

Abstract
Cytochrome P450 (CYP) epoxygenases and their arachidonic acid (AA) metabolites, the epoxyeicosatrienoic acids (EETs), have been shown to produce reductions in infarct size in canine myocardium following ischemia/reperfusion injury via opening of either the sarcolemmal KATP (sarc KATP) or mitochondrial KATP (mito KATP) channel. In the present study, we subjected intact rat hearts to 30 minutes of left coronary artery occlusion and 2 hours of reperfusion followed by tetrazolium staining to determine infarct size as a percent of the area at risk (IS/AAR,%). The results demonstrate that the two major regioisomers of the CYP epoxygenase pathway, 11,12-EET (2.5 mg/kg, iv) and 14,15-EET (2.5 mg/kg, iv) significantly reduced myocardial infarct size (IS/AAR,%) in rats as compared with control (41.9±2.3%, 40.9±1.2% vs. 61.5±1.6%, respectively), whereas, a third regioisomer, 8,9-EET (2.5 mg/kg,iv) had no effect (55.2±1.4). The protective effect of pretreatment with 11,12-and 14,15-EET was completely abolished (61.9±0.7%, 58.6±3.1%, HMR; 63.3±1.2%, 63.2±2.5%, 5-HD) in the presence of the selective sarc KATP channel antagonist, HMR 1098 (6 mg/kg, iv) or the selective mito KATP channel antagonist, 5-HD (10 mg/kg, iv) given 10 minutes after 11,12- or 14,15-EET administration but 5 minutes prior to index ischemia. Furthermore, concommitant pretreatment with 11,12- or 14,15-EET in combination with the free radical scavenger, 2-mercaptopropionyl glycine (2-MPG), at a dose (20 mg/kg,iv) that had no effect on IS/AAR (57.7±1.3%), completely abolished the cardioprotective effect of 11,12 and 14,15- EET (58.2±1.6%, 61.4±1.0%), respectively. These data suggest that part of the cardioprotective effects of EETs in rat hearts against infarction is the result of an initial burst of reactive oxygen species (ROS) and subsequent activation of both the sarc KATP and mito KATP channel.
Keywords: CYP450, 8, 9-EET, 11, 12-EET, 14, 15-EET, reactive oxygen species, mito-and sarc- KATP channels
Recently, the influence of the cytochrome P450 pathway (CYP) has been investigated in several animal models of ischemia-reperfusion injury (5,6,12,13,16,17). In an earlier study, Moffat et al.(11) examined the cardiac effects of all 4 regioisomers of the EETs in isolated guinea pig hearts and ventricular myocytes under conditions of normoxia and low-flow ischemia. The EETs had no effect on the contractile performance of the isolated hearts under normoxic conditions at 1–16 ng/ml. Similarly, under low-flow ischemia, 1 ng/ml of the EETs had no significant effect on the decreases in contractility, heart rate or perfusion pressure as compared to nontreated hearts during ischemia or at 30 minutes after reperfusion, although in the presence of 5,6 or 11,12-EET recovery of function was delayed for 10 min.. In isolated myocytes, the EETs had variable effects depending on the regioisomer studied, with 5,6 and 11,12-EET increasing cell shortening and cell calcium, whereas, 8,9 and 14,15-EET had no effect in isolated myocytes at the concentrations (100 pg/ml to 20 ng/ml) studied. Subsequently, , transgenic mice with cardiomyocyte-specific overexpression of CYP2J2, the predominant human CYP oxygenase, demonstrated an improvement in functional recovery after 20-min of global ischemia and 40-min of reperfusion(19). This beneficial action was hypothesized to be the result of an increased production of epoxyeicosatrienoic acids (EETs) since administration of exogenous 11,12-EET also produced a similar cardioprotective effect in control mice. More recently, treatment with several nonselective CYP inhibitors resulted in a reduction in myocardial infarct size in mice and rats and the reduction of infarct size was attributed to the ability of these compounds to decrease the formation and/or release of reactive oxygen species (ROS) at reperfusion (5). In contrast, a number of papers suggest that a small initial burst of ROS serve as a signaling element and trigger for ischemic preconditioning (IPC) and cardioprotection produced by several pharmacological agents including acetylcholine, opioids and bradykinin (2). However, the role of ROS as an integral part or trigger of EET-induced cardioprotection has not been addressed.
Additionally, several papers have suggested that the EETs are direct activators of sarcolemmal ATP-sensitive potassium channels (sarcKATP) in membrane patches isolated from rat myocardium (9,10) and have been shown to activate mitochondrial KATP channels (mitoKATP) in mouse hearts (16). Recent work from our laboratory (6) has also shown that the sarc KATP channel but not the mito KATP channel is a mediator of the protection observed in rat hearts following the administration of two CYP omega-hydroxylase inhibitors, miconazole and 17-ODYA, just prior to reperfusion. Since one or both of these channels have been shown to be important in mediating cardioprotection produced by the EETS, we decided to test the importance of these two KATP channels in mediating the cardioprotective effects of the EETs in our rat model of infarction.
Materials and Methods
Materials
8,9-, 11,12- and 14,15-EET were synthesized in the laboratory of Dr. J.R. Falck(12). 5-Hydroxydecanoic acid (5-HD) and 2-mercaptopropionyl-glycine (2-MPG) were obtained from Sigma Chemical Company. HMR1098 was kindly provided by Dr. Hans Gogelein, Aventis Pharmaceuticals. 8,9-EET, 11,12-EET and 14,15-EET were dissolved in a vehicle composed of 95% ethanol, polyethylene glycol 200 and 1 N sodium hydroxide (5:5:1). 5-HD, HMR 1098 and 2-MPG were dissolved in distilled water.
General preparation of rats
All experiments conducted in this study were in accordance with the “Position of the American Heart Association on Research and Animal Use” adopted by the American Heart Association and the guidelines of the Biomedical Resource Center of the Medical College of Wisconsin. The Medical College of Wisconsin is accredited by the American Association of Laboratory Animal Care (AALAC).
The protocol for the rat preparation and experiments has been previously described in detail (6). Briefly, male Sprague-Dawley rats, weighing 250–300 g were fasted overnight and anesthetized with Inactin (Sigma, 100 mg/kg, ip) and ventilated with room air supplemented with 100% oxygen. Body temperature was maintained at 37±1°C with a heating pad. Arterial blood pH, pCO2, and pO2 were monitored at selected intervals by an AVL automatic blood gas system and maintained within normal physiological limits (pH 7.35 to 7.45, pCO2 30 to 40 mmHg, and pO2 85 to 120 mmHg) by adjusting the respiration rate and oxygen flow or by intravenous administration of 1.5% sodium bicarbonate if necessary. Hemodynamics, heart rate, and coronary blood flow were monitored throughout the experiment. A cannula was placed into the jugular vein and carotid artery to administer drugs and measure peripheral hemodynamics, respectively.
Experimental design
Rats were sequentially assigned to 13 groups for the different treatments. Typically, 8 rats were included in each group of experiments. At 15 min before the 30-min LAD occlusion period, 8,9-EET (2.5 mg/kg), 11,12-EET (2.5 mg/kg) or 14,15-EET (2.5 mg/kg) or vehicle were administered by intravenous injection for 1–2 min as shown in Fig 1. The 2.5 mg/kg dose was chosen based on preliminary experiments in which 1 mg/kg of 11,12 and 14,15-EET was only modestly effective at reducing IS/AAR and 5 mg/kg was similar in efficacy in reducing IS/AAR as 2.5 kmg/kg (data not shown). In the KATP channel experiments either HMR 1098 (6 mg/kg, iv), the sarcolemmal selective antagonist or 5-HD (10 mg/kg, iv), the mitochondrial selective antagonist (1) were administered alone or 10 minutes after EET administration (5 minutes prior to occlusion). The 2-MPG (20 mg/kg, iv) was given 5 minutes prior to the EET administration. In all groups, hemodynamic measurements and blood gas analyses were performed at baseline and at 15 minutes into the 30-min occlusion period and at the end of 2 hr of reperfusion.
Figure 1.
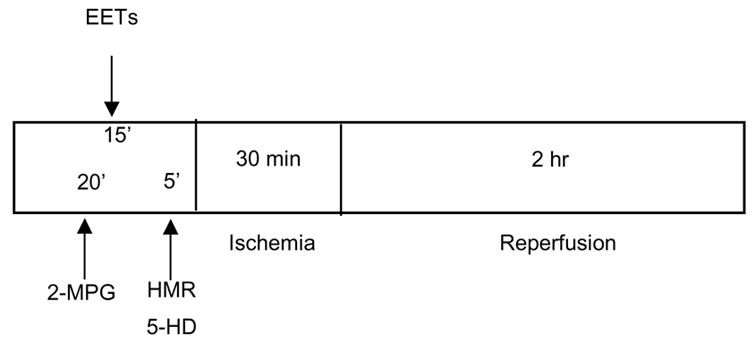
Experimental protocols for each series. Abbreviations:EETs:epoxyeicosa- trienoic acids; 2-MPG:2-mercaptopropionyl glycine; HMR: HMR 1098; 5-HD: 5- hydroxydecanoic acid.
Infarct size determination
Infarct size was determined as previously described (6,7). Briefly, at the end of the 2-h reperfusion period, the LAD was reoccluded. To determine the anatomic area at risk (AAR) and the nonischemic area, 1 mL of Patent blue dye was injected into the jugular vein and the heart was arrested by an iv injection of potassium chloride. The heart was then immediately removed and the left ventricle (LV) was dissected and sliced into serial transverse sections 1–2 mm in width. The nonstained ischemic area and the blue-stained normal area were separated and incubated with 1% 2,3,5-triphenyltetrazolium chloride (TTC, Sigma) in 0.1 mol/L phosphate buffer, pH 7.4 at 37°C for 15 min. After incubation overnight in 10% formaldehyde, the noninfarcted and infarcted tissues within the AAR were separated and determined gravimetrically under a dissecting microscope. Infarct size was expressed as a percent of the AAR.
Statistical analysis
All values are expressed as mean ± SEM. Differences between groups in hemodynamics were compared by using a two-way analysis of variance (ANOVA) followed by a Tukey’s post hoc test. Differences between groups in AAR and infarct size were compared by one-way ANOVA.. Differences between groups were considered significant if p < 0.05.
Results
Hemodynamics
Heart rate and mean arterial blood pressure at baseline between groups or at 30 minutes following occlusion or 2 hours following reperfusion were not significantly different (Data not shown). These data suggest that changes in infarct size were not the result of changes in peripheral hemodynamics.
Effects of 8,9-, 11,12- and 14,15-EET on myocardial infarct size: Importance of KATP channels
Myocardial infarct size (IS) data expressed as a percent of the area at risk (IS/AAR) in the 3 EET-treated groups are shown in Figure 2. 11,12-EET and 14,15-EET (2.5 mg/kg, iv) significantly reduced IS/AAR (41.9±2.3 and 40.9±1.2%, respectively) as compared to the control group (61.5±1.3%) when administered 15 minutes prior to occlusion. Interestingly, another regiosiomer, 8,9-EET, had no protective effect on IS/AAR. The beneficial effects of 11,12- and 14,15-EET pretreatment on IS/AAR were completely abolished by pretreatment with the sarc KATP channel blocker, HMR 1098 ( 61.9±0.7 and 58.6±3.1%, Fig. 3). Simliar effects were observed in the presence of the mito KATP channel inhibitor, 5-HD (63.3±1.2 and 63.3±2.5%, Fig. 4). HMR 1098 and 5-HD had no effect on IS/AAR when administered in the absence of the EETs (HMR=60.2±1.4%; 5-HD= 61.6± 1.7%).
Figure 2.
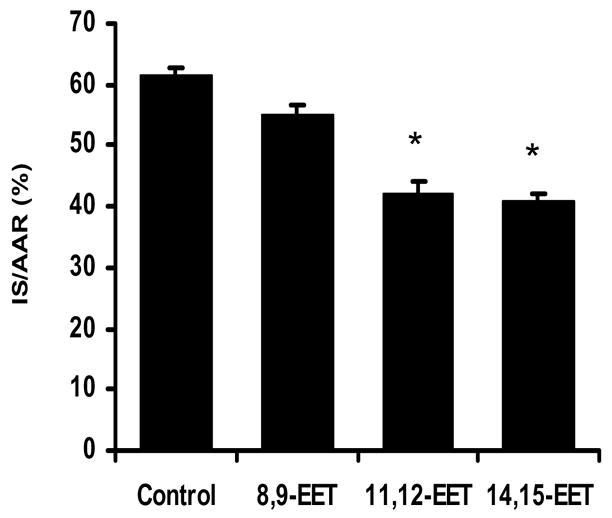
Effect of 3 regioisomers of the EETs, 8,9-, 11,12 and 14,15-EET, on myo- Myocardial infarct size expressed as a percent of the area at risk (IS/AAR)*p<0.01 versus the control group (N=8/group).
Figure 3.
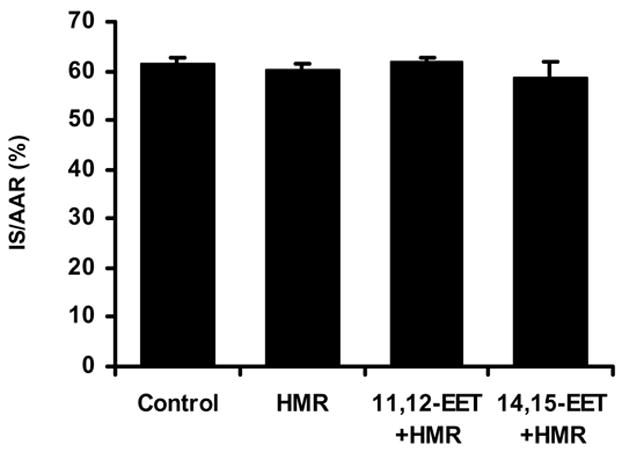
Effect of the sarcolemmal selective KATP channel antagonist HMR 1098 on the reduction of IS/AAR produced by 11,12- and 14,15-EET. HMR 1098 was given 10 minutes after the EETs 5 minutes prior to index ischemia (N=8/group).
Figure 4.
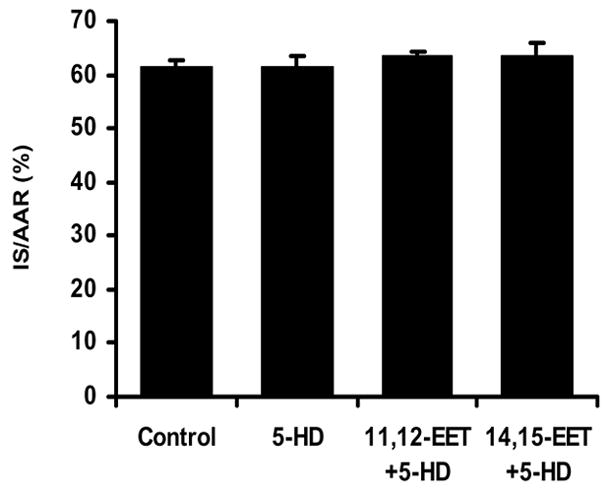
Effect of the mitochondrial selective KATP channel antagonist 5-HD on the reduction of IS/AAR produced by 11,12- and 14,15-EET. 5-HD was given 10 minutes after the EETs 5 minutes prior to index ischemia (N=8/group).
Effects of 2-MPG on EET-induced cardioprotection
To determine if reactive oxygen species (ROS) are involved in the triggering of cardioprotection by 11,12- or 14,15-EET, in 3 groups of rats we adminstered the ROS scavenger 2- MPG at 20 mg/kg, iv at 20 minutes prior to the occlusion/reperfusion protocol in control rats and 5 minutes prior to pretreatment with the EETs which were administered 15 minutes prior to occlusion as above (Fig. 1). By itself 2-MPG had no effect on IS/AAR (57.7±1.3%), however 2-MPG completely abolished cardioprotection produced by 11,12-EET (58.2±1.6%) and 14,15-EET (61.4±1.0%).
Discussion
We have previously demonstrated that CYP ω-hydroxylases and their AA metabolite, 20-HETE, have a detrimental role in enhancing myocardial ischemia-reperfusion injury in the canine and rat myocardium (6,14,16). In the present study, we demonstrate that exogenous administration of the 2 major CYP epoxygenase products (19), 11,12-EET and 14,15-EET but not 8,9-EET, significantly reduced infarct size in rat hearts subjected to a 30-min occlusion period followed by 2-h of reperfusion. The reduction in infarct size was observed when the EETs were administered 15 minutes prior to coronary artery occlusion. This observation, which agrees with our recently published data in the canine heart (13,14), suggests that EETs may reduce ischemia and/or reperfusion injury and may have important clinical implications (18). The present data suggest that the protective effects of the EETs are at least partially the result of opening sarcolemmal and mitochondrial KATP channels since both a selective sarcolemmal channel blocker HMR 1098 and the selective mitochondrial channel blocker, 5-HD, abolished the protective effects of the EETs. Our data also suggest that an initial burst of ROS may be necessary to trigger EET-induced cardioprotection since their protective effects were completely abrogated by pretreatment with the ROS scavenger, 2-MPG. These results agree with recent results from several other laboratories (2,3) which suggest that an initial burst of ROS triggers ischemic and pharmacological preconditioning to acetylcholine, bradykinin, opioids and to the direct mito KATP opener, diazoxide. In the study by Cohen et al. (2), 2-MPG was also used to assess the role of ROS in triggering cardioprotection, whereas, dichorofluorescein (DCF) was used to more directly assess ROS production by the mitochondria in the other study (3). Further sutdies, in which an antioxidant such as 2-MPG, is given after EET administration but prior to the prolonged ischemic time, are needed to prove that it is the initial burst of ROS elicited by the EETs which triggers the cardioprotected phenotype and not an effect manifest later on in the ischemia/reperfusion protocol. However, since the EETs have also been shown to be protective when administered just prior to reperfusion in rats (6), it is unlikely that an initial burst of ROS is necessaary for the cardioprotection observed under these conditions.
Interestingly, previous results obtained in dogs demonstrated that the protective effect of the EETs was completely blocked by the nonselective KATP channel blocker, glibenclamide (7). This finding in dogs was not that surprising since the EETs have been previously shown to activate sarcKATP channels in membrane patches (9,10) isolated from rat hearts and mitoKATP channels as assessed by changes in flavoprotein fluorescence in mitochondria isolated from mouse hearts (17). although several recent papers suggest that a mitochondrial KATP channel may not exist in a form similar to the sarc KATP channel and that 5-HD may be acting on fatty acid metabolism and not as a selective blocker of the mito KATP channel (9). In our previous studies in rats, we showed that the sarcolemmal selective blocker, HMR 1098, given 5 minutes prior to the CYP ω-hydroxylase inhibitor, DDMS, blocked the protective effect when administered 10 minutes prior to reperfusion, whereas, 5-HD was ineffective when given at the same time point (6 ). These data and the present data with the EETs indirectly suggest that the sarc KATP channel may be important later in the protective signaling pathway produced by EETs, perhaps at reperfusion, whereas, the mito KATP may serve its role in the more proximal triggering phase prior to index ischemia. Nevertheless, more experiments are needed to confirm or refute this hypothesis.
In conclusion, the present results indicate that exogenous administration of two of the major regioisomers of the CYP epoxygenase pathway, 11,12-EET and 14,15-EET, both produce equivalent significant reductions in IS/AAR in the rat heart when given prior to occlusion and that these effects are mediated by opening of both sarcKATP and mito KATP channels. The cardioprotective effects observed were nearly equivalent to that observed previously by several ω-hydroxylase inhibitors in rats (6). Although the cellular mechanism(s) responsible for the protection observed following EET administration are yet to be determined, the present data obtained with ROS scavenger, 2-MPG, suggest that a redox-dependent mechanism may be part of the signaling pathway involved as has been previously shown by several investigators studying IPC and pharmacological preconditioning (2,3,4). The site of ROS formation has yet to be identified and will be the focus of future studies.
Figure 5.
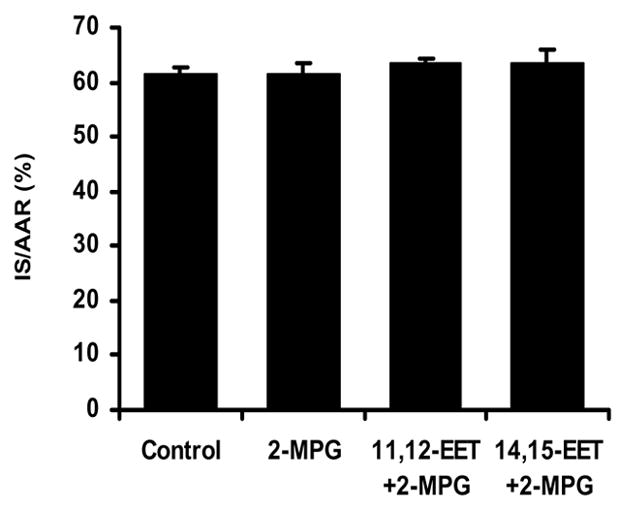
Effect of the free radical scavenger 2-MPG on the reduction of IS/AAR pro- duced by 11,12- and 14,15-EET. 2-MPG was administered 5 minutes prior to the EETs and 20 minutes before index ischemia (N=8/group).
Acknowledgments
This work was supported by National Heart, Lung, and Blood Institute Grant HL74314-01.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Garrett J. Gross, Department of Pharmacology and Toxicology, Medical College of Wisconsin 8701 Watertown Plank Road, Milwaukee, WI 53226
Anna Hsu, Department of Pharmacology and Toxicology, Medical College of Wisconsin 8701 Watertown Plank Road, Milwaukee, WI 53226.
John R. Falck, Departments of Biochemistry and Pharmacology, University of Texas Southwestern Medical Center, Dallas, TX 75390
Kasem Nithipatikom, Department of Pharmacology and Toxicology, Medical College of Wisconsin 8701 Watertown Plank Road, Milwaukee, WI 53226.
References
- 1.Auchampach JA, Grover GJ, Gross GJ. Blockade of ischemic preconditioning in dogs by the novel ATP dependent potassium channel antagonist sodium 5-hydroxydecanoate. Cardiovasc Res. 1992;26:1054–1062. doi: 10.1093/cvr/26.11.1054. [DOI] [PubMed] [Google Scholar]
- 2.Cohen MV, Yang XM, Liu GS, Heusch G, Downey JM. Acetylcholine, bradykinin, opioids, and phenylephrine but not adenosine, trigger preconditioning by generating free radicals and opening mitochondrial KATP channels. Circ Res. 2001;89:273–278. doi: 10.1161/hh1501.094266. [DOI] [PubMed] [Google Scholar]
- 3.Forbes RA, Steenbergen C, Murphy E. Diazoxide-induced cardioprotection requires signaling through a redox-sensitive mechanism. Circ Res. 2001;88:802–809. doi: 10.1161/hh0801.089342. [DOI] [PubMed] [Google Scholar]
- 4.Gottlieb RA. Cytochrome P450: major player in reperfusion injury. Arch Biochem Biophys. 2003;420:262–267. doi: 10.1016/j.abb.2003.07.004. [DOI] [PubMed] [Google Scholar]
- 5.Granville DJ, Tashakkor B, Takeuchi C, Gustafsson AB, Huang C, Sayen MR, Wentworth P, Jr, Yeager M, Gottlieb RA. Reduction of ischemia and reperfusion-induced myocardial damage by cytochrome P450 inhibitors. Proc Natl Acad Sci U S A. 2004;101:1321–1326. doi: 10.1073/pnas.0308185100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gross ER, Nithipatikom K, Hsu AK, Peart JN, Falck JR, Campbell WB, Gross GJ. Cytochrome P450 omega-hydroxylase inhibition reduces infarct size during reperfusion via the sarcolemmal KATP channel. J Mol Cell Cardiol. 2004;37:1245–1249. doi: 10.1016/j.yjmcc.2004.10.008. [DOI] [PubMed] [Google Scholar]
- 7.Gross GJ, Auchampach JA. Blockade of ATP-sensitive potassium channels prevents myocardial preconditioning in dogs. Circ Res. 1992;70:223–233. doi: 10.1161/01.res.70.2.223. [DOI] [PubMed] [Google Scholar]
- 8.Hanley PJ, Daut J. KATP channels and preconditioning: A re-examination of the role of mitochondrial KATP channels and an overview of alternative mechanisms. J Mol Cell Cardiol. 2005;39:17–50. doi: 10.1016/j.yjmcc.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 9.Lu T, Hoshi T, Weintraub NL, Spector AA, Lee HC. Activation of ATP-sensitive K(+) channels by epoxyeicosatrienoic acids in rat cardiac ventricular myocytes. J Physiol. 2001;537:811–827. doi: 10.1111/j.1469-7793.2001.00811.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lu T, VanRollins M, Lee HC. Stereospecific activation of cardiac ATP-sensitive K(+) channels by epoxyeicosatrienoic acids: a structural determinant study. Mol Pharmacol. 2002;62:1076–1083. doi: 10.1124/mol.62.5.1076. [DOI] [PubMed] [Google Scholar]
- 11.Moffat MP, Ward CA, Bend JR, Mock T, Farhangkhoee P, Karmazyn M. Effects of epoxyeicosatrienoic acids on isolated hearts and ventricular myocytes. Am J Physiol. 1993;264:H1154–H1160. doi: 10.1152/ajpheart.1993.264.4.H1154. [DOI] [PubMed] [Google Scholar]
- 12.Nithipatikom K, DiCamelli RF, Kohler S, Gumina RJ, Falck JR, Campbell WB, Gross GJ. Determination of cytochrome P450 metabolites of arachidonic acid in coronary venous plasma during ischemia and reperfusion in dogs. Anal Biochem. 2001;292:115–124. doi: 10.1006/abio.2001.5044. [DOI] [PubMed] [Google Scholar]
- 13.Nithipatikom K, Endsley MP, Moore JM, Isbell MA, Falck JR, Campbell WB, Gross GJ. Effects of selective inhibition of cytochrome P-450 {omega}-hydroxylases and ischemic preconditioning in myocardial protection. Am J Physiol Heart Circ Physiol. 2006;290:H500–H505. doi: 10.1152/ajpheart.00918.2005. [DOI] [PubMed] [Google Scholar]
- 14.Nithipatikom K, Grall AJ, Holmes BB, Harder DR, Falck JR, Campbell WB. Liquid chromatographic-electrospray ionization-mass spectrometric analysis of cytochrome P450 metabolites of arachidonic acid. Anal Biochem. 2001;298:327–336. doi: 10.1006/abio.2001.5395. [DOI] [PubMed] [Google Scholar]
- 15.Nithipatikom K, Gross ER, Endsley MP, Moore JM, Isbell MA, Falck JR, Campbell WB, Gross GJ. Inhibition of cytochrome P450omega-hydroxylase: a novel endogenous cardioprotective pathway. Circ Res. 2004;95:e65–71. doi: 10.1161/01.RES.0000146277.62128.6f. [DOI] [PubMed] [Google Scholar]
- 16.Seubert J, Yang B, Alyce Bradbury J, Graves J, Miller L, Gabel S, Gooch R, Foley J, Newman J, Mao L, Rockman HA, Hammock BD, Murphy E, Zeldin DC. Enhanced Postischemic Functional Recovery in CYP2J2 Transgenic Hearts Involves Mitochondrial ATP-Sensitive K+ Channels and p42/p44 MAPK Pathway. Circ Res. 2004;95:506–514. doi: 10.1161/01.RES.0000139436.89654.c8. [DOI] [PubMed] [Google Scholar]
- 17.Seubert J, Sinal CJ, Graves J, DeGraff LM, Bradbury A, Lee CR, Goralski K, Carey MA, Luria A, Newman JW, Hammock BD, Falck JR, Roberts H, Rockman HA, Murphy E, Zeldin DC. Role of soluble epoxide hydrolase in postischemic recovery of heart contractile function. Circ Res. 2006;99:442–450. doi: 10.1161/01.RES.0000237390.92932.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Spieker M, Liao JK. Cytochrome P450 epoxygenase CYP2J2 and the risk of coronary artery disease. Trends Cardiovasc Med. 2006;16:204–208. doi: 10.1016/j.tcm.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 19.Wu S, Chen W, Murphy E, Gabel S, Tomer KB, Foley J, Steenbergen C, Falck JR, Moomaw CR, Zeldin DC. Molecular cloning, expression, and functional significance of a cytochrome P450 highly expressed in rat heart myocytes. J Biol Chem. 1997;272:12551–12559. doi: 10.1074/jbc.272.19.12551. [DOI] [PubMed] [Google Scholar]