle compared with the MI control group, as determined by TTC staining and quantitative analysis (Figure 1A and B). Icatibant, however, abrogated kallikrein’s effect. Icatibant alone had no effect on myocardial infarct size as compared to the MI control (data not shown).
Figure 1.
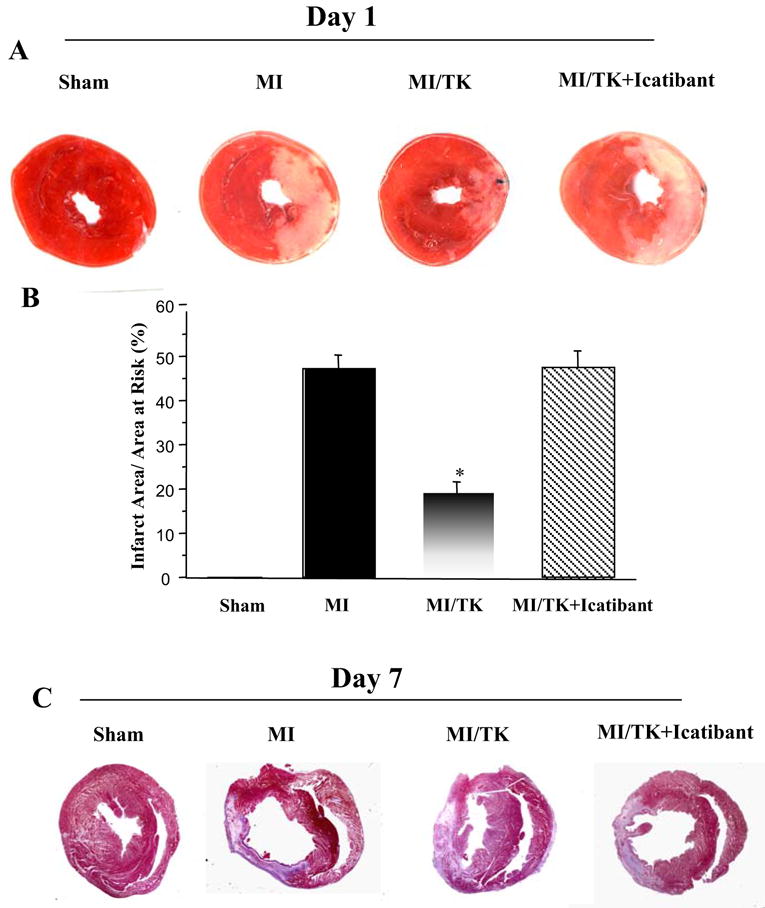
Tissue kallikrein treatment reduces infarct size after MI (A) Representative TTC staining of heart sections, magnification is 10×. (B) Quantitative analysis of infarct size. Infarct size is expressed as percentage of the infarct area vs. area at risk. (C) Representative Masson’s trichrome staining. Original magnification is 10×. Values are expressed as mean ± SEM (n=6–7, *P<0.01 vs. MI & MI/TK+Icatibant).
Apoptotic cardiomyocytes were detected by TUNEL staining in the infarcted myocardium 1 day after MI (Figure 2A). The ratio of TUNEL-positive cardiomyocytes to total number of cardiomyocytes in the kallikrein group was significantly reduced as compared to the control group (Figure 2B). However, kallikrein’s protective effect was blocked by icatibant. The effect of kallikrein on inhibiting cardiomyocyte apoptosis was further verified by Western blot of cleaved caspase-3 (Figure 2D). Moreover, kallikrein treatment resulted in a significant increase in cardiac NOx production compared with the MI control group, and this effect was abrogated by icatibant (Figure 2C). Kallikrein also caused increased phosphorylation of Akt and GSK-3β in the infarcted region as compared to the control (Figure 2D). Kallikrein’s effects on these signaling effectors were blocked by icatibant. The combined data indicate that activation of Akt and inactivation of GSK-3β and caspase-3 mediate the protective effect of tissue kallikrein in MI-induced myocardial apoptosis.
Figure 2.
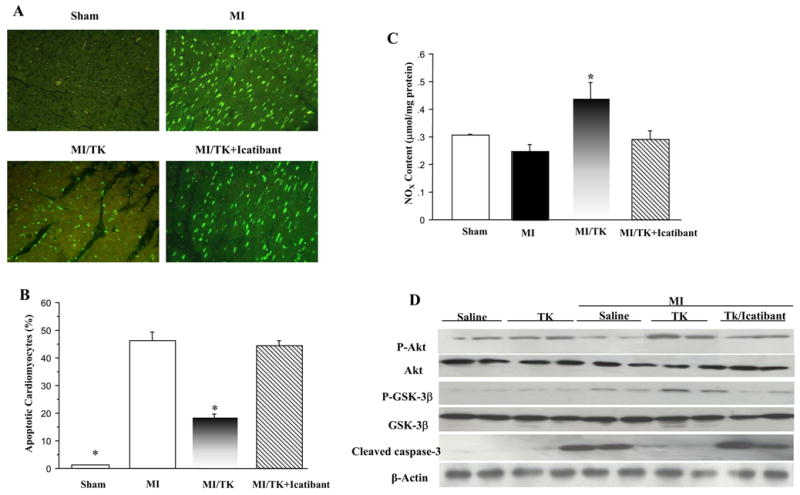
Kallikrein treatment reduces cardiomyocyte apoptosis after acute MI. (A) Representative photomicrographs show TUNEL-positive apoptotic cardiomyocytes from the infarcted region of rat hearts. Original magnification is 200×. (B) Quantitative analysis of apoptotic cardiomyocytes expressed as percentage of TUNEL-positive nuclei in cardiomyocytes. TUNEL-positive non-cardiomyocytes were excluded. (C) NOx levels. (D) Representative Western blots of phosphorylated and total Akt and GSK-3β, and cleaved caspase-3. Values are expressed as mean ± SEM (n=6–7, *P<0.05 vs. MI & MI/TK+Icatibant).
3.2. Kallikrein improves cardiac function and prevents remodeling
At 7 days after kallikrein infusion, we detected about 45% elevation of circulating tissue kallikrein levels measured by enzyme-linked immunosorbant assay (5.1 ± 0.2 ng/ml, n=6) in rats as compared to sham and control MI groups (3.4 ± 0.5 and 3.6 ± 0.5 ng/ml, n=6), suggesting that subcutaneous osmotic minipump implantation is sufficient in delivering tissue kallikrein into the bloodstream. However, it is impossible to determine whether tissue kallikrein injected in the border zone of the infarcted hearts is located there or diffuses to the peri-infarcted zone since the rat tissue kallikrein antibody can not distinguish between infused and endogenous rat tissue kallikrein. Table 1 shows the effect of kallikrein infusion on the physiological and hemodynamic parameters 7 days after MI. There were no significant differences in heart weight/body weight ratio or heart rate (HR) among the groups. MI resulted in reduced MAP as compared to the sham, kallikrein prevented the reduction as compared to the MI control group. MI induced a 5-fold increase of LVEDP compared to the sham group, whereas kallikrein significantly reduced LVEDP. Cardiac contractility was markedly reduced after MI, but was significantly increased by kallikrein. At 7 days after of MI, the survival rates of rats in MI, MI/kalikrein and MI/kallikrein/icartibant groups were 66.7%, 72.7% and 63.7%, respectively. Kallikrein’s cardioprotective effects were blocked by icatibant, indicating that kallikrein, through kinin B2 receptor activation, improves cardiac function.
Table 1.
Effects of Kallikrein Infusion on Cardiac Function 7 Days after MI
| Variable | Sham | MI | MI/TK | MI/TK+Icatibant |
|---|---|---|---|---|
| Infarct Size | 0 | 0.37 ± 0.03 | 0.19 ± 0.036* | 0.31 ± 0.05 |
| LV Wall Thickness (mm) | 3.5 ±10.15 | 1.3 ± 0.11 | 2.58 ± 0.27 * | 1.86 ± 0.32 |
| Heart Rate (bpm) | 382.3 ± 16.4 | 378.7 ± 17.6 | 368.1 ± 14.1 | 378.1 ± 13.3 |
| MAP (mmHg) | 101.0 ± 5.58 | 67.7 ± 2.76 | 85.4 ± 6.04 * | 67.6 ± 7.53 |
| LVEDP (mmHg) | 2.95 ± 0.87 | 16.27 ± 1.55 | 7.91 ± 1.02 * | 13.50 ± 2.02 |
| dP/dt max (mmHg/s) | 4163.2 ± 153.9 | 2196.5 ± 127.4 | 2842.4 ± 143.3* | 2133.7 ± 104.0 |
| dP/dt min (mmHg/s) | 3390.5 ± 139.1 | 1462.5 ± 121.4 | 2137.7 ± 154.6* | 1441.2 ± 100.2 |
Values are mean ± SEM.
P<0.05 vs. MI &MI/TK+Icatibant.
MI: myocardial infarction; MI/TK: MI with tissue kallikrein infusion; MI/TK+Icatibant: MI with tissue kallikrein and icatibant infusion.
Quantitative analysis of Masson’s trichrome staining showed that kallikrein infusion for 7 days reserved infarct size. Also, kallikrein preserved ventricular wall thickness, in contrast, significant thinning of the wall occurred in the MI and icatibant-treated animals. (Figure 1C and Table 1).
3.3. Kallikrein attenuates intramyocardial inflammation
Inflammatory cell accumulation in the infarcted region of the heart was identified by ED-1 immunostaining 7 days after MI (Figure 3A). ED-1 positive cells were counted for quantification of monocyte/macrophage number (Figure 3B). Increased inflammatory cell infiltration was detected in the infarct area of the heart after MI, but kallikrein infusion significantly decreased ED-1 positive cells compared with the control. Icatibant, however, blocked kallikrein’s effect. The protective effect of kallkrein on inflammatory cell accumulation was associated with decreased expression of the pro-inflammatory mediators VCAM-1 and MCP-1. Western blot (Figure 3C) and real-time PCR (Figure 3D) demonstrated that MI up-regulated VCAM-1 protein and mRNA levels, respectively, compared to the sham group. Kallikrein, however, reduced VCAM-1 levels, and this effect was reversed by icatibant. Similarly, a significant increase in MCP-1 mRNA expression was detected in the MI control group, which was attenuated by treatment of kallikrein, and icatibant blocked kallikrein’s effect (Figure 3E).
Figure 3.
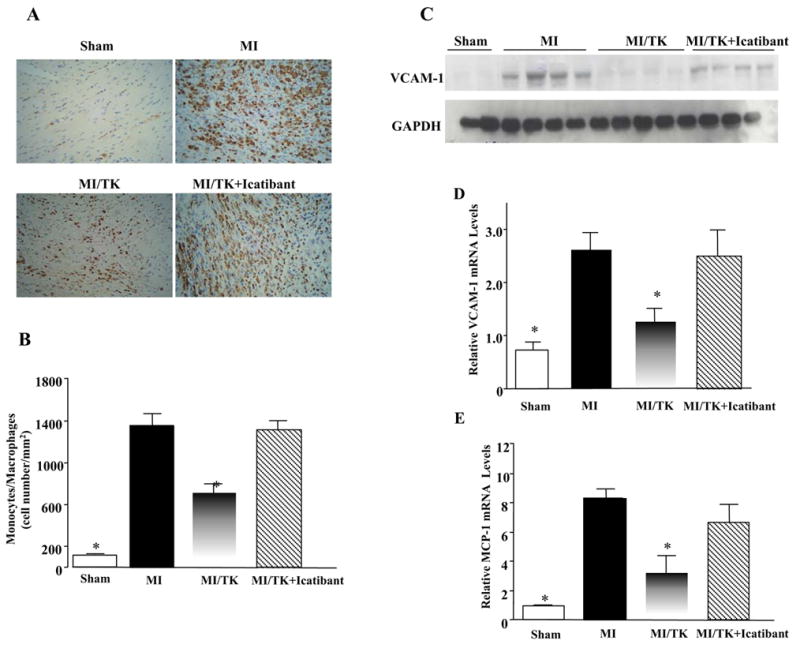
Kallikrein infusion reduces monocyte/macrophage accumulation and the expression of pro-inflammatory mediators in the infarcted myocardium. (A) Representative images of ED-1 immunostaining in the infarcted area. Original magnification is 200×. (B) Quantitative analysis of ED-1-positive cells. (C) Representative Western blots of cardiac VCAM-1 levels. (D) Relative VCAM-1 and (E) MCP-1 mRNA levels determined by real-time PCR. Values are expressed as mean ± SEM (n=6–7, *P<0.05 vs. MI & MI/TK+Icatibant).
3.4. Kallikrein reduces oxidative stress
NADH oxidase activity and superoxide levels were measured in the infarcted myocardium in order to evaluate the role of oxidative stress 7 days after MI injury. Figures 4A show that increases in NADH oxidative activity was significantly reduced by kallikrein. However, kallikrein’s effects were reversed by icatibant. The inhibitory effect of kallikein on NADH oxidase activity was further confirmed by reduced expression of p22phox, a subunit of NADH/NADPH oxidase. Real-time PCR showed that p22phox mRNA levels increased after MI and kallikein reduced the expression of p22phox, while icatibant blocked kallikrein’s effect (Figure 4C). Superoxide formation paralleled NADH oxidase activity. Superoxide levels were elevated in the MI group compared to the sham group. Kallikrein infusion significantly lowered MI-induced superoxide formation and icatibant diminished kallikrein’s effect (Figure 4D). Moreover, kallikrein completely prevented the increase in cardiac MDA levels induced by MI damage, and icatibant blocked the effect of kallikrein (Figure 4B).
Figure 4.
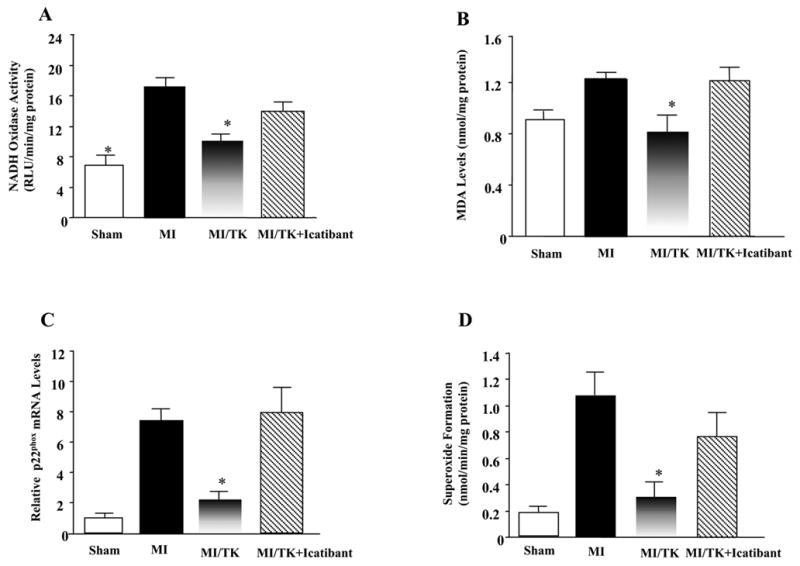
Effect of kallikrein infusion on (A) NADH oxidase activity, (B) MDA levels (C) relative p22phox mRNA levels, (D) superoxide formation. Values are expressed as mean ± SEM (n=6–7, *P<0.05 vs. MI & MI/TK+Icatibant).
3.5. Kallikrein inhibits JNK, p38MAPK and ERK1/2 phosphorylation
Phosphorylation of JNK, p38MAPK and ERK1/2 increased significantly 7 days after MI, as determined by Western blot analyses. However, kallikrein infusion prevented MI-induced MAPK activation, and icatibant abrogated kallikrein’s effects (Figure 5A). These results indicate that kallikrein inhibits MAPK activation during post-infarct remodeling through kinin B2 receptor activation.
Figure 5.
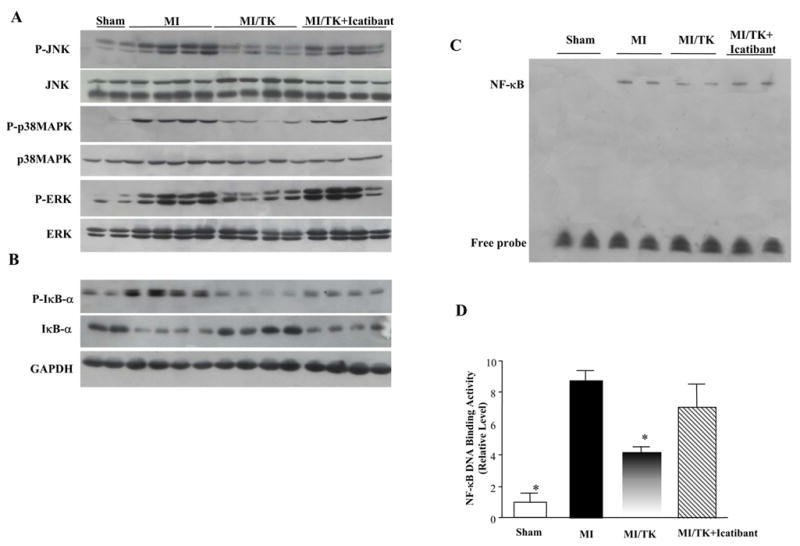
Kallikrein infusion reduces (A) JNK, p38MAPK and ERK phosphorylation. (B) Representative Western blot of phospho-IκB-α and IκB-α. GAPDH was used as an internal control. (C) Representative EMSA and (D) quantitative analyses of NF-κB DNA-binding activity. Values are expressed as mean ± SEM (n=6, *P<0.05 vs. MI & MI/TK+Icatibant).
3.6. Kallikrein reduces MI-Induced IκB-α phosphorylation and inhibits nuclear translocation and activation of NF-κB
Western blot analysis showed that MI induced IκB-α (Ser32) phosphorylation and reduced non-phosphorylated IκB-α levels at 7 days after MI, whereas kallikrein treatment reversed this effect (Figure 5B). Gel mobility shift assay indicated that nuclear NF-κB-DNA binding activity was increased in the infarcted heart after MI compared with the sham group. Administration of kallikrein markedly reduced NF-κB-DNA binding activity, but icatibant abolished this effect (Figure 5C). Quantitative analysis further confirmed the results from gel mobility shift assay (Figure 5D). These combined results indicate that kallikrein inhibits NF-κB activation by suppressing IκB-α phosphorylation and its degradation. These events in turn lead to the prevention of NF-κ B release from the I3B-3/NF-3B complex and its translocation to the nucleus.
4. Discussion
In the present study, we employed a rat model of myocardial infarction to investigate the protective effects and signaling mechanisms of tissue kallikrein in the ischemic myocardium at 24 hours and 7 days after coronary artery occlusion. Our results provides several novel findings. First, this is the first study to demonstrate that tissue kallikrein protein infusion protects against myocardial ischemia injury by inhibiting apoptosis and inflammation. The advantage of kallikrein protein infusion via osmotic milipumps is to provide a stable supply of the therapeutic protein or peptide without potential side effects. Second, in this study, kallikrein was administrated after coronary ligation rather than a prophylactic effect of local injection of the kallikrein gene several days prior to myocardial infarction. Third, this is the first study to demonstrated that kallikrein attenuated intramyocardial inflammation by inhibition of nuclear translocation and activation of NF-κB after MI. These findings provide significant insights regarding the role of tissue kallikrein in eliciting anti-oxidative, anti-apoptotic and anti-inflammatory effects to protect against myocardial damage.
Our previous study showed that local injection of adenovirus carrying the human tissue kallikrein into the heart protects against cardiomyocyte apoptosis after acute myocardial ischemia/reperfusion [19]. However, kallikrein protein infusion has advantages over adenovirus-mediated delivery. For example, protein infusion provides a stable supply of the therapeutic product and can be terminated at any time during the experiment, whereas the expression of recombinant gene product after adenovirus-mediated gene delivery is transient with highest levels around 3–5 days [20]. In addition, local injection of adenovirus can possibly produce an inflammatory response and is therefore no longer effective after a second injection.
Our present study showed that kallikrein treatment prevents cardiomyocyte apoptosis and increased Akt and GSK-3β phosphorylation at 1 day after MI. Akt is a key effector of in the survival pathway against apoptosis. Akt inactivates GSK-3β by phosphorylation, thereby blocking cytochrome c release and caspase-3 activation [21]. Akt-GSK-3β signaling also participates in the development of cardiac hypertrophy [22]. Active GSK-3β is a negative regulator of hypertrophy as decreased active GSK-3β or increased GSK-3β phosphorylation led to hypertrophy and active GSK-3β blunted hypertrophy. Indeed, our recent study showed that kallikrein gene delivery inhibited pressure overload-induced cardiac hypertrophy associated with increased GSK-3β activity and reduced Akt/GSK-3β phosphorylation [23]. In contrast to its role in hypertrophy, increased Akt/GSK-3β phosphorylation/GSK-3β inactivation plays a positive role in promoting cell survival. Our present results showed that kallikrein treatment significantly promotes cardiomyocyte survival at 1 day after MI in conjunction with increased Akt/GSK-3β phosphorylation and reduced GSK-3β activity. Prevention of cardiomyocyte apoptosis after acute MI would result in improving impaired cardiac function, reducing myocardial infarct size and preventing ventricular remodeling.
Nitric oxide, a potent anti-oxidant, has been shown to abolish mitochondrial oxidant damage in adult rat cardiomyocytes [24]. Nitric oxide is capable of inhibiting neutrophil superoxide anion production via a direct action on the membrane components of NADPH oxidase and the assembly of NADH/NADPH oxidase subunits [25]. However, NO has been considered to be a double-edged sword [26, 27]. It is well known that NO in the presence of superoxide can form the toxic peroxynitrite. The cofactor BH4 is highly sensitive to oxidation by peroxynitrite. Diminished levels of BH4 result in eNOS uncoupling and promote superoxide production by eNOS. Our results show that kallikrein protein infusion protects against MI-induced cardiac dysfunction in conjunction with increased NO formation in the infarcted heart. Kallikrein also significantly reduced oxidative stress by decreasing p22phox expression and NADH oxidase activity, superoxide production and MDA activity. However, all of kallikrein’s effects were blocked by icatibant. These results suggest that kallikrein through kinin B2 receptor activation acts as an anti-oxidant capable of improving cardiac function and limiting remodeling.
Sustained inflammatory responses after MI could contribute to ventricular remodeling and the development of heart failure. Kallikrein infusion reduced inflammatory cell accumulation in association with decreased MCP-1 and VCAM-1 expression. These combined results indicate kallikrein/kinin has a beneficial role in cardiac repair after MI by significantly reducing the inflammatory response in the infarcted myocardium.
JNK and p38MAPK play vital roles in cardiomyocyte apoptosis, inflammation, and may be activated by different cellular stress signals, such as oxidative stress and inflammatory cytokines [28]. Cardiac remodeling induced by isoproterenol infusion in rats has been shown to provoke cardiac oxidative stress, leading to phosphorylation of MAPKs [29]. Furthermore, ERK1/2 activation has been shown to mediate signaling events involved in cellular proliferation, differentiation and left ventricular remodeling [30]. Since cardiomyocyte hypertrophy and cardiac fibroblast proliferation contribute to ventricular remodeling, ERK1/2 activation may be vital to its progression after MI. We showed that kallikrein through the kinin B2 receptor inhibited IκB-α phosphorylation and degradation, and prevented NF-κB activation, leading to the down-regulation of MCP-1 and VCAM-1 and thus inhibition of inflammatory responses in the infarcted heart. It is likely that the anti-inflammatory effect produced by kallikrein involves initial NO formation, which in turn suppresses oxidative stress, NF-κB and MAPK activation, consequently reducing the monocyte infiltration in the infarcted area.
Although we found that kallikrein inhibited the inflammatory response after MI through kinin B2 receptor activation, kinins are well-known inducers of pro-inflammatory actions [31]. This may be due to distinctive roles of B1 and B2 receptors in different phase after MI, respectively. Up-regulation of the B1 receptor expression has been observed to reach its maximum at 24 hours and quickly reduced to a barely detectable level at 6 days after MI. However, expression of the B2 receptor in the left ventricle after MI reaches a peak at 24 hours and remains high for at least 6 days, suggesting the main role of the B2 receptor at the sub-acute remodeling phase after MI [32]. Neutrophil migration in inflamed tissues is reduced in kinin B1 knockout mice, indicating that the pro-inflammatory effect of kinin is mediated by the B1 receptor [33]. Furthermore, the inflammatory response after ischemia/reperfusion injury is significantly reduced in B1 receptor-deficient mice, and pretreatment with icatibant reverses this anti-inflammatory effect [34]. These combined results suggest that the B1 receptor is pro-inflammatory whereas the B2 receptor protects against tissue injury.
Acknowledgments
This work was supported by National Institutes of Health grants HL29397, DK066350, and C06 RR015455 from the Extramural Research Facilities Program of the National Center for Research Resources.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sia YT, Parker TG, Liu P, Tsoporis JN, Adam A. Improved post-myocardial infarction survival with probucol in rats: effects on left ventricular function, morphology, cardiac oxidative stress and cytokine expression. J Am Coll Cardiol. 2002;39:148–56. doi: 10.1016/s0735-1097(01)01709-0. [DOI] [PubMed] [Google Scholar]
- 2.Krijnen PA, Meischl C, Hack CE, Meijer CJ, Visser CA, Roos D, et al. Increased Nox2 expression in human cardiomyocytes after acute myocardial infarction. J Clin Pathol. 2003;56:194–99. doi: 10.1136/jcp.56.3.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Duranteau J, Chandel NS, Kulisz A, Shao Z, Schumacker PT. Intracellular signaling by reactive oxygen species during hypoxia in cardiomyocytes. J Biol Chem. 1998;273:11619–624. doi: 10.1074/jbc.273.19.11619. [DOI] [PubMed] [Google Scholar]
- 4.Cathcart MK. Regulation of superoxide anion production by NADPH oxidase in monocytes/macrophages: contributions to atherosclerosis. Arterioscler Thromb Vasc Biol. 2004;24:23–38. doi: 10.1161/01.ATV.0000097769.47306.12. [DOI] [PubMed] [Google Scholar]
- 5.Khaper N, Kaur K, Li T, Farahmand F, Singal PK. Antioxidant enzyme gene expression in congestive heart failure following myocardial infarction. Mol Cell Biochem. 2003;251:9–15. [PubMed] [Google Scholar]
- 6.Altavilla D, Deodato B, Campo GM, Arlotta M, Miano M, Squadrito G, et al. IRFI 042, a novel dual vitamin E-like antioxidant, inhibits activation of nuclear factor-kappaB and reduces the inflammatory response in myocardial ischemia-reperfusion injury. Cardiovasc Res. 2000;47:515–28. doi: 10.1016/s0008-6363(00)00124-3. [DOI] [PubMed] [Google Scholar]
- 7.Omura T, Yoshiyama M, Kim S, Matsumoto R, Nakamura Y, Izumi Y, et al. Involvement of apoptosis signal-regulating kinase-1 on angiotensin II-induced monocyte chemoattractant protein-1 expression. Arterioscler Thromb Vasc Biol. 2004;24:270–75. doi: 10.1161/01.ATV.0000112930.40564.89. [DOI] [PubMed] [Google Scholar]
- 8.Lu L, Chen SS, Zhang JQ, Ramires FJ, Sun Y. Activation of nuclear factor-kappaB and its proinflammatory mediator cascade in the infarcted rat heart. Biochem Biophys Res Commun. 2004;321:879–85. doi: 10.1016/j.bbrc.2004.07.048. [DOI] [PubMed] [Google Scholar]
- 9.Nolly HL, Saed G, Scicli G, Carretero OA, Scicli AG. The kallikrein-kinin system in cardiac tissue. Agents Actions Suppl. 1992;38:62–72. [PubMed] [Google Scholar]
- 10.Bhoola KD, Figueroa CD, Worthy K. Bioregulation of kinins: kallikreins, kininogens, and kininase. Pharmacol Rev. 1992;44:1–80. [PubMed] [Google Scholar]
- 11.Griol-Charhbili V, Messadi-Laribi E, Bascands JL, Heudes D, Meneton P, Giudicelli JF, et al. Role of tissue kallikrein in the cardioprotective effects of ischemic and pharmacological preconditioning in myocardial ischemia. FASEB J. 2005;19:1172–4. doi: 10.1096/fj.04-3508fje. [DOI] [PubMed] [Google Scholar]
- 12.Tschope C, Walther T, Escher F, Spillmann F, Du J, Altmann C, et al. Transgenic activation of the kallikrein-kinin system inhibits intramyocardial inflammation, endothelial dysfunction and oxidative stress in experimental diabetic cardiomyopathy. FASEB J. 2005;19:2057–59. doi: 10.1096/fj.05-4095fje. [DOI] [PubMed] [Google Scholar]
- 13.Pfeffer MA, Pfeffer JM, Fishbein MC, Fletcher PJ, Spadaro J, Kloner RA, et al. Myocardial infarct size and ventricular function in rats. Circ Res. 1979;44:503–12. doi: 10.1161/01.res.44.4.503. [DOI] [PubMed] [Google Scholar]
- 14.Chao J, Margolius HS. Isozymes of rat urinary kallikrein. Biochem Pharm. 1979;28:2071–79. doi: 10.1016/0006-2952(79)90226-0. [DOI] [PubMed] [Google Scholar]
- 15.Misko TP, Schilling RJ, Salvemini D, Moore WM, Currie MG. A fluorometric assay for the measurement of nitrite in biological samples. Anal Biochem. 1993;214:11–6. doi: 10.1006/abio.1993.1449. [DOI] [PubMed] [Google Scholar]
- 16.Griendling KK, Minieri CA, Ollerenshaw JD, Alexander RW. Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ Res. 1994;74:1141–48. doi: 10.1161/01.res.74.6.1141. [DOI] [PubMed] [Google Scholar]
- 17.de Cavanagh EM, Fraga CG, Ferder L, Inserra F. Enalapril and captopril enhance antioxidant defenses in mouse tissues. Am J Physiol. 1997;272:R514–18. doi: 10.1152/ajpregu.1997.272.2.R514. [DOI] [PubMed] [Google Scholar]
- 18.Deryckere F, Gannon F. A one-hour minipreparation technique for extraction of DNA-binding proteins from animal tissues. Biotechniques. 1994;16:405. [PubMed] [Google Scholar]
- 19.Yin H, Chao L, Chao J. Kallikrein/kinin protects against myocardial apoptosis after ischemia/reperfusion via Akt-glycogen synthase kinase-3 and Akt-Bad.14-3-3 signaling pathways. J Biol Chem. 2005;280:8022-1–30. doi: 10.1074/jbc.M407179200. [DOI] [PubMed] [Google Scholar]
- 20.Bledsoe G, Chao L, Chao J. Kallikrein gene delivery attenuates cardiac remodeling and promotes neovascularization in spontaneously hypertensive rats. Am J Physiol Heart Circ Physiol. 2003;285:H1479–88. doi: 10.1152/ajpheart.01129.2002. [DOI] [PubMed] [Google Scholar]
- 21.Zhang HM, Yuan J, Cheung P, Luo H, Yanagawa B, Chau D, et al. Overexpression of Interferon-γ-inducible GTPase Inhibits Coxsackievirus B3-induced Apoptosis through the Activation of the Phosphatidylinositol 3-Kinase/Akt Pathway and Inhibition of Viral Replication . J BiolChem. 2003;278:33011–19. doi: 10.1074/jbc.M305352200. [DOI] [PubMed] [Google Scholar]
- 22.Haq S, Choukroun G, Kang ZB, Ranu H, Matsui T, Rosenzweig A, et al. Glycogen synthase kinase-3beta is a negative regulator of cardiomyocyte hypertrophy. J Cell Biol. 2000;151:117–30. doi: 10.1083/jcb.151.1.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li HJ, Yin H, Yao YY, Shen B, Bader M, Chao L, et al. Tissue kallikrein protects against pressure overload-induced cardiac hypertrophy through kinin B2 receptor and glycogen synthase kinase-3β activation. Cardiovasc Res. 2006 doi: 10.1016/j.cardiores.2006.10.014. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu Z, Park SS, Mueller RA, Bagnell RC, Patterson C, Boysen PG. Adenosine produces nitric oxide and prevents mitochondrial oxidant damage in rat cardiomyocytes. Cardiovasc Res. 2005;65:803–12. doi: 10.1016/j.cardiores.2004.12.004. [DOI] [PubMed] [Google Scholar]
- 25.Fujii H, Ichimori K, Hoshiai K, Nakazawa H. Nitric oxide inactivates NADPH oxidase in pig neutrophils by inhibiting its assembling process. J Biol Chem. 1997;272:32773–778. doi: 10.1074/jbc.272.52.32773. [DOI] [PubMed] [Google Scholar]
- 26.Colasanti M, Suzuki H. The dual personality of NO. Trends Pharmacol Sci. 2000;21:249–52. doi: 10.1016/s0165-6147(00)01499-1. [DOI] [PubMed] [Google Scholar]
- 27.Förstermann U, Münzel T. Endothelial Nitric Oxide Synthase in Vascular Disease: From Marvel to Menace. Circulation. 2006;113:1708–14. doi: 10.1161/CIRCULATIONAHA.105.602532. [DOI] [PubMed] [Google Scholar]
- 28.Yoshiyama M, Omura T, Takeuchi K, Kim S, Shimada K, Yamagishi H, et al. Angiotensin Blockade Inhibits Increased JNKs, AP-1 and NF- κB DNA-binding Activities in Myocardial Infarcted Rats. J Mol Cell Cardiol. 2001;33:799–10. doi: 10.1006/jmcc.2001.1351. [DOI] [PubMed] [Google Scholar]
- 29.Zhang GX, Kimura S, Nishiyama A, Shokoji T, Rahman M, Yao L, et al. Cardiac oxidative stress in acute and chronic isoproterenol-infused rat. Cardiovasc Res. 2005;65:230–38. doi: 10.1016/j.cardiores.2004.08.013. [DOI] [PubMed] [Google Scholar]
- 30.Wang J, Xu N, Feng X, Hou N, Zhang J, Cheng X, et al. Targeted disruption of Smad4 in cardiomyocytes results in cardiac hypertrophy and heart failure. Circ Res. 2005;97:821–8. doi: 10.1161/01.RES.0000185833.42544.06. [DOI] [PubMed] [Google Scholar]
- 31.Calixto JB, Carbrini DA, Ferreira J, Compos MM. Kinins in pain and inflammation. Pain. 2000;87:1–5. doi: 10.1016/S0304-3959(00)00335-3. [DOI] [PubMed] [Google Scholar]
- 32.Tschope C, Heringer-Walther S, Walther T. Regulation of the kinin receptors after induction of myocardial infarction: a mini-review. Braz J Med Biol Res. 2000;33:701–08. doi: 10.1590/s0100-879x2000000600011. [DOI] [PubMed] [Google Scholar]
- 33.Araujo RC, Kettritz R, Fichtner I, Paiva AC, Pesquero JB, Bader M. Altered neutrophil homeostasis in kinin B1 receptor-deficient mice. Biol Chem. 2001;382:91–5. doi: 10.1515/BC.2001.014. [DOI] [PubMed] [Google Scholar]
- 34.Souza DG, Lomez ES, Pinho V, Pesquero JB, Bader M, Pesquero JL, et al. Role of bradykinin B2 and B1 receptors in the local, remote, and systemic inflammatory responses that follow intestinal ischemia and reperfusion injury. J Immunol. 2004;172:2542–48. doi: 10.4049/jimmunol.172.4.2542. [DOI] [PubMed] [Google Scholar]