

Abstract
It is well established that reperfusion of the heart is the optimal method of salvaging previously ischemic myocardium. However, the idea of reperfusion injury, i.e. injury caused by the process of reperfusion per se still remains a controversial issue. In this review, we present mounting evidence supporting the concept that reperfusion injury exists, based on work conducted with adenosine and opioid receptor ligands, and the discovery of two new concepts regarding reperfusion injury: ‘postconditioning’ (POC) and the reperfusion injury salvage kinase (RISK) signaling pathway.
Introduction
The concept of reperfusion injury has been a subject of debate for the past three decades, in which some investigators believe that all injury develops during the ischemic period whereas others argue that blood reflow extends tissue injury due to the release of oxygen-derived free radicals, dysregulation of intracellular and mitochondrial calcium, microvascular dysfunction leading to incomplete return of blood flow to areas of the microcirculation (the no-reflow phenomenon), an overzealous inflammatory reaction involving influx of various populations of immune cells, and delayed cell death due to apoptosis. However, several key discoveries in recent years have bolstered the concept of reperfusion injury. First, several different pharmacological agents, including adenosine and opioid receptor ligands, have been shown to attenuate myocardial injury when applied at the time of reperfusion [1-3]. Second, Vinten-Johansen's group has introduced a novel method of reperfusion that provides marked cardioprotection involving reinstitution of blood flow in a stuttering fashion [4, 5]. This phenomenon, termed ‘postconditioning’ (POC), is distinctly different and clearly more clinically relevant than that of ischemic ‘preconditioning’, (IPC) whereby intermittent ischemia/reperfusion is applied prior to a prolonged coronary occlusion [4, 5]. Finally, a pro-survival signaling pathway termed the reperfusion injury salvage kinase (RISK) pathway has been uncovered in the myocardium [6]. Recent evidence suggests that this signaling cascade may provide a molecular mechanism by which pharmacological agents as well as IPC and POC may, in part, reduce reperfusion injury. This pathway (Figure 1) includes several anti-apoptotic pro-survival signaling kinases (phosphatidylinositol-3-OH kinase [PI-3 kinase] - Akt, mammalian target of rapamycin [mTOR], p70s6 kinase, glycogen synthase kinase 3β [GSK3β], p42/p44 extracellular signal-regulated kinases [ERK 1/2]), ATP sensitive potassium (KATP) channels, and the mitochondrial permeability transition pore (MPTP), which may serve as a major convergence point that determines whether a cell survives or not. In this article, we review current evidence to suggest that reperfusion injury exists, based on recent discoveries in the field of cardioprotection with adenosine, opioids, and POC.
Figure 1.
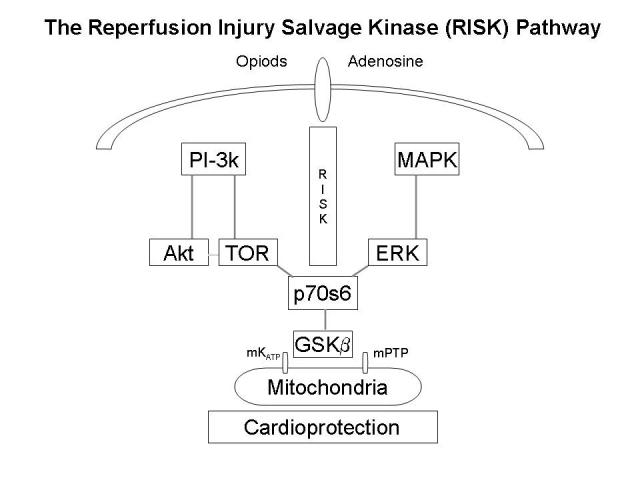
Schematic illustration of anti-apoptotic pro-survival signaling mechanisms, termed the reperfusion injury salvage kinase (RISK) pathway, that have been proposed to protect the heart from reperfusion injury. Reperfusion is believed to promote opening of the mitochondrial permeability transition pore, which induces both apoptotic and necrotic cell death due to the release of mitochondrial proteins and loss of ATP-generating capacity. Activation of G protein-coupled receptors (opioid and potentially adenosine receptors) or growth factor receptors during reperfusion is believed to initiate signaling mechanisms involving phosphatidyl inositol-3 kinase (PI-3k), akt, p42/p44 extracellular signal-regulated kinase (ERK), mammalian target of rapamycin (TOR), p70s6 kinase, and glycogen synthase kinase β (GSKβ) that prevents opening of the MPTP. It has been proposed that an isoform of the ATP sensitive potassium channel (KATP) may also be expressed in the mitochondria that regulates MPTP opening.
Adenosine and Reperfusion Injury
Studies with adenosine arguably provide the strongest evidence to suggest that reperfusion promotes tissues injury and that treatment with pharmacological agents can be used effectively to diminish it. In 1987, Olafsson and colleagues [7] first demonstrated that treatment with adenosine during reperfusion reduced infarct size in a dog model of left anterior descending coronary artery (LAD) occlusion and reperfusion. These investigators [7] infused adenosine directly into the coronary circulation of anesthetized dogs at a rate of 3.75 mg/min for the first hour of reperfusion after 90 min of total occlusion of the LAD coronary artery. After 24 hours of reperfusion, adenosine treatment was shown to reduce infarct size ∼75% and to improve both regional and global indices of ventricular function; adenosine treatment was also shown to reduce the degree of neutrophil infiltration and capillary plugging and preserve endothelial function. Although some suggested that adenosine was only effective if administered in conjunction with lidocaine [8], subsequent work by others essentially confirmed this initial finding by Olafsson and further observed that treatment with adenosine is only effective if the period of occlusion is relatively short (<3 hrs; [9, 10]). Using a similar dog model, Pitarys and colleagues [11] demonstrated that adenosine is also effective if administered systemically. These investigators [11] infused adenosine intravenously for the first hour of reperfusion in dogs subjected to 90 min of LAD occlusion, and found that infarct size was reduced ∼50% by adenosine treatment after 72 hours of reperfusion. In this study [11], adenosine was administered at a rate of 0.15 mg/kg/min, a dose that in anesthetized dogs did not decrease heart rate or blood pressure. Collectively, these results provided strong evidence that reperfusion injury exists, and that it comprises 50-75% of the final infarct size after reperfusion. Furthermore, since many of these initial studies involved assessment of infarct size or function after long periods of reperfusion (24-72 h), the results of these studies suggest that the protection against tissue injury provided by adenosine treatment is permanent.
Based on these positive preclinical experiments, the AMISTAD (Acute Myocardial Infarction Study of Adenosine) trials were initiated to determine whether intravenous treatment with adenosine provides benefit to patients with an acute myocardial infarction undergoing reperfusion therapy [12, 13]. The AMISTAD-I trial (236 patients) was designed to determine whether adenosine reduces infarct size in patients with an evolving myocardial infarction (ST-segment elevation myocardial infarction; STEMI) subjected to reperfusion therapy with thrombolytic agents [13]. The AMISTAD-II trial was a larger follow-up study (2,118 patients) focused on determining whether treatment with adenosine during reperfusion with either pharmacological or mechanical therapy improves clinical outcomes, defined as development of new congestive heart failure or death within 6 months [12]. In both of these multicenter prospective studies, adenosine was infused intravenously at a dose of 50 - 70 μg/kg/min for 3 hrs beginning no more than 6 hrs after the onset of symptoms, and infarct size was assessed by single-photon emission computed tomography (SPECT) myocardial perfusion imaging with technetium-99 m sestamibi*. Although there are varying interpretations of the results of the AMISTAD trials, it is generally considered that treatment with adenosine in both studies produced a significant reduction (∼67% in the AMISTAD-I trial and ∼60% in the AMISTAD-II trial with the 70 mg/kg/min dose) in infarct size in patients with anterior wall infarction, but that it did not significantly improve clinical outcomes [12, 13]. However, this apparent lack of a beneficial effect of adenosine treatment on clinical outcomes was explained by the fact that both of the studies were underpowered. Subsequent subset analysis of the AMISTAD-II trial revealed that treatment with adenosine produced a significant improvement in clinical outcomes if the analyses were confined to those patients that were successfully reperfused or were administered adenosine within 3 hrs of symptom onset, results that are consistent with the known cardioprotective profile of adenosine established previously in preclinical trials in dogs [14]. Overall, the AMISTAD studies support the contention that treatable reperfusion injury also exists in humans.
The AMISTAD trials also demonstrated that treating acute myocardial infarction patients with intravenous adenosine exerts no serious adverse clinical effects, although it tended to slightly increase the incidence of hypotension, bradycardia, heart block, and ventricular arrhythmias, likely due to activation of A1 adenosine receptors (AR) in conducting tissue and A2AARs in vascular smooth muscle cells [12-14]. In fact, an increased incidence in adverse events contributed to the decision to test a lower dose of adenosine (50 μg/kg/min) in the second AMISTAD trial. Thus, the cardioprotective effectiveness of adenosine, which is a non-selective agonist with equal potency at activating A1, A2A, and A3ARs†, appears to be limited by side effects when administered by the intravenous route.
For this reason, preclinical efforts are currently focused on testing the cardioprotective efficacy of subtype-selective adenosine receptor agonists in models of reperfusion injury. By targeting specific adenosine receptor subtypes, it has been hypothesized that the cardioprotective efficacy of selective agonists may not only produce fewer side-effects, but also be more efficacious since it has been speculated that activation of some adenosine receptor subtypes during reperfusion may promote tissue injury [15, 16]. Particular attention has focused on A2AAR agonists, since current evidence suggests that this receptor subtype mediates the protective effects of adenosine during reperfusion injury. Similar to results obtained with adenosine, intracoronary infusion with the A2AAR-selective agonist CGS 21680 has been reported by several groups to effectively reduce infarct size in canine and swine models by 50-60%, but only at doses (0.15 – 2.0 μg/kg/min) that produced either hypotension or reflex tachycardia [17-20]. Considering the importance of the A2AAR in regulating vascular tone, these initial studies therefore suggested that the usefulness of A2AAR agonists may also be confounded by undesirable hemodynamic effects. More recently, however, Glover and associates [21] showed that intravenous infusion of the A2AAR agonist ATL 146e effectively reduced infarct size in anesthetized dogs subjected to 90 min of LAD occlusion and 2 h of reperfusion when administered at a low dose (0.01 μg/kg/min) that did not alter heart rate, systemic blood pressure, or coronary blood flow. Since ATL 146e was infused prior to occlusion, during occlusion, and throughout reperfusion in this study [21], it is unclear whether it was effective by specifically reducing reperfusion injury. Nevertheless, this study supports the concept that non-vasoactive doses of A2AAR agonists may be effective at reducing reperfusion injury, suggesting that the cardioprotective potency of certain A2AAR agonists may be greater than their potency to dilate blood vessels.
In additional to A2AAR agonists, A3AR agonists have also been tested for their efficacy at protecting against myocardial reperfusion injury. The A3AR is the most recent member of the adenosine receptor family that, like the A1AR, is coupled to inhibitory Gi proteins [22]. Although most previous work has focused on the involvement of the A3AR in IPC, we have recently reported that intravenous bolus administration of 100 μg/kg of the A3AR agonist IB-MECA immediately before reperfusion reduced infarct size by 50% in dogs subjected to 60 min of LAD occlusion and 3 h of reperfusion, suggesting that activation of the A3AR can also effectively reduce reperfusion injury [23]. Administration of IB-MECA in this study did not alter systemic hemodynamic parameters or coronary blood flow [23]. Similar findings have been reported by Tracey and colleagues [24] in dogs using the highly selective A3AR agonist CP-602,903. Since the A3AR does not appear to be involved in regulating vascular tone or heart contractile function, A3 agonists may have less potential for producing adverse hemodynamic effects compared to adenosine or A2AAR agonists.
Regarding specific mechanisms, compelling evidence suggests that adenosine and A2AAR agonists are effective by suppressing inflammation associated with reperfusion. Reperfusion induces a vigorous inflammatory response characterized by a dramatic increase in neutrophil adherence, which leads to capillary plugging, edema, and a reduction in coronary blood flow [25]. A2AARs are highly expressed on cells involved in the inflammatory response including neutrophils, macrophages, and endothelial cells [3, 26-30]. Stimulation of A2AARs on neutrophils inhibits oxidase activity, degranulation, and adhesion molecule engagement [3, 26-30]. Activation of A2AARs on endothelial cells and macrophages inhibits pro-inflammatory cytokine/chemokine production and adhesion molecule expression [3, 26-30]. Most of these actions of the A2AAR are mediated by the Gs protein-cAMP signaling axis [3, 26-30]. Thus, A2AARs clearly play a critical role in inhibiting pro-inflammatory responses and reducing immune cell-mediated injury. Numerous in vivo studies have provided supportive evidence to suggest that adenosine and A2AAR agonists suppress indices of inflammation during reperfusion injury, including neutrophil accumulation and pro-inflammatory protein expression [3]. Interestingly, Yang and colleagues [31] recently conducted an elegant study examining the mechanism by which ATL 146e reduces reperfusion injury using A2AAR gene ‘knock-out’ mice and immunocompromised Rag-1 ‘knock-out’ mice lacking B and T lymphocytes. These investigators [31] found that ATL 146e (5 or 10 μg/kg bolus given at the onset of reperfusion) reduced infarct size in wild-type mice, but not in global A2AAR gene knock-out mice, chimeric mice lacking the expression of A2AARs only on bone marrow-derived cells, or RAG-1 ‘knock-out’ mice. These findings provided additional evidence supporting the theory that A2AAR activation reduces reperfusion injury by an anti-inflammatory mechanism, but further pointed towards interactions with A2AARs expressed in bone marrow-derived T and B lymphocytes [31]. Importantly, these results are the first to implicate involvement of lymphocytes (probably T lymphocytes) in injury associated with myocardial ischemia and reperfusion. Although it remains to be determined, it has been speculated that neutrophils and monocytic cells may be recruited to the reperfused myocardium by cytokines generated by specific subsets of natural killer T cells activated by tissue injury.
Current evidence also suggests that A3AR agonists effectively reduce reperfusion injury by suppressing inflammatory responses. Ge and colleagues [32] recently presented preliminary data showing that the A3AR agonist Cl-IB-MECA effectively reduced infarct size in wild-type mice, but not in A3AR gene knock-out mice or in bone marrow chimeric mice lacking the expression of A3ARs on bone marrow-derived cells. Although the A3AR was initially labeled as a pro-inflammatory receptor because it stimulates degranulation of mast cells from rodent specifies‡, more recent work has uncovered that this receptor subtype may also serve anti-inflammatory roles. For instance, it has been proposed that the A3AR inhibits cytokine expression from certain cell types, although this remains controversial [27]. More recently, we have demonstrated that activation of the A3AR inhibits superoxide production and chemotaxis of mouse neutrophils [33]. Although most data suggests that A3ARs are effective by inhibiting inflammatory responses, Park and colleagues [34] have recently suggested that the A3AR agonist IBMECA reduces reperfusion injury by inhibiting opening of the mitochondrial permeability transition pore via pro-survival kinase signaling. In this study using rats [34], however, it is unlikely that IB-MECA functioned via the A3AR since it was used at a very high concentration and its effects were blocked by the human A3AR antagonist MRS 1334, which displays very low potency for rodent A3ARs [35].
Opioid Receptors and Reperfusion Injury
Endogenous opioid peptides and their respective receptors have been studied extensively in the central nervous system in relation to pain perception. However, it is becoming increasingly appreciated that the opioid system also functions in the myocardium where it regulates normal and disease states. Of the three known opioid receptors (μ, δ, and κ), the Gi protein-coupled δ and κ receptors have been shown to be expressed in ventricular myocytes; these receptors appear to be involved in the regulation of contractile force [36]. Large stores of endogenous opioid peptide precursors are also found in the myocardium [37-40]. In this regard, it has been shown that the heart has the ability to synthesize and release all three major opioid peptides including the enkephalins, endorphins, and dynorphins [37-40]. Surprisingly, mRNA expression of preproenkaphalin, the precursor peptide to the endogenous δ opioid receptor ligand enkephalin, is highest in the heart compared to all other organs, including the brain [41]. Several studies have been conducted over the past several years supporting the concept that opioid peptides are released in response to ischemia, which act upon δ and κ receptors to contribute to both the early and late phases of IPC [37, 40]. Furthermore, it is well established that exogenous administration of opioid receptor agonists, including high doses of morphine§, reduces myocardial ischemia/reperfusion injury when administered prior to the ischemic insult [37, 40].
More recent studies have focused on addressing whether opioid receptor ligands are also cardioprotective when administered at the time of reperfusion. Gross and associates [42-44] have shown that morphine, the δ opioiod receptor agonists BW373U86, or the irreversible δ opioid receptor agonist fentanyl isothiocyanate reduced infarct size in rats subjected to 30 min of coronary occlusion and 2 h of reperfusion to a similar extent when administered prior to the coronary occlusion or 5 min before release of the occlusion. In contrast, the κ opioid receptor agonist U50,488 was not effective at reducing reperfusion injury [1]. Further mechanistic studies showed that the protection provided by both morphine and the δ opioid receptor agonists was blocked by inhibitors of components of the RISK pathway including wortmannin/LY294002 (PI-3 kinase inhibitors) and rapamycin (mTOR) and a p70s6 kinase inhibitor, and that activation of opioid receptors increased phosphorylation of GSKβ within the ischemic zone at serine9 during early reperfusion [42]. Collectively, these data provide the first evidence to suggest that activation of opioid receptors (most likely δ opioid receptors) reduces reperfusion injury by interacting with anti-apoptotic pro-survival kinase signaling pathways [42]. Furthermore, these data support the continued use of morphine to treat patients with acute ischemia/reperfusion injury as well as efforts to develop selective peripherally active δ opioid receptor agonists for treating reperfusion injury.
Postconditioning
The existence of POC is the newest evidence that has emerged to support the concept of reperfusion injury. The term postconditioning refers to the phenomenon in which multiple brief periods of reperfusion interspersed with brief periods of ischemia (10 – 60 s) results in a reduction in infarct size [4, 5]. Generally, 3 cycles of ischemia/reperfusion are required to produce a maximal POC effect, although 4 and 6 cycles have been shown to be effective by some investigators [4, 5]. However, it is the interval of reperfusion and ischemia that is the most critical factor in determining how efficacious POC will be. POC protocols shown to be maximally effective at reducing infarct size range from 10 – 60 s depending on the specific species being studied (10 – 15 s in mice or rats and 30 s in dogs [4, 5]). These ischemia/reperfusion cycles need to be performed precisely, since any variation for a given species results in the failure of POC to be effective. It is also important to note that, like IPC, POC is only effective at reducing infarction induced by relatively short periods of ischemia (30-45 min in rodents/rabbits and 60-90 min in dogs/swine; [4, 5]).
The initial studies by Zhao et al in Vinten-Johansen's laboratory was the first to discovery POC [45], which was appropriately named to distinguish it from the more commonly studied phenomenon IPC. The initial studies by Zhao et al [45] were carried out in anesthetized dogs in which a 1-h coronary artery occlusion followed by 3 h of reperfusion was performed in the presence or absence of POC induced by subjecting the hearts to 3 cycles of 30 s reperfusion/30 s ischemia applied at the start of reperfusion. In this study [45], two additional groups were included in the study that were subjected to IPC alone or in combination with POC. The results revealed that POC produced an equivalent reduction in infarct size of ∼40-50% compared to IPC, and that POC combined with IPC produced no additive or synergistic protective effect on infarct size [45]. Additional analyses demonstrated that POC reduced endothelial dysfunction, apoptosis, lipid peroxidation, superoxide production, and neutrophil activation/infiltration into the reperfused myocardium and that this effect was not a delay in injury, but rather a sustained reduction in injury that persisted for at least 72 h [4, 5]. Since these initial studies, a number of other investigators have demonstrated the existence of POC in other species including rodents, rabbits, and swine [4, 5].
Importantly, evidence for the existence of POC has also been obtained in humans. Laskey et al [46] and Staat et al [47] recently published two landmark studies in which POC was shown to reduce ischemia/reperfusion injury in patients experiencing an acute myocardial infarction. In the study by Staat et al [47], 30 patients with acute myocardial infarction being reperfused by balloon angioplasty were subjected to either standard reperfusion therapy or a POC protocol consisting of four cycles of 1-min reperfusion/1-min occlusion by catheter balloon inflation. In this study [47], tissue necrosis was estimated by creatine kinase release over the ensuing 72 h. Interestingly, all of the standard determinants of infarct size including the size of the risk region and collateral function were similar in the two treatment groups. However, the area-under-the-curve calculated from plasma creatine kinase levels over time was significantly smaller (36%) in the POC group [47]. This value is similar to the infarct size reduction observed in the first report of POC in dogs by Zhao and colleagues [45]. These results in humans are quite exciting and suggest that POC could become a routine procedure for use in patients undergoing elective surgery including coronary bypass or angioplasty, or in patients with an acute myocardial infarction. Certainly additional clinical studies are necessary to further assess the efficacy of POC in various clinical conditions.
Interestingly, the mechanism of POC seems to involve enhanced release of endogenous adenosine, which may reduce reperfusion injury by mechanisms similar to that provided by exogenous administration of receptor agonists during reperfusion. Using an isolated buffer-perfused rabbit heart model, Yang et al [48] demonstrated that the reduction in infarct size induced by POC induced by 6 cycles of 10 s reperfusion/10 s occlusion was blocked by the non-selective AR antagonist 8-SPT. Subsequent studies by Kin and colleagues [49] using an isolated buffer-perfused mouse heart model demonstrated that protection provided by POC appeared to be mediated via A2A and or A3ARs, since it was blocked by administration of ZM 241385 or the rodent A3AR antagonist MRS 1523. This study also showed that POC delayed washout of adenosine during reperfusion, which presumably would result in enhanced activation of myocardial ARs [49]. Interestingly, evidence has been provided suggesting that the release of endogenous opioid peptides may also be enhanced by POC. Preliminary results recently demonstrated that administration of two different non-selective opioid receptor antagonists naloxone or quaternary naloxone (peripherally acting), the selective δ opioid receptor antagonist naltrindole, or the selective κ opioid receptor antagonist nor-BNI all blocked the protective effect of POC in an in vivo rat model of infarction [4].
Conclusions
In summary, with the discovery of POC as well as new data showing a beneficial effect of administering adenosine receptor and opioid receptor ligands at the time of reperfusion greatly strengthen the concept of lethal reperfusion injury. It is envisioned that the use of POC protocols and pharmacological agents for treating reperfusion injury will soon become routine in the clinical setting. Even though POC may be old wine in a new bottle, as suggested by Heusch [50], the reintroduction of POC is undoubtedly one of the most exciting new concepts in cardiology in recent years, providing a prime example of bench-to-bedside translational research. Further studies are needed to decipher the cellular mechanisms by which POC and RISK signaling produce cellular protection, and studies of POC in other organs such as the brain and kidney where reperfusion injury is also a significant problem are warranted.
Acknowledgements
The studies conducted in the authors' laboratories were supported by NIH grants R01 HL08311 (GJG), R01 HL60051 (JAA), and R01 HL07707 (JAA).
Footnotes
Infarct size was also assessed in a subset of 266 patients in the AMISTAD-II trial.
Adenosine is also an agonist of the fourth adenosine receptor subtype, the A2B adenosine receptor, at 20-100-fold higher concentrations.
The A2BAR promotes degranulation of mast cells from non-rodent species.
Although morphine is considered to be a μ opioid receptor agonist, it is effective in models of myocardial ischemia/reperfusion injury at high doses most likely by activating δ and/or κ opioid receptors.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gross ER, Gross GJ. Ligand triggers of classical preconditioning and postconditioning. Cardiovasc Res. 2006;70:212–21. doi: 10.1016/j.cardiores.2005.12.019. [DOI] [PubMed] [Google Scholar]
- 2.Ely SW, Berne RM. Protective effects of adenosine in myocardial ischemia. Circulation. 1992;85:893–904. doi: 10.1161/01.cir.85.3.893. [DOI] [PubMed] [Google Scholar]
- 3.Vinten-Johansen J, Thourani VH, Ronson RS, Jordan JE, Zhao ZQ, Nakamura M, et al. Broad-spectrum cardioprotection with adenosine. Ann Thorac Surg. 1999;68:1942–8. doi: 10.1016/s0003-4975(99)01018-8. [DOI] [PubMed] [Google Scholar]
- 4.Zhao ZQ, Vinten-Johansen J. Postconditioning: reduction of reperfusion-induced injury. Cardiovasc Res. 2006;70:200–11. doi: 10.1016/j.cardiores.2006.01.024. [DOI] [PubMed] [Google Scholar]
- 5.Vinten-Johansen J, Zhao ZQ, Jiang R, Zatta AJ. Myocardial protection in reperfusion with postconditioning. Expert Rev Cardiovasc Ther. 2005;3:1035–45. doi: 10.1586/14779072.3.6.1035. [DOI] [PubMed] [Google Scholar]
- 6.Hausenloy DJ, Yellon DM. New directions for protecting the heart against ischaemia-reperfusion injury: targeting the Reperfusion Injury Salvage Kinase (RISK)-pathway. Cardiovasc Res. 2004;61:448–60. doi: 10.1016/j.cardiores.2003.09.024. [DOI] [PubMed] [Google Scholar]
- 7.Olafsson B, Forman MB, Puett DW, Pou A, Cates CU, Friesinger GC, et al. Reduction of reperfusion injury in the canine preparation by intracoronary adenosine: importance of the endothelium and the no-reflow phenomenon. Circulation. 1987;76:1135–45. doi: 10.1161/01.cir.76.5.1135. [DOI] [PubMed] [Google Scholar]
- 8.Homeister JW, Hoff PT, Fletcher DD, Lucchesi BR. Combined adenosine and lidocaine administration limits myocardial reperfusion injury. Circulation. 1990;82:595–608. doi: 10.1161/01.cir.82.2.595. [DOI] [PubMed] [Google Scholar]
- 9.Babbitt DG, Virmani R, Forman MB. Intracoronary adenosine administered after reperfusion limits vascular injury after prolonged ischemia in the canine model. Circulation. 1989;80:1388–99. doi: 10.1161/01.cir.80.5.1388. [DOI] [PubMed] [Google Scholar]
- 10.Babbitt DG, Virmani R, Vildibill HD, Jr., Norton ED, Forman MB. Intracoronary adenosine administration during reperfusion following 3 hours of ischemia: effects on infarct size, ventricular function, and regional myocardial blood flow. Am Heart J. 1990;120:808–18. doi: 10.1016/0002-8703(90)90196-5. [DOI] [PubMed] [Google Scholar]
- 11.Pitarys CJ, 2nd, Virmani R, Vildibill HD, Jr., Jackson EK, Forman MB. Reduction of myocardial reperfusion injury by intravenous adenosine administered during the early reperfusion period. Circulation. 1991;83:237–47. doi: 10.1161/01.cir.83.1.237. [DOI] [PubMed] [Google Scholar]
- 12.Ross AM, Gibbons RJ, Stone GW, Kloner RA, Alexander RW. A randomized, double-blinded, placebo-controlled multicenter trial of adenosine as an adjunct to reperfusion in the treatment of acute myocardial infarction (AMISTAD-II) J Am Coll Cardiol. 2005;45:1775–80. doi: 10.1016/j.jacc.2005.02.061. [DOI] [PubMed] [Google Scholar]
- 13.Mahaffey KW, Puma JA, Barbagelata NA, DiCarli MF, Leesar MA, Browne KF, et al. Adenosine as an adjunct to thrombolytic therapy for acute myocardial infarction: results of a multicenter, randomized, placebo-controlled trial: the Acute Myocardial Infarction STudy of ADenosine (AMISTAD) trial. J Am Coll Cardiol. 1999;34:1711–20. doi: 10.1016/s0735-1097(99)00418-0. [DOI] [PubMed] [Google Scholar]
- 14.Kloner RA, Forman MB, Gibbons RJ, Ross AM, Alexander RW, Stone GW. Impact of time to therapy and reperfusion modality on the efficacy of adenosine in acute myocardial infarction: the AMISTAD-2 trial. Eur Heart J. 2006 doi: 10.1093/eurheartj/ehl094. in press; ePUB available. [DOI] [PubMed] [Google Scholar]
- 15.Forman MB, Vitola JV, Velasco CE, Murray JJ, Dubey RK, Jackson EK. Sustained reduction in myocardial reperfusion injury with an adenosine receptor antagonist: possible role of the neutrophil chemoattractant response. J Pharmacol Exp Ther. 2000;292:929–38. [PubMed] [Google Scholar]
- 16.Auchampach JA, Jin X, Moore J, Wan TC, Kreckler LM, Ge ZD, et al. Comparison of three different A1 adenosine receptor antagonists on infarct size and multiple cycle ischemic preconditioning in anesthetized dogs. J Pharmacol Exp Ther. 2004;308:846–56. doi: 10.1124/jpet.103.057943. [DOI] [PubMed] [Google Scholar]
- 17.Zhao ZQ, Budde JM, Morris C, Wang NP, Velez DA, Muraki S, et al. Adenosine attenuates reperfusion-induced apoptotic cell death by modulating expression of Bcl-2 and Bax proteins. J Mol Cell Cardiol. 2001;33:57–68. doi: 10.1006/jmcc.2000.1275. [DOI] [PubMed] [Google Scholar]
- 18.Schlack W, Schafer M, Uebing A, Schafer S, Borchard U, Thamer V. Adenosine A2-receptor activation at reperfusion reduces infarct size and improves myocardial wall function in dog heart. J Cardiovasc Pharmacol. 1993;22:89–96. doi: 10.1097/00005344-199307000-00015. [DOI] [PubMed] [Google Scholar]
- 19.Lasley RD, Jahania MS, Mentzer RM., Jr. Beneficial effects of adenosine A2A agonist CGS-21680 in infarcted and stunned porcine myocardium. Am J Physiol Heart Circ Physiol. 2001;280:H1660–6. doi: 10.1152/ajpheart.2001.280.4.H1660. [DOI] [PubMed] [Google Scholar]
- 20.Jordan JE, Zhao ZQ, Sato H, Taft S, Vinten-Johansen J. Adenosine A2 receptor activation attenuates reperfusion injury by inhibiting neutrophil accumulation, superoxide generation and coronary endothelial adherence. J Pharmacol Exp Ther. 1997;280:301–9. [PubMed] [Google Scholar]
- 21.Glover DK, Riou LM, Ruiz M, Sullivan GW, Linden J, Rieger JM, et al. Reduction of infarct size and postischemic inflammation from ATL-146e, a highly selective adenosine A2A receptor agonist, in reperfused canine myocardium. Am J Physiol Heart Circ Physiol. 2005;288:H1851–8. doi: 10.1152/ajpheart.00362.2004. [DOI] [PubMed] [Google Scholar]
- 22.Linden J. Cloned adenosine A3 receptors: pharmacological properties, species differences and receptor functions. Trends Pharmacol Sci. 1994;15:298–306. doi: 10.1016/0165-6147(94)90011-6. [DOI] [PubMed] [Google Scholar]
- 23.Auchampach JA, Ge ZD, Wan TC, Moore J, Gross GJ. The A3 adenosine receptor agonist IB-MECA reduces myocardial ischemia/reperfusion injury in dogs. Am J Physiol Heart Circ Physiol. 2003;10:H607–H13. doi: 10.1152/ajpheart.01001.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tracey WR, Magee WP, Oleynek JJ, Hill RJ, Smith AH, Flynn DM, et al. Novel N6-substituted adenosine 5′-N-methyluronamides with high selectivity for human adenosine A3 receptors reduce ischemic myocardial injury. Am J Physiol Heart Circ Physiol. 2003;285:H2780–7. doi: 10.1152/ajpheart.00411.2003. [DOI] [PubMed] [Google Scholar]
- 25.Frangogiannis NG, Smith CW, Entman ML. The inflammatory response in myocardial infarction. Cardiovasc Res. 2002;53:31–47. doi: 10.1016/s0008-6363(01)00434-5. [DOI] [PubMed] [Google Scholar]
- 26.Linden J. Molecular approach to adenosine receptors: receptor-mediated mechanisms of tissue protection. Annu Rev Pharmacol Toxicol. 2000;41:775–87. doi: 10.1146/annurev.pharmtox.41.1.775. [DOI] [PubMed] [Google Scholar]
- 27.Bouma MG, van den Wildenberg FA, Buurman WA. The anti-inflammatory potential of adenosine in ischemia-reperfusion injury: established and putative beneficial actions of a retaliatory metabolite. Shock. 1997;8:313–20. doi: 10.1097/00024382-199711000-00001. [DOI] [PubMed] [Google Scholar]
- 28.Sullivan GW, Linden J. Role of A2A Adenosine Receptors in Inflammation. Drug Dev Res. 1998;45:103–12. [Google Scholar]
- 29.Sitkovsky MV, Lukashev D, Apasov S, Kojima H, Koshiba M, Caldwell C, et al. Physiological control of immune response and inflammatory tissue damage by hypoxia-inducible factors and adenosine A2A receptors. Annu Rev Immunol. 2004;22:657–82. doi: 10.1146/annurev.immunol.22.012703.104731. [DOI] [PubMed] [Google Scholar]
- 30.Hasko G, Cronstein BN. Adenosine: an endogenous regulator of innate immunity. Trends Immunol. 2004;25:33–9. doi: 10.1016/j.it.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 31.Yang Z, Day YJ, Toufektsian MC, Ramos SI, Marshall M, Wang XQ, et al. Infarct-sparing effect of A2A-adenosine receptor activation is due primarily to its action on lymphocytes. Circulation. 2005;111:2190–7. doi: 10.1161/01.CIR.0000163586.62253.A5. [DOI] [PubMed] [Google Scholar]
- 32.Ge ZD, Wan TC, Auchampach JA. The A3 adenosine receptor agonist Cl-IB-MECA reduces myocardial infarct size in mice when administered during reperfusion: Mechanistic studies with A3AR gene ‘knock-out’ and bone marrow chimeric mice. Circulation. 2004;110:III–29. [Google Scholar]
- 33.van der Hoeven D, Auchampach JA. Characterization of expression and function of adenosine receptors in mouse neutrophils. FASEB J. 2006;20:A249. [Google Scholar]
- 34.Park SS, Zhao H, Jang Y, Mueller RA, Xu Z. N6-(3-iodobenzyl)-adenosine-5′-N-methylcarboxamide confers cardioprotection at reperfusion by inhibiting mitochondrial permeability transition pore opening via glycogen synthase kinase 3β. J Pharmacol Exp Ther. 2006;318:124–31. doi: 10.1124/jpet.106.101477. [DOI] [PubMed] [Google Scholar]
- 35.Jiang J, van Rhee AM, Chang L, Patchornik A, Ji XD, Evans P, et al. Structure-activity relationships of 4-(phenylethynyl)-6-phenyl-1,4-dihydropyridines as highly selective A3 adenosine receptor antagonists. J Med Chem. 1997;40:2596–608. doi: 10.1021/jm970091j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pugsley MK. The diverse molecular mechanisms responsible for the actions of opioids on the cardiovascular system. Pharmacol Ther. 2002;93:51–75. doi: 10.1016/s0163-7258(02)00165-1. [DOI] [PubMed] [Google Scholar]
- 37.Peart JN, Gross ER, Gross GJ. Opioid-induced preconditioning: recent advances and future perspectives. Vascul Pharmacol. 2005;42:211–8. doi: 10.1016/j.vph.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 38.Barron BA. Opioid peptides and the heart. Cardiovasc Res. 1999;43:13–6. doi: 10.1016/s0008-6363(99)00112-1. [DOI] [PubMed] [Google Scholar]
- 39.Barron BA. Cardiac opioids. Proc Soc Exp Biol Med. 2000;224:1–7. doi: 10.1046/j.1525-1373.2000.22358.x. [DOI] [PubMed] [Google Scholar]
- 40.Gross GJ. Role of opioids in acute and delayed preconditioning. J Mol Cell Cardiol. 2003;35:709–18. doi: 10.1016/s0022-2828(03)00135-4. [DOI] [PubMed] [Google Scholar]
- 41.Howells RD, Kilpatrick DL, Bailey LC, Noe M, Udenfriend S. Proenkephalin mRNA in rat heart. Proc Natl Acad Sci U S A. 1986;83:1960–3. doi: 10.1073/pnas.83.6.1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gross ER, Hsu AK, Gross GJ. Opioid-induced cardioprotection occurs via glycogen synthase kinase beta inhibition during reperfusion in intact rat hearts. Circ Res. 2004;94:960–6. doi: 10.1161/01.RES.0000122392.33172.09. [DOI] [PubMed] [Google Scholar]
- 43.Gross ER, Hsu AK, Gross GJ. Acute aspirin treatment abolishes, whereas acute ibuprofen treatment enhances morphine-induced cardioprotection: role of 12-lipoxygenase. J Pharmacol Exp Ther. 2004;310:185–91. doi: 10.1124/jpet.103.064667. [DOI] [PubMed] [Google Scholar]
- 44.Gross ER, Peart JN, Hsu AK, Auchampach JA, Gross GJ. Extending the cardioprotective window using a novel δ-opioid agonist fentanyl isothiocyanate via the PI3-kinase pathway. Am J Physiol Heart Circ Physiol. 2005;288:H2744–9. doi: 10.1152/ajpheart.00918.2004. [DOI] [PubMed] [Google Scholar]
- 45.Zhao ZQ, Corvera JS, Halkos ME, Kerendi F, Wang NP, Guyton RA, et al. Inhibition of myocardial injury by ischemic postconditioning during reperfusion: comparison with ischemic preconditioning. Am J Physiol Heart Circ Physiol. 2003;285:H579–88. doi: 10.1152/ajpheart.01064.2002. [DOI] [PubMed] [Google Scholar]
- 46.Laskey WK. Brief repetitive balloon occlusions enhance reperfusion during percutaneous coronary intervention for acute myocardial infarction: a pilot study. Catheter Cardiovasc Interv. 2005;65:361–7. doi: 10.1002/ccd.20397. [DOI] [PubMed] [Google Scholar]
- 47.Staat P, Rioufol G, Piot C, Cottin Y, Cung TT, L'Huillier I, et al. Postconditioning the human heart. Circulation. 2005;112:2143–8. doi: 10.1161/CIRCULATIONAHA.105.558122. [DOI] [PubMed] [Google Scholar]
- 48.Yang XM, Philipp S, Downey JM, Cohen MV. Postconditioning's protection is not dependent on circulating blood factors or cells but involves adenosine receptors and requires PI3-kinase and guanylyl cyclase activation. Basic Res Cardiol. 2005;100:57–63. doi: 10.1007/s00395-004-0498-4. [DOI] [PubMed] [Google Scholar]
- 49.Kin H, Zatta AJ, Lofye MT, Amerson BS, Halkos ME, Kerendi F, et al. Postconditioning reduces infarct size via adenosine receptor activation by endogenous adenosine. Cardiovasc Res. 2005;67:124–33. doi: 10.1016/j.cardiores.2005.02.015. [DOI] [PubMed] [Google Scholar]
- 50.Heusch G. Postconditioning: old wine in a new bottle? J Am Coll Cardiol. 2004;44:1111–2. doi: 10.1016/j.jacc.2004.06.013. [DOI] [PubMed] [Google Scholar]