

Summary
Meiotic recombination is initiated by Spo11-generated DNA double-strand breaks (DSBs) [1]. A fraction of total DSBs is processed into crossovers (CRs) between homologous chromosomes, which promotes their accurate segregation at meiosis I (MI) [2]. The coordination of recombination-associated events and MI progression is governed by the “pachytene checkpoint” [3]. In budding yeast Rad17, a component of PCNA clamp-like complex, and Pch2, an AAA-ATPase, have been implicated in this checkpoint [3–7]. We show that RAD17 and PCH2 are required for two genetically separable pathways that monitor the presence of different meiotic recombination-associated lesions: First, deletion of RAD17 or SAE2, encoding a DSB-end processing factor [8, 9], suppresses delay or arrest of MI timing when DNA repair intermediates are present. Second, deletion of PCH2 suppresses delay of MI timing when aberrant synaptonemal complex (SC) is present. Importantly, ZIP1, encoding the central element of the SC [10] is required for activation of the PCH2-dependent checkpoint pathway. Analysis of the rad17Δ pch2Δ double mutant revealed a redundant role for these genes in regulating inter-homolog CR formation. These findings suggest a link between the surveillance of distinct recombination-associated lesions, control of CR formation kinetics and regulation of MI timing. A PCH2-ZIP1 dependent meiotic checkpoint is likely conserved among synaptic organisms from yeast to human [6, 11].
Results and Discussion
Two independent pathways control timing of meiosis I and crossover formation
We previously showed that deletion of NDJ1, a gene encoding a meiosis-induced telomere binding protein in budding yeast, delays the turnover of SPO11-initiated recombination intermediates during meiosis and the timing of chromosome mass separation (referred to here as MI timing) compared to wild type [12]. The MI delay conferred by ndj1Δ was partially suppressed by rad17Δ (Figure 1A; [12]). By contrast, the ndj1Δ delay was exacerbated by deletion of PCH2, which by itself exhibited a SPO11-dependent delay of MI (Figure 1B, 1C). Analogously, the MI arrest phenotype conferred by deletion of DMC1, encoding a recA homolog involved in an early strand-invasion event during meiotic prophase [13], is also partially suppressed by rad17Δ, but was not suppressed by pch2Δ (Figure S1A, S1B) [5, 7]. The difference in the ability of rad17Δ and pch2Δ to suppress the meiotic delay conferred by ndj1Δ and dmc1Δ suggests that the rad17Δ and pch2Δ are defective in distinct surveillance mechanisms. These results are consistent with a previous suggestion that Rad17 and Pch2 serve in different pathways to monitor lesions associated with meiotic recombination [4, 5].
Figure 1.
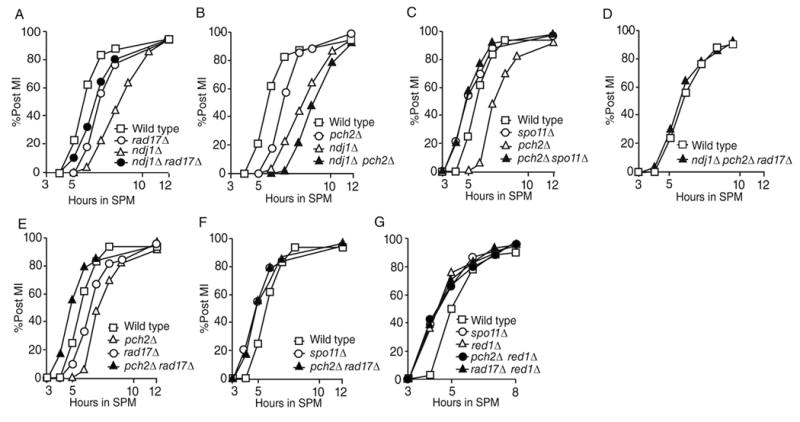
MI timing in mutants with pch2Δ and rad17Δ mutations and other lesions (A)~(G) The number of DAPI staining foci per cell (n=200) was counted for each time point following transfer to sporulation media (SPM). Panel (A) is adapted from [12]. Results shown in panel (A) and (B) are from the same time course. Similarly, results shown in panel (C), (E) and (F) are from the same time course. Subsets of data are shown in individual panels for clarity. All experiments presented in this paper have been replicated one or more times and give similar results. While the absolute timing of meiotic division may vary by half hour between time courses, the relative differences between wild type and mutant strains remain consistent for each time course carried out in parallel.
Notably, deletion of RAD17 alone leads to a delay in meiotic progression and a similar delay in meiosis timing was observed in the ndj1Δ rad17Δ and dmc1Δ rad17Δ mutations (Figure 1A, S1A) [14, 15]. Since the meiotic delay conferred by rad17Δ is dependent on the initiation of meiotic recombination (i.e. SPO11) [14], it is likely that a recombination-association lesion(s) produced in the rad17Δ background activates an alternative checkpoint pathway.
We tested whether or not RAD17 and PCH2 might be required for the MI delay conferred by pch2Δ and rad17Δ, respectively. Intriguingly, we found that MI timing in the pch2Δ rad17Δ double mutant was accelerated, even compared with wild type (Figure 1E; data not shown, (t50 pch2Δ rad17Δ - t50 WT) = −0.55 ± 0.26 hours; n = 5; p = 0.0009). Timing in the double mutant was indistinguishable (p=0.28) from the MI kinetics exhibited by spo11Δ compared with wild type [Figure 1F and data not shown, (t50 spo11Δ - t50 WT) = −0.70 ± 0.09 hours; n = 4; p = 0.003] [16, 17]. The meiotic delay conferred by ndj1Δ rad17Δ and dmc1Δ rad17Δ is also further suppressed by deleting PCH2 (Figure 1D and Figure S1C). Combined, these data suggest that PCH2 and RAD17 act independently as negative regulators of MI division.
Crossovers formed in the pch2Δ rad17Δ mutant are not sufficient to promote accurate meiosis I segregation
We examined the physical products of meiotic recombination at the well-characterized HIS4::LEU2 recombination hot spot [18] in wild type, pch2Δ, rad17Δ and pch2Δ rad17Δ mutants. Interestingly, while the rad17Δ and pch2Δ mutants each gave a delay in crossover formation as observed previously [5, 14, 15], the pch2Δ rad17Δ double mutant exhibited wild-type kinetics of crossover formation, albeit with reduced levels similar to rad17Δ (~30% of wild type; Figure 2A~2D). This result suggests that RAD17 functions as a negative regulator of crossover timing in a pch2Δ mutant and vice versa. DSB turnover observed in the pch2Δ rad17Δ double mutant is consistent with faster DSB repair compared with the rad17Δ mutant [14], alternatively, hyperresection of DSB ends may preclude detection by Southern analysis.
Figure 2.
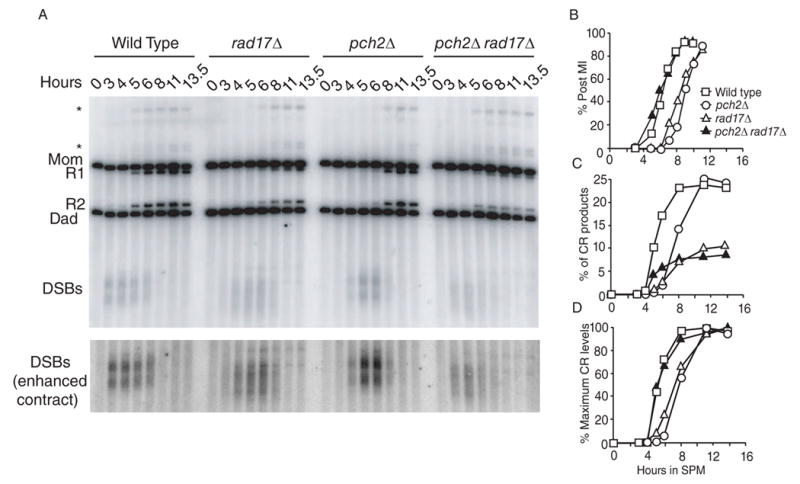
Meiotic time course showing DSB and CR formation in the wild-type, pch2Δ, rad17Δ and pch2Δ rad17Δ strains at the his4x::LEU2-URA3 (Dad)/HIS4::LEU2 (Mom) locus [18]. (A) Southern blot. Restriction with XhoI gives different sized fragments of the DSBs, reciprocal interhomolog CRs (R1 and R2) and parental bands (Mom and Dad). The * denotes a band that likely represents a slow migrating DSB intermediate [18, 37]. (B) Kinetics of MI timing in cells from culture used in (A). (C) Percent of CRs. Percent final levels of CRs [100*(R1+R2)/total probed DNA signals] were: wild type- 23.7%, pch2Δ-25.4%, rad17Δ- 10.4% and pch2Δ rad17Δ- 8.5%. (D) CR levels normalized to maximum levels observed in each strain.
The pch2Δ rad17Δ double mutant also exhibited a synergistic defect in spore viability; the pch2Δ rad17Δ double mutant gave only 0.9% viable spores while the spore viability of the pch2Δ (93.6%) and rad17Δ (33.8%) single mutants agreed with previously reported values (Table 1) [6, 7, 14].
Table 1.
Spore viability of mutants analyzed in this study
| Strains | Viability | # spores analyzed* |
|---|---|---|
| WT | 96.8 | 688 |
| pch2Δ rad17Δ | 0.9 | 544 |
| pch2Δ | 93.6 | 644 |
| rad17Δ | 33.8 | 728 |
| spo13Δ | 31.3 | 192 |
| spo13Δ pch2Δ rad17 | 32.9 | 252 |
| spo13Δ rad17 | 21.9 | 256 |
| spo13Δ pch2Δ | 37.5 | 128 |
| rad9Δ rad17Δ | 33.0 | 264 |
| rad9Δ pch2Δ | 98.3 | 60 |
| zip1Δ | 40.8 | 444 |
| zip1Δ rad17Δ | 4.2 | 1060 |
| zip1Δ pch2Δ | 23.1 | 576 |
| ndj1Δ | 75.2 | 648 |
| ndj1Δ pch2Δ | 49.2 | 624 |
| rad17Δ ndj1Δ | 2.8 | 668 |
Asci containing 4 spores were dissected for all strains except for spo13Δ containing strains, in which case dyads were dissected
Taken together, the synergistic effect of combining the pch2Δ and rad17Δ mutations on MI timing, spore viability and DSB detection supported two possible models: First, the accelerated progression through prophase I exhibited by pch2Δ rad17Δ could lead to catastrophic spore inviability due to premature separation of broken chromosomes at MI. Alternatively, CR products or CR-associated events generated in the pch2Δ rad17Δ double mutant could be intrinsically different than those formed in the rad17Δ cells so that they can not promote the formation of viable spore products. This may be due to non-disjunction arising from defects in recombination partner choice (sister vs. homolog) or loss of crossover interference.
To distinguish between these two possibilities, we assayed spore viability in a spo13Δ strain background. The spo13Δ mutant forms diploid dyads after a single round of chromosome segregation. Viability of the spore products in a spo13Δ strain background does not depend on the formation of crossing over between homologous chromosomes, but instead depends on genome integrity [19]. If pch2Δ rad17Δ gave low levels of spore inviability due to the illicit repair of DSBs (i.e. using sister chromatids) then pch2Δ rad17Δ spo13Δ would be expected to give viable spores. By contrast, if genome instability from accelerated division timing is the cause of spore inviability, then the triple mutant should phenocopy the pch2Δ rad17Δ double mutant and give very low spore viability. We found that spore viabliltiy in the pch2Δ rad17Δ mutant was similar to PCH2 RAD17 in a spo13Δ background (32.9% vs. 31.3%, respectively, Table 1), consistent with the idea that the DSBs in the pch2Δ rad17Δ mutant are repaired by using sister chromatids as templates or by forming interhomolog non-crossover product. The pch2Δ rad17Δ double mutant likely bypasses recombination-associated surveillance due to the absence of presenting lesions. It is thus conceivable that checkpoint surveillance and control of interhomolog crossover timing are naturally integrated and inseparable during meiotic recombination.
The combined phenotypes conferred by pch2Δ rad17Δ are reminiscent of those conferred by deletion of RED1, which encodes a major component of the meiotic chromosome axis [20, 21]. Crossovers formed in a red1Δ mutant do not ensure homolog disjunction [22] and DSBs formed in this mutant are repaired largely from sister chromatids [23, 24]. Also like pch2Δ rad17Δ, MI arrest conferred by dmc1Δ is suppressed by red1Δ, and MI divisions are accelerated compared to wild type [25]. In addition, we found that red1Δ in combination with either pch2Δ or rad17Δ mutations gives accelerated MI timing (Figure 1G). Therefore, RED1 can act either upstream or downstream to govern PCH2 and RAD17-dependent checkpoint function.
SAE2 functions in a RAD17-dependent surveillance pathway
Rad17, together with Ddc1 and Mec3, form the PCNA-like clamp that binds RPA-coated single-stranded DNA (ssDNA) generated during resection of the 5’ ends of breaks [26]. Sae2 is required for generating the single-stranded DNA substrate that activates the RAD17-dependent checkpoint pathway [8, 9]. We thus reasoned that the sae2Δ mutation might behave similarly to rad17Δ in combination with pch2Δ. Indeed, we found the sae2Δ mutation was able to suppress the MI delay conferred by pch2Δ (Figure 3A). Thus SAE2 functions in a checkpoint to detect lesions generated in pch2Δ. This is likely the same checkpoint pathway governed by RAD17.
Figure 3.
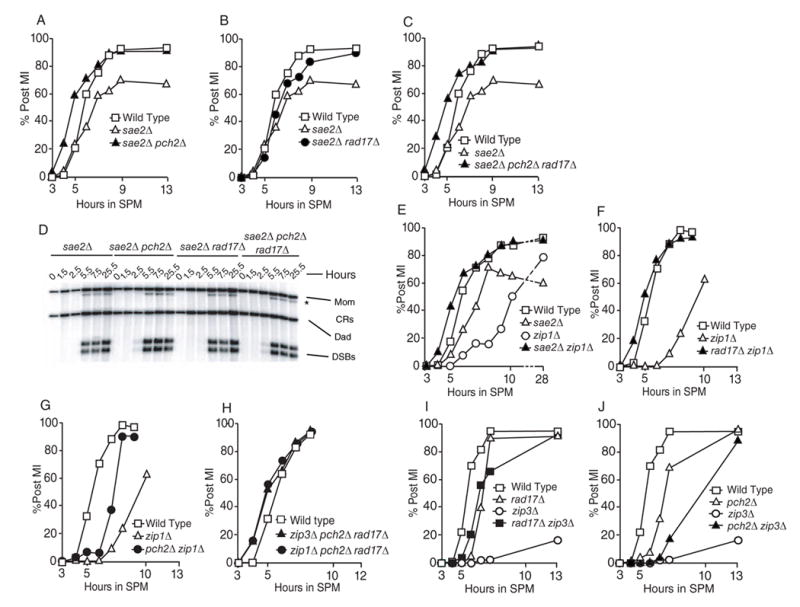
Epistasis analysis of sae2Δ zip1Δ and zip3Δ with mutations affecting recombination and/or checkpoint functions. All Post MI data is depicted as in Figure 1. The data presented in panels (A), (B), (C), (F), (G), (I) and (J) are from one time course. (D) Southern blot of the sae2Δ, pch2Δ sae2Δ, rad17Δ sae2Δ and pch2Δ rad17Δ sae2Δ strains as described in Figure 2.
The sae2Δ mutant also confers an MI arrest phenotype (only 60 to 70% go through MI), which, surprisingly, was suppressed by rad17Δ (Figure 3B). Nevertheless, the sae2Δ pch2Δ rad17Δ triple mutant exhibited MI timing with accelerated kinetics (Figure 3C). Although we cannot fully explain the suppression of sae2Δ meiotic arrest by rad17Δ, we suggest that the sae2Δ mutant presents more than one substrate for checkpoint activation.
We tested whether or not the pch2Δ, rad17Δ or the pch2Δ rad17Δ mutations in combination with sae2Δ allow for unresected DNA to be repaired, thereby eliminating a checkpoint signal (similar to pch2Δ rad17Δ above). We found unresected meiotic DNA breaks at sae2Δ levels in all strains evaluated (Figure 3D). This result indicates that PCH2 is required for delaying meiotic progression in the presence of unresected DSBs thereby defining it more precisely as a recombination-associated checkpoint factor [27]. That both pch2Δ and rad17Δ confer defects in MI timing recombination is not unusual for factors that also function in bona fide checkpoint pathways [28].
A RAD9-dependent checkpoint pathway is not activated by lesions generated in either pch2Δ or rad17Δ mutants
It has been shown that the rad50S-induced delay in meiotic division depends on TEL1 and RAD9, two mitotic DNA-damage checkpoint proteins [29]. Since the meiotic delay and arrest conferred by sae2Δ is dependent on PCH2, and both sae2Δ and rad50S mutants accumulate unresected DSBs, it is reasonable to speculate that PCH2 is one component of the RAD9 and TEL1-dependent checkpoint pathway. To address this possibility, we compared spore viability in a rad9Δ rad17Δ double mutant with corresponding single mutants. If the RAD9-dependent pathway is also responsible for monitoring lesions generated in rad17Δ, the rad9Δ rad17Δ double mutant would strongly reduce spore viability like pch2Δ rad17Δ. By contrast, we found the rad9Δ rad17Δ double mutant gave spore viability similar to the rad17Δ single mutant (33% and 33.8%, respectively, Table 1). Likewise the rad9Δ pch2Δ double mutant exhibited spore viability (98.3%) similar to pch2Δ and wild type (93.6% and 96.8%, respectively, Table 1). The failure to produce synergistic spore inviability in the double mutant strains argues against the hypothesis that RAD9-dependent checkpoint pathway is activated by lesions generated in pch2Δ or rad17Δ. However, our result cannot rule out a possibility that the PCH2 and RAD9-dependent checkpoint functions overlap prior to DNA strand resection.
A SC-dependent checkpoint in budding yeast
At least two independent meiotic surveillance mechanisms function to monitor chromosomal lesions during meiosis in higher eukaryotes [11, 30, 31]. PCH-2, a homolog of yeast Pch2, has been implicated in a synapsis checkpoint in C. elegans [11]. Through a comparative genomic analysis, we found that potential orthologs of C. elegans PCH-2 and budding yeast Pch2 exist in organisms known to undergo synaptic meioses (e.g. human, mouse, Arabidopsis and fruit fly, Table 2, Figure S2), but are absent from asynaptic organisms (e.g. S. pombe, A. nidulans and Tetrahymena thermophila) (Table 2, Figure S2; data not shown) [32–36]. Thus, a conserved Pch2-dependent checkpoint may have evolved specifically to monitor aberrant or incomplete structure(s) comprising the synaptonemal complex (SC).
Table 2.
Summary of the BLASTP search against C. elegans PCH-2 and budding yeast Pch2 proteins*
| C. elegans PCH-2 BLAST | ||||
|---|---|---|---|---|
| Species | Protein | E-Value | Identities | Synapsis |
| Human | TRIP13 | 2e-63 | 141/321 (43%) | + |
| Mouse | TRIP13 | 3e-63 | 138/313 (44%) | + |
| Arabidopsis | AT4G24710.1 | 5e-61 | 138/313 (44%) | + |
| Fruit fly | CG31453-PA | 5e-55 | 132/301 (43%) | + |
| Budding yeast | Pch2 | 2e-34 | 94/238 (39%) | + |
| Fission yeast | Sec18 # | 2e-12 | 61/202 (30%) | − |
| A. nidulans | AN7254.2 § | 3e-10 | 62/215 (28%) | − |
| S. cerevisiae Pch2 BLAST | ||||
| Species | Protein | E-Value | Identities | Synapsis |
| Arabidopsis | AT4G24710.1 | 2e-40 | 105/238 (44%) | + |
| Human | TRIP13 | 2e-38 | 98/263 (37%) | + |
| Mouse | TRIP13 | 4e-38 | 101/263 (38%) | + |
| Fruit fly | CG31453-PA | 4e-37 | 100/237 (42%) | + |
| C. elegans | Pch2 | 2e-34 | 94/238 (39%) | + |
| Fission yeast | Sec18 # | 6e-11 | 52/168 (30%) | − |
| A. nidulans | AN7254.2 § | 1e-9 | 50/172 (29%) | − |
Only the best hit for each organism was listed. The Aspergillus nidulans AN7254.2 (#) and a fragment of the fission yeast Schizosaccharomyces pombe Sec18 (§) protein shows limited homology at the AAA+ (ATPases associated with various cellular activities) ATPase domain [41]. Both the fission yeast Sec18 and the A. nidulans AN7254.2 protein contain two AAA+ ATPase domains and show homology to P97/Cdc48 at the N terminus. Sec18 is required for ER to Golgi vesicle-mediated transport. Cdc48 is involved in retrotranslocation of ubiquitinated proteins from the ER to the cytosol for degradation [42]. These data suggest that the PCH-2 homologs are absent in these two organisms.
If a checkpoint exists in budding yeast to detect aberrant or incomplete synapsis, we would expect a mutation that fails to form the checkpoint-activating lesion would also bypass checkpoint-mediated delay and/or arrest. Similar logic has been applied to explain spo11Δ suppression of the arrest/delay phenotype conferred by mutations defective in meiotic recombination [9]. To test this notion, we asked whether or not the elimination of ZIP1, would act to bypass the MI-delay phenotype conferred by either sae2Δ or rad17Δ. Indeed, we found that sae2Δ zip1Δ and rad17Δ zip1Δ double mutants gave accelerated MI timing compared with wild type (Figure 3E and 3F). Moreover, spore viability in the rad17Δ zip1Δ double mutant, like rad17Δ pch2Δ, was synergistically reduced (4.2%) compared to either single mutant (zip1Δ 40.8%; rad17Δ 33.8%; Table 1, see also [7]).
PCH2 was originally identified as a suppressor of zip1Δ arrest in a non-SK1 strain background [6]. We observed similar results using SK1 (Figure 3G , see also [5]). Suppression, however, is not complete, suggesting that a complex signal gives rise to the MI delay exhibited by the zip1Δ mutation in SK1. Nonetheless, the pch2Δ zip1Δ mutations together impose a RAD17-dependent MI delay since the rad17Δ pch2Δ zip1Δ triple mutant gave accelerated MI divisions kinetics (Figure 3H).
The role of ZIP1 in crossover formation is distinct from its role in an SC-dependent checkpoint pathway
Zip1 has been shown to play roles during meiosis in addition to its function as the transverse element of the SC. For example, the ZMM epistasis group (comprising Zip1~4, Msh5 and Mer3) promotes the development of crossover intermediates during meiosis I prophase [37–39]. To understand whether it is ZMM function per se, or if the presence of SC is required for the budding yeast PCH2-dependent checkpoint function, we took advantage of the zip3Δ mutation. Like the zip1Δ mutant, zip3Δ exhibits MI delay and defects in crossover formation that lead to the accumulation of DSBs [37, 40]. By contrast, zip3Δ exhibits partial SC formation, while zip1Δ gives none [37]. Thus, our prediction was that the zip3Δ mutant would present both ssDNA and incomplete SC and thus activate both RAD17-SAE2-dependent and PCH2-ZIP1-dependent checkpoint pathways. Consistent with this notion, we found that both rad17Δ and pch2Δ mutations gave partial suppression of the zip3Δ MI timing delay (Figure 3I, 3J). By contrast, the zip3Δ pch2Δ rad17Δ triple mutant undergoes accelerated MI timing (Figure 3H). This result indicates a unique role of ZIP1 in the surveillance of meiotic chromosome metabolism compared to ZIP3 and probably other ZMM class proteins.
We suggest that an SC checkpoint operating in budding yeast monitors the integrity of synapsis and coordinates meiotic progression with the timing of crossover formation. Alternatively, it is also conceivable that SC must be completely removed before the segregation of meiotic chromosomes at anaphase. A mechanism to monitor the presence of residual SC might be important to prevent chromosome breakage during segregation.
Conclusion
The major challenge in dissecting the function(s) of the pachytene checkpoint in budding yeast is the apparent dual roles of genes required for both checkpoint response signaling and in the process of recombination and/or SC formation [5, 6, 14, 15, 25]. We overcame this issue by showing different effects of rad17Δ and pch2Δ on MI timing when combined with mutations affecting different aspects of meiotic chromosome metabolism including recombination-associated checkpoints. Our results demonstrate that the presence of recombination intermediates (i.e. in dmc1Δ, ndj1Δ, pch2Δ, zip1Δ and zip3Δ) activates a RAD17-PCH2-dependent checkpoint to arrest/delay meiotic progression. By contrast, the presence of aberrant SC intermediates (i.e. rad17Δ, sae2Δ and zip3Δ) activates a PCH2-ZIP1-dependent checkpoint pathway to delay the progression of meiosis I (Figure 4). The timely formation of crossover products and the synergistic effect of spore inviability in the pch2Δ rad17Δ double mutant compared with either single mutant suggest that the functions of these genes act in different pathways to promote recombination-dependent homolog segregation. Taken together, our results indicate a link between checkpoint surveillance and crossover timing during meiosis. Our data along with recent findings describing the function of the PCH-2 homologs in C. elegans [11] demonstrate that surveillance of SC structure is an evolutionary conserved mechanism among synaptic organisms.
Figure 4.
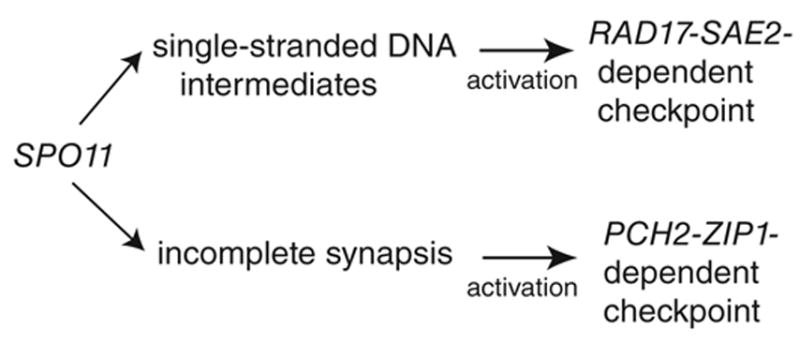
The pachytene checkpoint comprises two separable pathways Spo11 initiates meiotic recombination and is required for single-strand DNA and SC formation, which is required for generating substrates for both checkpoint pathways. Single-strand DNA activates RAD17-SAE2 dependent pathway. Incomplete SC is required for the activation of PCH2-ZIP1 dependent pathway. The signals from both pathways are likely governed by RED1 (see text).
Supplementary Material
Supplementary data include two figures, one table and Experimental Procedures can be found on line at:
Acknowledgments
We are grateful to JoAnne Engebrecht, Wolf-Dietrich Heyer, Neil Hunter, Joshua Chang Mell, Tamara Peoples-Holst and anonymous reviewers for discussion and/or critical reading of the manuscript. This study was supported by American Cancer Society Research Scholar’s Grant to S.M.B. (RSG-01-053-01-CCG) and the Arnold and Mabel Beckman Foundation to S.M.B.
Abbreviations footnote
- DSB
DNA double strand break
- SC
synaptonemal complex
- MI
Meiosis I
References
- 1.Keeney S. Mechanism and control of meiotic recombination initiation. Curr Top Dev Biol. 2001;52:1–53. doi: 10.1016/s0070-2153(01)52008-6. [DOI] [PubMed] [Google Scholar]
- 2.Page SL, Hawley RS. Chromosome Choreography: The Meiotic Ballet. Science. 2003;301:785–789. doi: 10.1126/science.1086605. [DOI] [PubMed] [Google Scholar]
- 3.Roeder GS, Bailis JM. The pachytene checkpoint. Trends Genet. 2000;16:395–403. doi: 10.1016/s0168-9525(00)02080-1. [DOI] [PubMed] [Google Scholar]
- 4.Hochwagen A, Amon A. Checking your breaks: surveillance mechanisms of meiotic recombination. Curr Biol. 2006;16:R217–228. doi: 10.1016/j.cub.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 5.Hochwagen A, Tham WH, Brar GA, Amon A. The FK506 binding protein Fpr3 counteracts protein phosphatase 1 to maintain meiotic recombination checkpoint activity. Cell. 2005;122:861–873. doi: 10.1016/j.cell.2005.07.010. [DOI] [PubMed] [Google Scholar]
- 6.San-Segundo PA, Roeder GS. Pch2 links chromatin silencing to meiotic checkpoint control. Cell . 1999;97:313–324. doi: 10.1016/s0092-8674(00)80741-2. [DOI] [PubMed] [Google Scholar]
- 7.Lydall D, Nikolsky Y, Bishop D, Weinert T. A meiotic recombination checkpoint controlled by mitotic checkpoint genes. Nature. 1996;383:840–843. doi: 10.1038/383840a0. [DOI] [PubMed] [Google Scholar]
- 8.Prinz S, Amon A, Klein F. Isolation of COM1, a new gene required to complete meiotic double-strand break-induced recombination in Saccharomyces cerevisiae. Genetics. 1997;146:781–795. doi: 10.1093/genetics/146.3.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McKee AH, Kleckner N. A general method for identifying recessive diploid-specific mutations in Saccharomyces cerevisiae, its application to the isolation of mutants blocked at intermediate stages of meiotic prophase and characterization of a new gene SAE2. Genetics. 1997;146:797–816. doi: 10.1093/genetics/146.3.797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sym M, Engebrecht J, Roeder GS. ZIP1 is a synaptonemal complex protein required for meiotic chromosome synapsis. Cell. 1993;72:365. doi: 10.1016/0092-8674(93)90114-6. [DOI] [PubMed] [Google Scholar]
- 11.Bhalla N, Dernburg AF. A conserved checkpoint monitors meiotic chromosome synapsis in Caenorhabditis elegans. Science. 2005;310:1683–1686. doi: 10.1126/science.1117468. [DOI] [PubMed] [Google Scholar]
- 12.Wu HY, Burgess SM. Ndj1, a Telomere-Associated Protein, Promotes Meiotic Recombination in Budding Yeast. Mol Cell Biol. 2006;26:3683–3694. doi: 10.1128/MCB.26.10.3683-3694.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bishop DK. RecA homologs Dmc1 and Rad51 interact to form multiple nuclear complexes prior to meiotic chromosome synapsis. Cell. 1994;79:1081–1092. doi: 10.1016/0092-8674(94)90038-8. [DOI] [PubMed] [Google Scholar]
- 14.Shinohara M, Sakai K, Ogawa T, Shinohara A. The Mitotic DNA Damage Checkpoint Proteins Rad17 and Rad24 Are Required for Repair of Double-Strand Breaks During Meiosis in Yeast. Genetics. 2003;164:855–865. doi: 10.1093/genetics/164.3.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grushcow JM, Holzen TM, Park KJ, Weinert T, Lichten M, Bishop DK. Saccharomyces cerevisiae Checkpoint Genes MEC1, RAD17 and RAD24 Are Required for Normal Meiotic Recombination Partner Choice. Genetics. 1999;153:607–620. doi: 10.1093/genetics/153.2.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Malone RE, Haring SJ, Foreman KE, Pansegrau ML, Smith SM, Houdek DR, Carpp L, Shah B, Lee KE. The signal from the initiation of meiotic recombination to the first division of meiosis. Eukaryotic Cell. 2004;3:598–609. doi: 10.1128/EC.3.3.598-609.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kee K, Keeney S. Functional Interactions Between SPO11 and REC102 During Initiation of Meiotic Recombination in Saccharomyces cerevisiae. Genetics. 2002;160:111–122. doi: 10.1093/genetics/160.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hunter N, Kleckner N. The Single-End Invasion: An Asymmetric Intermediate at the Double-Strand Break to Double-Holliday Junction Transition of Meiotic Recombination. Cell. 2001;106:59–70. doi: 10.1016/s0092-8674(01)00430-5. [DOI] [PubMed] [Google Scholar]
- 19.Klapholz S, Esposito RE. Isolation of SPO12-1 and SPO13-1 from a natural variant of yeast that undergoes a single meiotic division. Genetics. 1980;96:567–588. doi: 10.1093/genetics/96.3.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blat Y, Protacio RU, Hunter N, Kleckner N. Physical and Functional Interactions among Basic Chromosome Organizational Features Govern Early Steps of Meiotic Chiasma Formation. Cell. 2002;111:791–802. doi: 10.1016/s0092-8674(02)01167-4. [DOI] [PubMed] [Google Scholar]
- 21.Smith AV, Roeder GS. The Yeast Red1 Protein Localizes to the Cores of Meiotic Chromosomes. J Cell Biol. 1997;136:957–967. doi: 10.1083/jcb.136.5.957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rockmill B, Roeder GS. RED1: a yeast gene required for the segregation of chromosomes during the reductional division of meiosis. Proc Natl Acad Sci U S A. 1988;85:6057–6061. doi: 10.1073/pnas.85.16.6057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Niu H, Wan L, Baumgartner B, Schaefer D, Loidl J, Hollingsworth NM. Partner Choice during Meiosis Is Regulated by Hop1-promoted Dimerization of Mek1. Mol Biol Cell. 2005 doi: 10.1091/mbc.E05-05-0465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schwacha A, Kleckner N. Interhomolog Bias during Meiotic Recombination: Meiotic Functions Promote a Highly Differentiated Interhomolog-Only Pathway. Cell. 1997;90:1123–1135. doi: 10.1016/s0092-8674(00)80378-5. [DOI] [PubMed] [Google Scholar]
- 25.Xu L, Weiner B, Kleckner N. Meiotic cells monitor the status of the interhomolog recombination complex. Genes Dev. 1997;11:106–118. doi: 10.1101/gad.11.1.106. [DOI] [PubMed] [Google Scholar]
- 26.Zou L, Liu D, Elledge SJ. Replication protein A-mediated recruitment and activation of Rad17 complexes. Proc Natl Acad Sci U S A. 2003;100:13827–13832. doi: 10.1073/pnas.2336100100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hartwell LH, Weinert TA. Checkpoints: controls that ensure the order of cell cycle events. Science. 1989;246:629–634. doi: 10.1126/science.2683079. [DOI] [PubMed] [Google Scholar]
- 28.Zhou BB, Elledge SJ. The DNA damage response: putting checkpoints in perspective. Nature. 2000;408:433–439. doi: 10.1038/35044005. [DOI] [PubMed] [Google Scholar]
- 29.Usui T, Ogawa H, Petrini JHJ. A DNA Damage Response Pathway Controlled by Tel1 and the Mre11 Complex. Molecular Cell. 2001;7:1255–1266. doi: 10.1016/s1097-2765(01)00270-2. [DOI] [PubMed] [Google Scholar]
- 30.Barchi M, Mahadevaiah S, Di Giacomo M, Baudat F, de Rooij DG, Burgoyne PS, Jasin M, Keeney S. Surveillance of different recombination defects in mouse spermatocytes yields distinct responses despite elimination at an identical developmental stage. Mol Cell Biol. 2005;25:7203–7215. doi: 10.1128/MCB.25.16.7203-7215.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Odorisio T, Rodriguez TA, Evans EP, Clarke AR, Burgoyne PS. The meiotic checkpoint monitoring synapsis eliminates spermatocytes via p53-independent apoptosis. Nat Genet. 1998;18:257–261. doi: 10.1038/ng0398-257. [DOI] [PubMed] [Google Scholar]
- 32.Page SL, Hawley RS. The genetics and molecular biology of the synaptonemal complex. Annual Review of Cell and Developmental Biology. 2004;20:525–558. doi: 10.1146/annurev.cellbio.19.111301.155141. [DOI] [PubMed] [Google Scholar]
- 33.Egel-Mitani M, Olson LW, Egel R. Meiosis in Aspergillus nidulans: another example for lacking synaptonemal complexes in the absence of crossover interference. Hereditas. 1982;97:179–187. doi: 10.1111/j.1601-5223.1982.tb00761.x. [DOI] [PubMed] [Google Scholar]
- 34.Olson LW, Edén U, Mitani ME, Egel R. Asynaptic meiosis in fission yeast? Hereditas. 1978;89:189–199. [Google Scholar]
- 35.Wolfe J, Hunter B, Adair WS. A cytological study of micronuclear elongation during conjugation in Tetrahymena. Chromosoma. 1976;55:289–308. doi: 10.1007/BF00292827. [DOI] [PubMed] [Google Scholar]
- 36.Loidl J, Scherthan H. Organization and pairing of meiotic chromosomes in the ciliate Tetrahymena thermophila. J Cell Sci. 2004;117:5791–5801. doi: 10.1242/jcs.01504. [DOI] [PubMed] [Google Scholar]
- 37.Borner GV, Kleckner N, Hunter N. Crossover/noncrossover differentiation, synaptonemal complex formation, and regulatory surveillance at the leptotene/zygotene transition of meiosis. Cell. 2004;117:29–45. doi: 10.1016/s0092-8674(04)00292-2. [DOI] [PubMed] [Google Scholar]
- 38.Perry J, Kleckner N, Borner GV. Bioinformatic analyses implicate the collaborating meiotic crossover/chiasma proteins Zip2, Zip3, and Spo22/Zip4 in ubiquitin labeling. PNAS. 2005;102:17594–17599. doi: 10.1073/pnas.0508581102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tsubouchi T, Zhao H, Roeder GS. The Meiosis-Specific Zip4 Protein Regulates Crossover Distribution by Promoting Synaptonemal Complex Formation Together with Zip2. Developmental Cell. 2006;10:809–819. doi: 10.1016/j.devcel.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 40.Agarwal S, Roeder GS. Zip3 provides a link between recombination enzymes and synaptonemal complex proteins. Cell. 2000;102:245–255. doi: 10.1016/s0092-8674(00)00029-5. [DOI] [PubMed] [Google Scholar]
- 41.Hanson PI, Whiteheart SW. AAA+ proteins: have engine, will work. Nat Rev Mol Cell Biol. 2005;6:519–529. doi: 10.1038/nrm1684. [DOI] [PubMed] [Google Scholar]
- 42.Ye Y, Meyer HH, Rapoport TA. Function of the p97-Ufd1-Npl4 complex in retrotranslocation from the ER to the cytosol: dual recognition of nonubiquitinated polypeptide segments and polyubiquitin chains. J Cell Biol. 2003;162:71–84. doi: 10.1083/jcb.200302169. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data include two figures, one table and Experimental Procedures can be found on line at: