

Abstract
Endoglin is predominantly expressed on endothelium and is mutated in hereditary hemorrhagic telangiectasia (HHT) type 1 (HHT1). We report the analysis of endoglin in tissues of a newborn (family 2), who died of a cerebral arteriovenous malformation (CAVM), and in a lung specimen surgically resected from a 78-year-old patient (family 5), with a pulmonary AVM (PAVM). The clinically affected father of the newborn revealed a novel mutation that was absent in his parents and was identified as a duplication of exons 3 to 8, by quantitative multiplex polymerase chain reaction. The corresponding mutant protein (116-kd monomer) and the missense mutant protein (80-kd monomer) present in family 5 were detected only as transient intracellular species and were unreactive by Western blot analysis and immunostaining. Normal endoglin (90-kd monomer) was reduced by 50% on peripheral blood-activated monocytes of the HHT1 patients. When analyzed by immunostaining and densitometry, presumed normal blood vessels of the newborn lung and brain and vessels adjacent to the adult PAVM showed a 50% reduction in the endoglin/PECAM-1 ratio. A similar ratio was observed in the CAVM and PAVM, suggesting that all blood vessels of HHT1 patients express reduced endoglin in situ and that AVMs are not attributed to a focal loss of endoglin.
Hereditary hemorrhagic telangiectasia (HHT), also known as Rendu-Osler-Weber syndrome, is an autosomal dominant vascular dysplasia that affects 1:10,000 individuals. This disorder is associated with epistaxis and telangiectases in the majority of patients and with pulmonary and cerebral arteriovenous malformations (PAVM and CAVM, respectively) in 15 to 20% of cases. 1,2 These abnormalities are caused primarily by the dilatation of postcapillary venules, which eventually connect directly to arterioles without intervening capillaries. 3 Direct shunting of blood through CAVMs may lead to ischemic and/or hemorrhagic infarctions. 4
Only a small proportion of CAVMs observed in the general population is associated with HHT. CAVMs have been observed predominantly in newborns and children and may present with either high-output congestive heart failure, migraine-like headaches, or rupture, which may lead to death. 5-9 Such lesions are thought to originate from a disordered mesodermal differentiation occurring between 3 and 8 weeks of gestation. 10 The expression of factors such as vascular endothelial growth factor and the vascular tyrosine kinase receptor Tie-1 in CAVMs suggests active angiogenesis in these malformations. 11,12
Of PAVM cases, at least 70% are associated with HHT. 2,13,14 These range from diffuse telangiectases to large complex structures consisting of a bulbous aneurysmal sac between dilated feeding arteries and draining veins. 14 Current data suggest that HHT1 families have a much higher incidence of PAVM than HHT2 families. 15-18 CAVMs often cluster in families with a higher prevalence of PAVM and are thus likely also to be more frequent in HHT1 families.
HHT is a heterogeneous disorder in terms of its clinical manifestations. This is explained in part at the molecular level by the involvement of at least two different loci. 16,19 The candidate gene for HHT1 was mapped to chromosome 9q33–34 19,20 and was identified as endoglin, 16 which codes for a homodimeric integral membrane glycoprotein expressed at high levels on vascular endothelial cells 21 and previously mapped to 9q33–34. 22 Endoglin was first shown to be a component of transforming growth factor (TGF)-β1 and TGF-β3 receptor complexes. 23 More recently, it was demonstrated to interact with the ligand-binding receptor for several members of the TGF-β superfamily, including activin and bone morphogenic proteins. 24 Thirty-nine distinct mutations in the endoglin gene have now been reported in HHT1 patients, 14,16,25-30 and most families carry a distinct mutation.
The ALK-1 gene, coding for an activin-like kinase receptor type I of the TGF-β receptor super family, maps to chromosome 12q and is mutated in HHT2. 31,32 The clinical HHT2 phenotype is characterized by a later onset of disease and less penetrance; 18 distinct mutations in the ALK-1 gene have been described. 33-35
With human umbilical vein endothelial cells (HUVEC) and activated monocytes in culture obtained from HHT1 patients, we have demonstrated that mutated forms of endoglin are transient intracellular species that do not reach the cell surface. 24,28,30 This suggests that a reduction in the level of functional endoglin (haploinsufficiency) rather than a dominant negative effect of the mutant protein is responsible for HHT1. In the current study, we were able to examine tissues of a newborn, who died subsequent to the rupture of a CAVM, and a lung specimen resected from an elderly patient with a PAVM. This permitted us to establish that reduced endoglin levels can be observed in situ in seemingly normal vessels. We also studied vessels of the vascular lesions, CAVM and PAVM respectively, and determined that they still expressed endoglin.
Materials and Methods
Clinical Evaluation and Patient Samples
Informed consent was obtained from all participants for blood samples and surgical specimens. All procedures were reviewed and approved by the Research Ethics Board of the Research Institute at the Hospital for Sick Children. All members of families with HHT are given a number, which is referred to with the prefix H (for HHT).
In family 2 (Figure 1A) ▶ , patient H9 died at 7 days of a cerebral hemorrhage due to the rupture of one of the two CAVMs present, followed by heart failure; autopsy was performed 8 hours after death. Paraffin-embedded sections (5–7 μm) of CAVMs and unaffected vascular beds were obtained from the Pathology Department, Hospital for Sick Children, Toronto. Control samples were prepared similarly from newborns who died of unrelated causes. Blood samples were received from patients H300, H262, H283, H299, H3, H4, and from placenta and umbilical cord from newborn H11.
Figure 1.
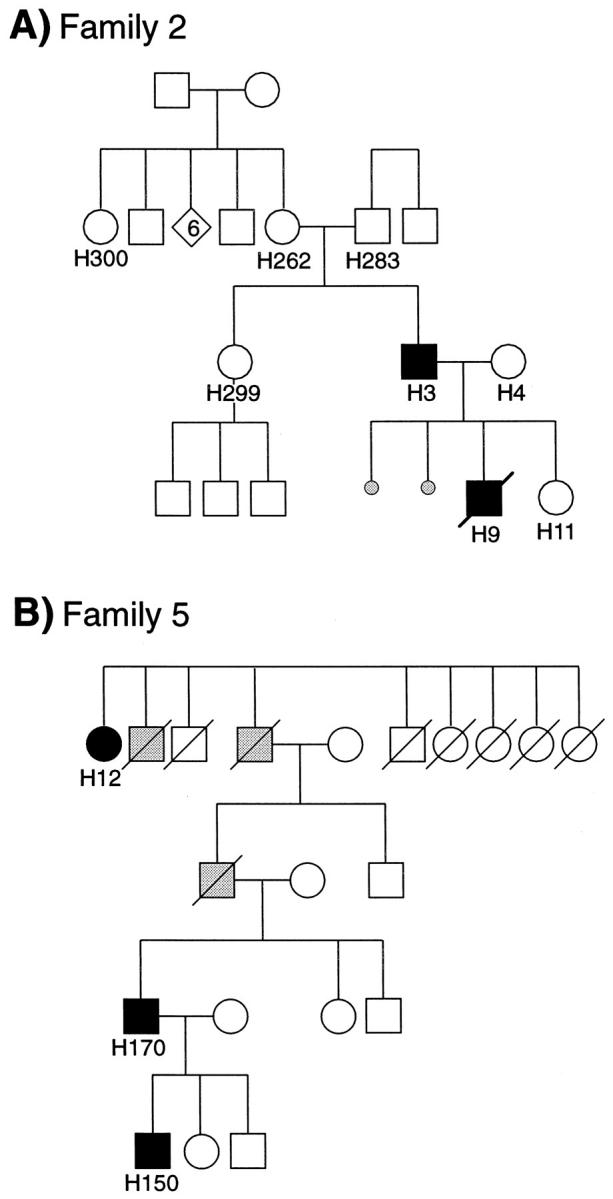
A: Pedigrees of HHT Family 2. Patients H283 and H262 were both unaffected and are the biological parents of patient H3, who presented with telangiectases, nosebleeds, and PAVM. His wife, patient H4, was unaffected and had two miscarriages at 7 weeks of gestation. Their first child, patient H9, died at 7 days of a cerebral hemorrhage followed by heart failure caused by rupture of one of the two CAVMs present. Their second child, patient H11, was born normally and was confirmed unaffected. B: Pedigree of family 5 with a history of HHT1 for four generations. Patient H12 had nosebleeds, telangiectases, and a PAVM. Patient H150 presented with a PAVM and telangiectases, whereas patient H170 had nosebleeds and telangiectases. Black symbols, HHT1-affected individuals; gray symbols, HHT1-suspected individuals; white symbols, unaffected individuals.
Family 5 has four generations of clinically affected individuals, as illustrated in Figure 1B ▶ . A lung PAVM was surgically resected from patient H12, a 78-year-old, nonsmoking woman. This specimen was dissected into the lesion itself and adjacent areas. Fresh tissues were used for preparation of DNA, protein extracts, and formalin-fixed, paraffin-embedded tissue blocs. Sections (5–7 μm) of PAVM and unaffected lung adjacent areas were obtained by J. Tessitore, Pathology Department, University of Vermont College of Medicine, Burlington. Blood samples were obtained from patients H150 and H170. Normal lung samples from a 68-year-old, nonsmoking woman were obtained by B. Mullen, Pathology Department, Mount Sinai Hospital, Toronto.
Cell Culture, Metabolic Labeling, and Immunoprecipitation
Activated monocytes were prepared from peripheral blood of patients and controls, by adherence to plastic and culture for 16 hours at 37°C as previously described. 28,36 Cells were washed with serum-free media and incubated for 30 minutes in methionine-free Dulbecco’s modified Eagle medium before labeling with 100 μCi/ml 35S-methionine (trans-label; ICN Pharmaceuticals, Montreal, Quebec, Canada) for 3.5 hours. Cells were then solubilized in 0.01 mol/L Tris, pH 7.5, 0.128 mol/L NaCl, 1 mmol/L ethylenediaminetetraacetic acid, 1% Triton X-100 plus protease inhibitors (lysis solution) and immunoprecipitated with saturating amounts of monoclonal antibody (mAb) P3D1 or P4A4. mAb P3D1 (and mAb SN6h used in Western blot and immunostaining) recognize epitopes in the first 204 amino acids of the extracellular domain of endoglin (corresponding to exons 1–5). mAb P4A4 reacts with an epitope encoded by exon 7 and located between amino acids 277 and 331. 37 To quantify endoglin expression and correct for differences in yield between samples, aliquots of total lysates were precipitated with 10% trichloracetic acid, and total incorporation into proteins was determined. Equivalent amounts of labeled proteins (in counts per minute) were fractionated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE; 4–12%; Novex Experimental Technology, San Diego, CA) under reducing (0.05 mol/L dithiothreitol) and nonreducing conditions. Gels were exposed on X-OMAT AR film (Eastman Kodak Co., Rochester, NY), and radioactivity in each band was quantified by Phosphorimager and ImageQuant software (Molecular Dynamics, Sunnyvale, CA).
Western Blot
Peripheral blood-activated monocytes were solubilized in lysis solution. Protein concentration was estimated with the Bio-Rad assay (Bio-Rad Laboratories Ltd., Mississauga, ON, Canada), and known amounts were fractionated on SDS-PAGE (8%) under nonreducing conditions. Proteins were transferred electrophoretically onto nitrocellulose membranes, which were then blocked for 1 hour in Tris-buffered saline-T (0.02 mol/L Tris, pH 7.5, 0.137 mol/L NaCl, 0.1% Tween 20) containing 5% skim milk. Membranes were incubated for 1 hour with mAb SN6h (ascites, diluted 30,000-fold) or mAb P4A4 (1 μg/ml), followed by 1 hour with horseradish peroxidase-conjugated rabbit anti-mouse immunoglobulin G (IgG; H&L, 10,000-fold dilution; Jackson Immunolabs, Bio-Can, Mississauga, ON, Canada). Endoglin was detected by enhanced chemiluminescence (ECL detection kit, Amersham Life Sciences, Oakville, ON, Canada).
Mutation Analysis by Quantitative Multiplex Polymerase Chain Reaction and Sequencing
Genomic DNA was extracted from blood lymphocytes, placenta, HUVEC, and lung specimens using DNAZOL (Life Technologies Inc.). Purity and quality of the DNA and accurate estimation of the concentration are critical for fragment analysis by quantitative multiplex polymerase chain reaction (QMPCR). All 15 exons of endoglin were amplified in five PCR reactions using one selected Cy 5.5 fluorescent conjugated primer for each exon as previously described. 30 Quantitative amplification was achieved with 18 to 22 cycles, annealing T° ranging from 51 to 55°C, optimized primer concentrations ranging from 80 to 800 nmol/L, and 150 ng of genomic DNA. The amplification of a c4 fragment (329 or 282 bp) derived from a gene on chromosome 15 was included as internal standard in each of the reactions. The multiplex PCR pools and fragment sizes (in base pairs) were as follows: reaction 1, exon 9b (149), exon 4 (283) c4 standard (329), exon 2 (363), exon 6 (389) and exon 11 (426); reaction 2, exon 12 (154), c4 standard (282), exon 10 (304), exon 1 (314), and exon 8 (373); reaction 3, exon 9a (222), exon 5 (238), exon 13 (255), and c4 standard (282); reaction 4, exon 14 (269), exon 7 (289), and c4 standard (329); reaction 5, exon 3 (251) and c4 standard.(329)
Thermocycling was performed using DNAEngine (MJ Research), and QMPCR products were run on a MicroGene Blaster Sequencer (Visible Genetics Inc., Toronto, Ontario, Canada) for 30 to 40 minutes. The data were analyzed using GeneObjects DNA analysis software (Visible Genetics Inc.). The ratio of the peak area for each endoglin exon was calculated relative to that of the c4 internal standard for each patient sample and compared with the normal two-copy control DNA samples.
The exons to be sequenced were first amplified with nonlabeled primers that were identical to those used in QMPCR and then were sequenced as described above. 30
Immunohistochemical Staining
Paraffin-embedded sections of lung, spinal cord, and CAVM from patient H9 and age-matched controls and from lung specimens from patient H12 and age-matched controls were dewaxed by standard procedures, blocked with 5% normal goat serum (Dako, Mississauga, ON, Canada) in Tris-buffered saline-T (0.01 mol/L Tris, pH 7.4, 0.16 mol/L NaCl, 0.2% Tween 20) for 20 minutes and incubated at 4°C for 2 hours with optimal concentrations of primary antibody. These were mAb JC70A to PECAM-1(CD31; hybridoma supernatant diluted eightfold; D. Mason, Oxford, U.K.), mAb 1A4 to α-smooth muscle cell actin (ascites diluted 2000-fold; Sigma Chemical Co., Oakville, ON, Canada), nonimmune murine IgG1 (10 μg/ml; Coulter, Burlington, ON, Canada), and mAb SN6h to endoglin (ascites diluted 8000-fold; obtained from B. Seon, through the VI Leukocyte International Workshop). mAb SN6h is the best of about 40 mAbs tested at detecting human endoglin in paraffin-embedded tissue sections. 39-41 Slides were washed and incubated for 1 hour at 4°C with an alkaline phosphatase goat anti-mouse IgG Fab′2 (diluted 400-fold; Jackson Immunolabs, Bio-Can). In some experiments, the labeled streptavidin biotin-positive Dako amplification system was used by the manufacturer’s instructions (Dako). The enzymatic reaction (1 hour at 23°C) was initiated by adding 0.35 mmol/L 5-bromo-4-chloro-3-indolylphosphate toluidinium, 0.45 mmol/L nitroblue tetrazolium (Boehringer Mannheim, Montreal, Quebec, Canada), and 0.2 mmol/L levamisole (Sigma Chemical Co., St-Louis, MO) to block endogenous alkaline phosphatase activity. To facilitate image analysis, sections were not counterstained.
Tissue morphology was assessed with the hematoxylin and eosin stain. The elastin histochemical stain was performed by the manufacturer’s instructions (Accustain; Sigma).
Image Analysis
Images were acquired in black and white directly from the stained tissue sections with an Olympus BX50 microscope linked to a charge-coupled device video camera, using Image Pro software analysis (Carsen Medical Scientific Co., Ltd., Markham, ON, Canada) and digitization on a Power Macintosh G3 computer. Staining intensity on endothelial cells of arteries, veins, and capillaries was quantified as follows, using the software NIH Image 1.61/fat for Power Macintosh (http://rsb.info.nih.gov/nih-image/Default.html) and Image Pro-Plus. For each large vessel or group of capillaries, images were enlarged, and 300 measurements were taken on the endothelial layer with the cross-hair tool counting the average gray value of the selected pixel (scale of 0–256 shades of gray; 0 = white, and 256 = black). Mean density values and SEM were calculated for PECAM-1, endoglin, and IgG1 of both control and patient tissue sections stained in the same experiment. Note that the background mean intensity values, determined with IgG1 control were less than 15 in all sections. The ratio of the mean density values of endoglin and PECAM-1 was calculated for each vessel. The endoglin/PECAM-1 ratio of the HHT patient was then compared with that of the control and expressed as relative endoglin levels (%).
Results
Description of a New Endoglin Mutant Protein in Family 2
The pedigree of family 2 is illustrated in Figure 1A ▶ . The clinical diagnosis of HHT was made on examination of patient H3, who presented with daily nosebleeds since childhood and also had telangiectases. Further investigations revealed two small PAVMs but no CAVMs. His son, H9, died at 7 days of a cerebral hemorrhage due to rupture of one of two CAVMs present followed by heart failure, rendering the diagnosis of HHT in this child likely. A healthy daughter, H11, was born 2 years later. No definite diagnosis of HHT was made for other family members, although patient H262, mother of H3, and her sister (H300), had frequent nosebleeds in childhood and adolescence, which subsequently subsided.
We first examined the level of endoglin and the presence of potential mutant proteins in peripheral blood-activated monocytes from members of this family. Figure 2A ▶ demonstrates that fully glycosylated endoglin monomer (E; 90 kd) and a partially glycosylated precursor (P; 80 kd) were immunoprecipitated from control lysates (lanes 1 and 3) with both mAb P3D1 and P4A4, which recognize distinct regions of endoglin. In patient H3 samples, E and P were expressed at reduced levels, and a mutant protein M (116 kd) was detected (lanes 2 and 4). In the absence of dithiothreitol, the normal dimers of endoglin (E; 160 kd) and precursor (P; 140 kd) were observed in control lanes 9 and 11. With patient H3, these normal homodimers were present at lower levels (lanes 10 and 12); additional dimers were seen at 200 and 240 kd, as well as traces of monomers (lanes 10 and 12). The level of surface endoglin (E) was estimated by Phosphorimager and ImageQuant software analysis, at 50 ± 4% in H3 activated monocytes compared with control (mean of 4 values; Table 1 ▶ ). The mutant form (M in Figure 2 ▶ ) represented 45% of the total endoglin (E + P + M) seen in patient H3.
Figure 2.

Analysis of peripheral blood-activated monocytes shows reduced levels of normal endoglin and presence of a mutant protein in patient H3 but not in his parents. A: Cells from the clinically affected patient H3 (family 2), and a normal control (C) were labeled with 35S-methionine, solubilized in Triton X-100, and immunoprecipitated with mAb P3D1 or P4A4 to endoglin. Equivalent amounts of labeled proteins were fractionated on 4 to 12% gradient SDS-PAGE, under reducing (lanes 1–4) and nonreducing (lanes 9–12) conditions. Fully processed endoglin (E) and partially glycosylated precursor (P) were observed in patient H3 at reduced levels compared with control, and a mutant protein (M) is noted. Cells from individuals H262 and H283, mother and father of patient H3, were also analyzed in the same manner (lanes 5–8 and 13–16), normal E and P forms of endoglin were seen at normal levels, and no mutant protein was observed. B: Activated monocytes from patient H3 were lysed in Triton X-100, and various amounts of proteins were fractionated on 8% sodium dodecyl sulfate-polyacrylamide gels, under nonreducing conditions (without dithiothreitol). Gels were transferred and analyzed by Western blot analysis, using mAb SN6h to endoglin. The control (C) was an extract from HUVEC. Only the normal dimers of endoglin (E) are detected by Western blot analysis. Molecular mass markers in kilodaltons are indicated.
Table 1.
Clinical and Molecular Data on HHT Families 2 and 5
| Family | Patient | Clinical manifestations | Endoglin levels on monocytes (mean ± SD) | Mutation identified at DNA level |
|---|---|---|---|---|
| 2 | 3 | Nosebleeds, telangiectases, PAVM | 50 ± 4 | Yes |
| Duplication of exon 3–8 | ||||
| 2 | 4 | — | nd* | No |
| 2 | 9 | CAVMs | nd | nd |
| 2 | 11 | — | 85 ± 13† | No |
| 2 | 262 | Nosebleeds before puberty‡ | 121 ± 17 | No |
| 2 | 283 | — | 83 ± 9 | No |
| 2 | 299 | Nosebleeds before puberty‡ | 97 ± 11 | No |
| 2 | 300 | — | 126 ± 20 | No |
| 5 | 12 | Telangiectases, nosebleeds, PAVM | nd | Yes |
| Missense G447C, exon 4 | ||||
| 5 | 150 | Telangiectases, PAVM | 34 ± 8 | Yes |
| Missense G447C, exon 4 | ||||
| 5 | 170 | Telangiectases, nosebleeds | nd | Yes |
| Missense G447C, exon 4 |
*nd, not done; —, unaffected family member.
†Levels of endoglin measured on HUVEC for newborn H11, rather than on activated monocytes as for adults.
‡Although patients 262 and 299 had nosebleeds in childhood, they had no other signs of HHT and were confirmed not to have the mutation.
Protein analysis was performed on several other family members, including parents of patient H3, the presumed affected mother (H262) and control father (H283). To our surprise, normal levels of endoglin were found in H262, and no mutant protein was present (Figure 2A ▶ , lanes 5–8 and 13–16). Additional family members tested had normal levels of endoglin, and no detectable mutant, including patient H11, sister of the proband for whom HUVEC analysis was performed (Table 1 ▶ and Figure 1A ▶ ).
Because only pathological specimens were available from newborn H9, we needed to establish that the mutant protein, if present, would not be detected by immunostaining, which reflects steady-state levels of protein expression. The relative stability of mutant endoglin was thus analyzed by Western blot using mAb SN6h, which gives optimal detection of endoglin on paraffin-embedded sections. 37,39 Only normal endoglin dimers were reactive with SN6h in H3 samples; no trace of mutant protein was observed (Figure 2B) ▶ . Furthermore, the mutant protein was not reactive with mAb P4A4 by Western blot, whereas it was detected as a newly synthesized mutant by metabolic labeling (Figure 2A) ▶ . We also demonstrated by cell surface biotinylation of activated monocytes from patient H3 that no mutant protein was detected at the cell surface (data not shown). Therefore, this novel mutant form of endoglin, despite its larger molecular mass, is only a transient intracellular species not expressed at the cell surface and not detected at steady-state level, either by Western blot or immunostaining.
Identification of a Novel Endoglin Mutation Arising in Family 2
DNA samples were subjected to QMPCR to screen for an endoglin mutation that could account for the generation of a larger-than-normal protein, as well as resolve the question of a new mutation arising in patient H3 and absent from his parents. Figure 3 ▶ demonstrates that DNA from patient H3, fractionated in five different reactions so that each exon could be analyzed, contained an additional copy of exons 3 to 8. The area under each peak is proportional to the allele copy number when QMPCR is truly quantitative, as optimized in preliminary studies. 30 The ratio of the peak area for exons 3 to 8 from H3 DNA ranged from 1.3 to 1.6, compared with the mean values derived from 12 controls that had been run in the same analysis. This implies that three copies of these exons were present compared with the two-copy control, presumably due to an intronic mutation that included the duplication of exons 3 to 8. Each exon of the endoglin gene was amplified individually from the DNA of patient H3 and sequenced, but no mutation was found at splice junctions or in the exons, further supporting an intronic mutation. This mutation was not seen in the parents (H283 and H262) of patient H3, confirming that they were not affected and that the endoglin mutation had arisen in patient H3. DNA was also analyzed from H4, H11, H299, and H300 individuals (see Figure 1A ▶ ), and no mutation was found, confirming that they were not affected.
Figure 3.
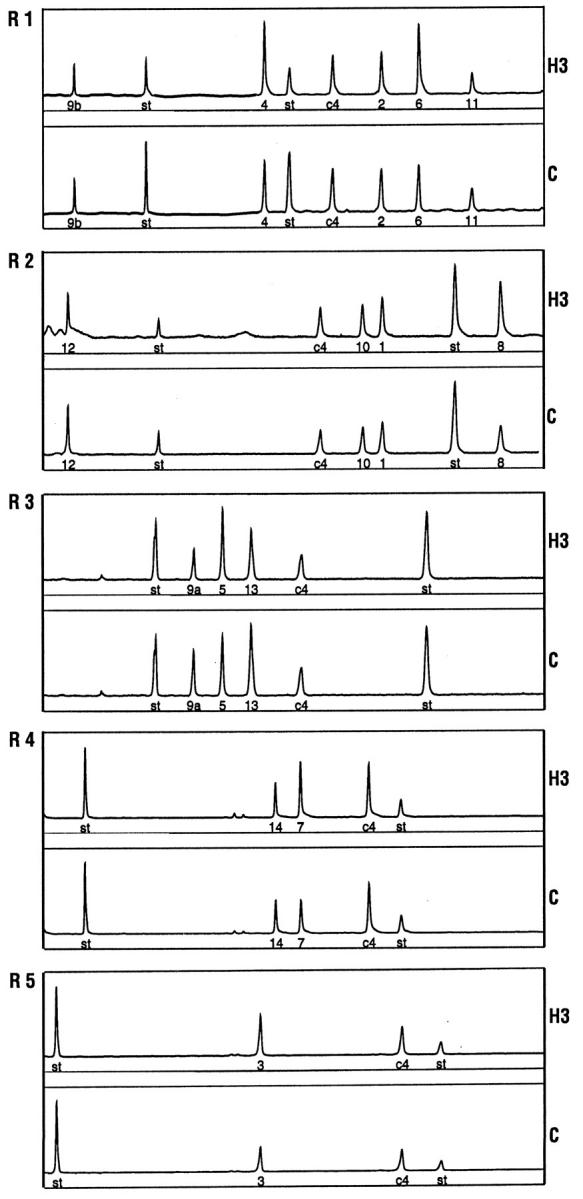
QMPCR fragment analysis reveals a duplication of exons 3 to 8 in patient H3 of family 2. DNA samples were analyzed by QMPCR using five different reactions (R1–R5), optimized so that peak area is proportional to copy number for each exon. Illustrated here is DNA from patient H3 and control (c) for each of the five reactions. The exon number is indicated under the peak, and “st” refers to internal size standards. The ratio of each exon is expressed relative to c4 fragment, an internal copy number standard. In each experiment, the peak area was estimated for H3 relative to the mean ratio observed for 12 control samples. The ratios of H3 relative to these controls were 1.0 for exons 1 and 2 and ranged from 1.3 to 1.6 for exons 3 to 8 and from 0.84 to 1.0 for exons 9a to 14. Accordingly, peak surface area is greater for exons 3 to 8 in patient H3.
DNA fingerprinting with 9 different markers confirmed that individual H3 and his parents, H262 and H283, shared the expected alleles and established that this mutation arose in patient H3. DNA isolated from shavings of the paraffin-embedded CAVM and adjacent brain tissue of newborn H9 was of sufficient quality to confirm 5 markers shared with his parents, H3 and H4, but not pure enough to demonstrate the mutation for QMPCR analysis.
Analysis of Endoglin DNA and Protein in Family 5
Family 5 shows four generations of individuals affected with HHT (Figure 1B) ▶ . Patient H12, age 78, underwent resection of the right lung middle lobe because she was unfit to undergo transcatheter embolization of her PAVM. Patient H150, also with a PAVM, was shown to express reduced levels of endoglin in peripheral blood-activated monocytes (34 ± 8%; Table 1 ▶ ).
The mutation in this family was reported previously, as a missense mutation in exon 4, a G to C substitution at bp 447 of the endoglin cDNA. 29 In the current study, DNA was isolated from the PAVM lesion itself of patient H12, and, when analyzed, it revealed the expected mutation (Figure 4) ▶ . Sequencing also revealed that the normal copy of endoglin was still present in the lesion, ruling out a loss of heterozygosity (Figure 4) ▶ .
Figure 4.

Normal and mutant copies of endoglin DNA are present in the PAVM of patient H12. DNA extracted from the PAVM of patient H12, (family 5) was sequenced for exon 4. At position 447, G of the normal allele and C of the mutant allele are both present. This missense mutation was absent in the control DNA sample.
This mutation leads to a tryptophan-to-cysteine conversion at amino acid 149. The mutant protein was detectable only by metabolic labeling and as a transient intracellular precursor of 80 kd, as described previously. 36 This transient protein was not seen by Western blot analysis of protein extracts from PAVM and adjacent areas in samples of lung from patient H12. However, normal endoglin was readily detectable in both the highly vascularized PAVM and the adjacent tissues (data not shown).
Immunostaining of the Vasculature of a Newborn with CAVM (Family 2)
Normal Vasculature
Lung and spinal cord resected from newborn H9, at the time of autopsy, and from age-matched controls were analyzed by immunostaining. We first demonstrated that endoglin and PECAM-1, both classified as endothelial surface antigens, were specifically detected in formalin-fixed tissues, using mAb SN6h 39,41 and mAb JC70A, 42 respectively (Figure 5) ▶ . Both antibodies stained endothelial cells of arteries, veins, and capillaries in control and patient H9; no other cell type was reactive. Specificity of staining was confirmed by the absence of reaction with control murine IgG1, and differential staining of arteries, veins, and capillaries was achieved. Staining with mAb 1A4 to α-smooth muscle cell actin was also performed to differentiate veins and arteries (data not shown).
Figure 5.
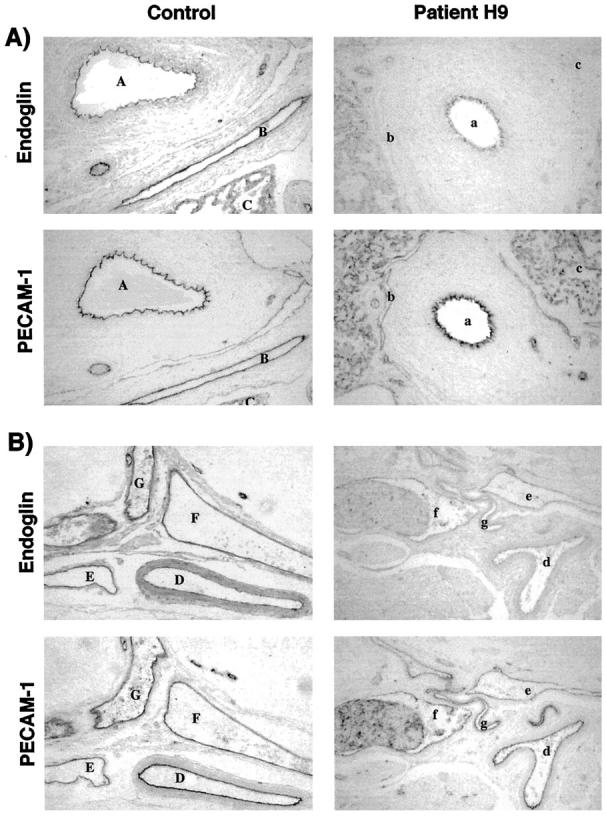
Vessels of lung and central nervous system from the HHT1 newborn express less endoglin than control, compared with PECAM-1. Formalin-fixed, paraffin-embedded lung (A) and spinal cord (B) sections from control and patient H9 were stained with mAbs SN6h to endoglin and JC70A to PECAM-1, followed by an alkaline phosphatase-conjugated antibody. A: Endoglin and PECAM-1 staining on an artery (A, control; a, patient H9), vein (B; b) and capillaries (C; c) of lung tissues are illustrated for control and patient H9. B: Sections of dural vessels of spinal cord are shown. Two arteries (D; d and E; e), two veins (F; f and G; g) are represented for age-matched control and patient H9. PECAM-1 was also detected on platelets in vessels with blood infiltrates. Original magnification, ×194.
We next compared the levels of expression of endoglin and PECAM-1 in lung sections of patient H9 and control (Figure 5A) ▶ . In the control newborn, endoglin and PECAM-1 were observed at equivalent levels on the endothelium of artery (areas A in Figure 5A ▶ ), vein (areas B in Figure 5A ▶ ), and capillaries (areas C in Figure 5A ▶ ). With patient H9, the staining intensity of endoglin, compared with PECAM-1, was much reduced on the artery (areas a in Figure 5A ▶ ), vein (areas b in Figure 5A ▶ ), and capillaries (areas c in Figure 5A ▶ ), compared with control sections. We quantified, by image digitization and densitometry analysis, the staining intensity of vessels illustrated in Figure 5A ▶ and of additional surrounding lung vessels (Table 2) ▶ . The PECAM-1 mean densities were relatively similar between vessel types and between patient and control samples stained in the same experiment. Values ranged from 107 to 171 in control sections and from 95 to 182 in patient H9. Endoglin mean density was also similar between vessel types, but clearly lower in patient H9 (30 to 81) than control (106 to 145). To standardize data analysis, the endoglin/PECAM-1 ratio was calculated for each vessel, to allow comparison between patient and control (Table 2) ▶ . The relative endoglin levels (endoglin/PECAM-1 ratio of patient to control) were then calculated and shown to be reduced in all lung vessels, including 46 to 61% in arteries, between 31% and 52% in veins, and between 47% and 52% in capillaries (Table 2) ▶ .
Table 2.
Densitometry Analysis of Endoglin and PECAM-1 on Endothelial cells.
| Vessel type | Control | Patient H9 | Relative endoglin levels (%) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Endoglin mean density | PECAM-1 mean density | E/P ratio | Endoglin mean density | PECAM-1 mean density | E/P ratio | ||||
| Lung artery | A | 112 ± 1.7 | 153 ± 1.6 | 0.73 | a | 69 ± 1.4 | 155 ± 1.9 | 0.45 | 61 ± 6% |
| Lung vein | B | 145 ± 1.4 | 142 ± 1.4 | 1.02 | b | 30 ± 0.5 | 95 ± 1.5 | 0.32 | 31 ± 5% |
| Lung capillaries | C | 128 ± 1.5 | 144 ± 2.1 | 0.89 | c | 80 ± 1.7 | 171 ± 2.1 | 0.47 | 52 ± 6% |
| Lung artery | 125 ± 2.1 | 171 ± 2.0 | 0.73 | 75 ± 1.7 | 182 ± 2.2 | 0.41 | 57 ± 6% | ||
| Lung artery | 106 ± 1.8 | 139 ± 2.1 | 0.76 | 58 ± 1.3 | 163 ± 2.6 | 0.36 | 46 ± 7% | ||
| Lung vein | 133 ± 1.7 | 126 ± 2.6 | 1.06 | 81 ± 1.6 | 146 ± 2.8 | 0.55 | 52 ± 7% | ||
| Lung capillaries | 113 ± 1.9 | 107 ± 1.8 | 1.06 | 69 ± 1.1 | 138 ± 1.5 | 0.50 | 47 ± 6% | ||
| Spinal cord artery | D | 123 ± 1.5 | 179 ± 2.2 | 0.69 | d | 51 ± 0.8 | 121 ± 1.7 | 0.42 | 61 ± 5% |
| Spinal cord artery | E | 119 ± 1.2 | 121 ± 1.6 | 0.98 | e | 59 ± 0.8 | 87 ± 1.1 | 0.68 | 69 ± 5% |
| Spinal cord vein | F | 116 ± 1.4 | 141 ± 1.8 | 0.82 | f | 61 ± 1.0 | 109 ± 1.6 | 0.56 | 68 ± 6% |
| Spinal cord vein | G | 137 ± 1.5 | 147 ± 1.7 | 0.93 | g | 42 ± 0.8 | 103 ± 1.2 | 0.41 | 44 ± 5% |
| CAVM feeder artery | N.A. | N.A. | N.A. | 97 ± 2.4 | 195 ± 1.4 | 0.50 | N.A. | ||
| CAVM venous pouch | N.A. | N.A. | N.A. | 27 ± 0.8 | 46 ± 1.5 | 0.58 | N.A. |
The mean density values were evaluated for selected vessels indicated by corresponding letters in Figures 5 to 7 ▶ ▶ ▶ , as well as additional ones in adjacent tissues, and are expressed as mean ± SEM of 300 measurements per vessel. The relative endoglin levels (%) are estimated as endoglin mean density/PECAM-1 mean density of patient H9 versus the endoglin level of a newborn control. E/P, Endoglin/PECAM-1 ratio; N.A., not applicable.
To extend our observations to other unaffected vascular beds, we examined dural vessels of spinal cord of child H9 and control. Endoglin and PECAM-1 were detectable on endothelium of all vessels, as shown in Figure 5B ▶ for selected arteries (areas D, d, E, e) and veins (F, f; G, g). Endoglin levels were noticeably reduced, compared with PECAM-1, in patient H9. Mean density values of PECAM-1 and endoglin on endothelial cells in control dural vessels were similar to those observed in lung vessels (Table 2) ▶ . Densitometry analysis (Table 2) ▶ revealed that the relative endoglin levels of patient H9 were significantly reduced on dural arteries (61% and 69%) and veins (44% and 68%). Although not present on the sections analyzed by densitometry, capillaries of the central nervous system showed reduced expression of endoglin in patient H9. Thus, in situ arteries, veins, and capillaries of normal vasculature of the newborn with HHT1 have significantly reduced endoglin levels.
CAVM
To determine whether vascular lesions in HHT1 are associated with further reduction or focal loss of endoglin, we examined, by immunostaining, the CAVM from patient H9. Sections from a major feeding vessel to the CAVM, the right middle cerebral artery, were stained for elastin, endoglin, and PECAM-1 (Figure 6) ▶ . The elastin stain demonstrates an abnormally large, dilated, and tortuous vessel. Mesenchymal cells, connective tissue, and collagen fibers were disorganized in the adventitia, whereas a thick and compact layer of smooth muscle cells characterized the media. The intima was separated from the media by a continuous internal elastica lamina (Figure 6 ▶ , dark black line) and a thin subendothelial layer lining this feeding vessel. On the uninterrupted endothelial lining of the intima, the endoglin/PECAM-1 ratio was estimated at 0.5 (Table 2) ▶ . Therefore, the feeding vessel to the AVM of patient H9, despite its highly abnormal structure, expressed 50% endoglin on its endothelium, as observed with unaffected vessels of this patient.
Figure 6.
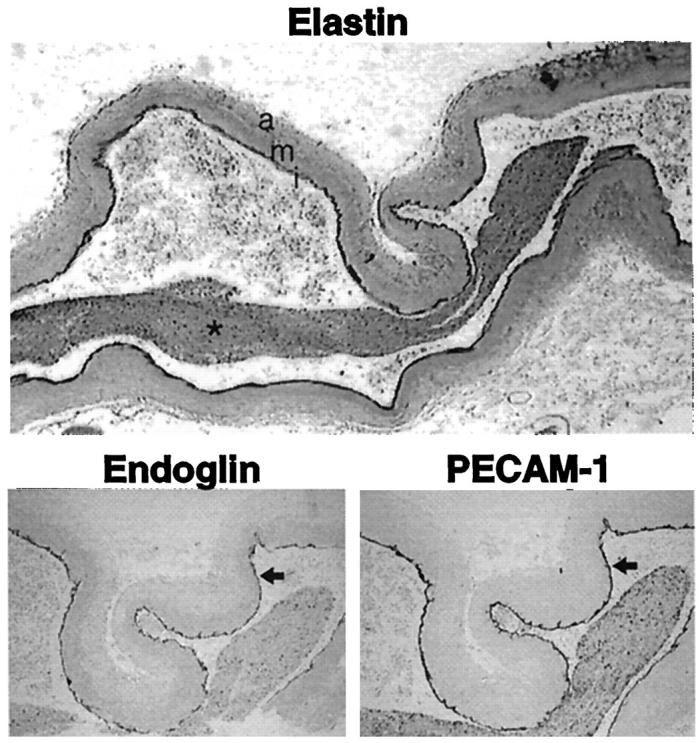
Endoglin is expressed on the large abnormal feeding vessel of the CAVM in patient H9. Sections from the right middle cerebral artery feeding the CAVM that ruptured in patient H9 were stained with Van Giesen elastin stain. The adventitia (a), media (m), and intima (i) of the artery are marked. The lumen of this vessel is filled with blood (*). Sections stained for endoglin and PECAM-1 by alkaline phosphatase immunostaining are shown at higher magnification. The arrow outlines endoglin and PECAM-1 staining on the endothelium. Original magnifications: ×31 and ×78.
AVMs consist of a tangled network of vascular channels, which form dilatations. In patient H9, the aneurysmic dilatation downstream from the right middle cerebral artery was examined (Figure 7) ▶ . Staining for α-smooth muscle cell actin revealed a thin and tortuous vessel filled with blood, at the site of rupture (Figure 7A) ▶ . Intense remodeling of the vessel wall can be seen at higher magnification, where α-smooth muscle cell actin showed variable thickness and a complex network of disorganized smooth muscle cells (Figure 7B) ▶ . Endoglin was barely detectable on the endothelial layer but was seen on mesenchymal-like cells in the small and very compact adventitia. Such a pattern may reflect the ongoing vascular remodeling process in the malformation. The venous pouch (Figure 7C) ▶ formed in the aneurysmic dilatation showed an extremely thin layer of endothelium outlined by PECAM-1 staining. There was minimal endoglin left on these endothelial cells. The extreme dilatation led to a dispersed endothelium and to lower mean density values for both PECAM-1 (46 ± 1.5) and endoglin (27 ± 0.8), evaluated by image analysis (Table 2) ▶ . In the dilatation, the endoglin/PECAM-1 ratio (0.58) was, however, not significantly distinct from that observed in the unaffected vessels of the patient (0.47), suggesting that CAVMs in HHT1 patients are not associated with a complete absence or a focal loss of endoglin in the vascular lesion.
Figure 7.
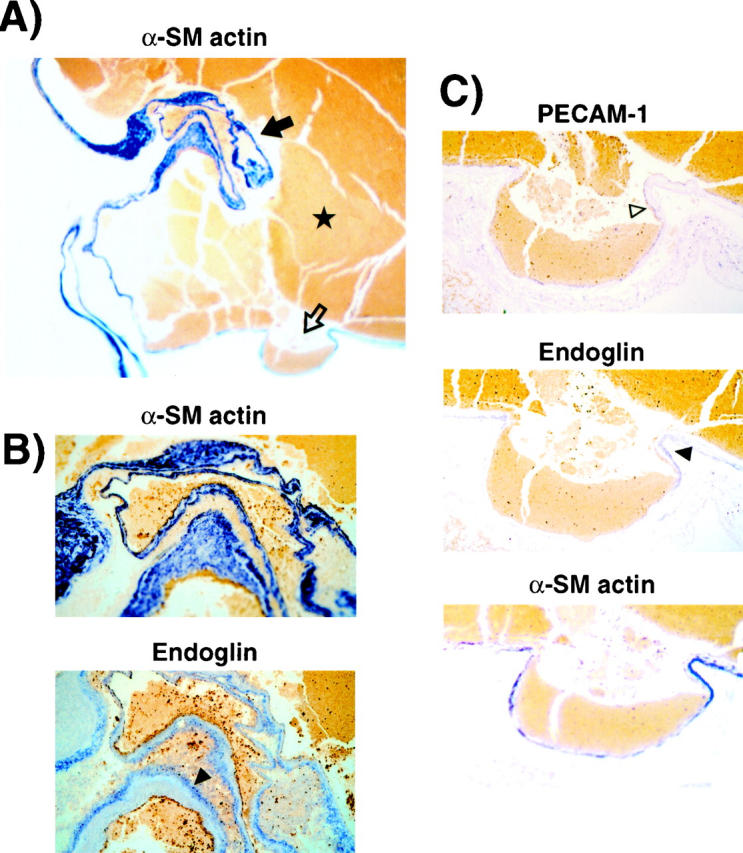
Endoglin is detected on the endothelium and the adventitia of the aneurysmic dilatation of the CAVM. A: Sections of the aneurysm of patient H9, downstream from the right middle cerebral feeding artery, were stained for α-smooth muscle cell actin. ★, hemorrhage in the ruptured vessel. Original magnification, ×22. B: An area showing intense remodeling of the vessel wall in A (solid arrow) is shown at higher magnification (original magnification, ×194). The subsequent section stained for endoglin revealed expression on mesenchymal cells (solid arrowhead). C: The venous pouch shown in A (open arrow) was stained for PECAM-1, endoglin, and α-smooth muscle cell actin. The dispersed endothelium is indicated by the open arrowhead. Original magnification, ×194.
Immunostaining of PAVM and Adjacent Tissues of Patient H12 (Family 5)
Normal Vasculature
To confirm and extend our observations that endoglin is present at reduced levels on all types of vessels, in HHT1 patients, we studied a 78-year-old patient (H12) who presented with a PAVM. Surgical specimens of unaffected lung tissues adjacent to the PAVM were immunostained and compared with those of an age-matched control woman. Endoglin and PECAM-1 were detectable on the endothelium of all vessels, and their mean densities were carefully measured for three arteries and three veins in sections of uninvolved lung in patient H12 and control. The endoglin/PECAM-1 ratios ranged from 0.62 to 0.80 for the control and from 0.35 to 0.45 for patient H12 (Table 3) ▶ . The relative endoglin levels in unaffected lung tissues of patient H12 versus control ranged from 52% to 66% on arteries and from 46% to 62% on veins (Table 3) ▶ .
Table 3.
Densitometry Analysis of Endoglin and PECAM-1 on Endothelial Cells
| Vessel type | Control | Patient H12 | Relative endoglin levels (%) | ||||
|---|---|---|---|---|---|---|---|
| Endoglin mean density | PECAM-1 mean density | E/P ratio | Endoglin mean density | PECAM-1 mean density | E/P ratio | ||
| Lung artery | 139 ± 2.3 | 173 ± 2.3 | 0.80 | 55 ± 1.3 | 122 ± 1.8 | 0.45 | 56 ± 7% |
| Lung artery | 106 ± 1.9 | 158 ± 1.7 | 0.67 | 51 ± 1.2 | 114 ± 1.6 | 0.45 | 66 ± 7% |
| Lung artery | 105 ± 2.3 | 140 ± 1.9 | 0.75 | 50 ± 1.9 | 127 ± 2.0 | 0.39 | 52 ± 9% |
| Lung vein | 109 ± 1.9 | 161 ± 1.8 | 0.68 | 57 ± 1.4 | 136 ± 1.8 | 0.42 | 62 ± 7% |
| Lung vein | 90 ± 1.7 | 146 ± 1.6 | 0.62 | 39 ± 1.1 | 111 ± 1.8 | 0.35 | 57 ± 7% |
| Lung vein | 115 ± 1.8 | 153 ± 1.8 | 0.75 | 42 ± 1.2 | 121 ± 1.8 | 0.35 | 46 ± 7% |
| PAVM | N.A. | N.A. | N.A. | 24 ± 2.5 | 68 ± 2.0 | 0.35 | N.A. |
The mean density values were evaluated and are expressed as mean ± SEM of 300 measurements per vessel. The relative endoglin levels (%) are estimated as endoglin mean density/PECAM-1 mean density of patient H12 versus that of age-matched control. E/P, Endoglin/PECAM-1 ratio; N.A., not applicable.
PAVM
The PAVM located in the lung right middle lobe of patient H12 was stained for elastin, endoglin and PECAM-1 (Figure 8) ▶ . The elastin stain demonstrated an abnormally dilated vascular space, with very dispersed and disorganized mesenchymal cells, connective tissue, and collagen fibers in the adventitia. A variable thickness of smooth muscle cells was found in the media of the vessel wall, suggesting attempts at remodeling. The intima was separated from the media by a discontinuous internal elastica lamina (Figure 8 ▶ , dark black line). Elastin was also abundant in the parenchyma, adjacent to the malformation. The endothelial cells of the intima, outlined by PECAM-1 staining, were dispersed as the vessel was highly dilated. Endoglin was still detectable on the endothelium of the PAVM. Image analysis revealed that the endoglin/PECAM-1 ratio was 0.35 in the lesion, a value not significantly different from that observed on unaffected vessels of this patient (0.40; Table 3 ▶ ). Thus, the PAVM is not due to a focal loss of endoglin on the endothelium of the lesion.
Figure 8.

Endoglin is still detectable on the endothelial cells of the PAVM. Sections from the resected lung right middle lobe of patient H12, containing a PAVM, were stained with Van Giesen elastin stain. The lung parenchyma (p) and the three layers of the abnormally dilated vessel—adventitia (a), media (m), and intima (i)—are shown. Subsequent sections of the PAVM were stained for endoglin and PECAM-1 by alkaline phosphatase immunostaining. The arrow shows the staining of the endothelial cells with both markers. Original magnifications: ×30 and ×165.
Discussion
In the current study, we report a novel endoglin mutation arising in the father of a newborn, who died subsequent to rupture of a CAVM. This mutation was characterized at DNA and protein levels. A duplication of exons 3 to 8 led to expression of a transient mutant protein of higher molecular mass than normal, detectable only by metabolic labeling. Relative levels of endoglin, quantified in several vascular beds of the newborn, were reduced by half, indicating that only the normal allele was expressed on endothelium in situ. Similar results were obtained in the vasculature adjacent to a PAVM resected from an elderly patient with a known HHT1 mutation. In both cases, endoglin was still expressed in the AVM, indicating that the lesions were not caused by a loss of heterozygosity.
The endoglin mutation present in patient H3, but absent from his biological parents, constitutes the first demonstration of a new mutation arising in an HHT1 family. Furthermore, such a large insertion has not been reported previously for endoglin. However, we have also observed a duplication of exons 3 to 8 in another family of different ethnicity. This novel type of mutation was detected using fragment analysis by QMPCR, which was optimized such that peak height is proportional to the number of copies of each exon present. 30 This mutation codes for a mutant monomer of 116 kd, larger than the normal monomer of 90 kd and seen only by metabolic labeling. This mutant form of endoglin was not found at the cell surface, despite the presence of a transmembrane region. It exists only as a transient intracellular species and not at steady-state level, and it is consequently not detectable by Western blot and immunostaining.
In this study, the level of normal endoglin was reduced on activated monocytes of patient H3 (family 2) to 50 ± 4%, and in patient H150 (family 5), to 34 ± 8% of control. Endoglin levels are considered reduced when they are less than 70%, as determined from the analysis of affected members from 67 HHT1 families. 14,30 Reduced levels of endoglin, which were revealed here by in vitro analyses in patients with a novel insertion mutation (family 2) and with a missense mutation (family 5), support a model of haploinsufficiency for HHT1, as previously proposed and demonstrated. 14,27,28,30,36
Data presented in this paper show that all types of blood vessels in the two HHT1 patients for which pathological specimens were available (H9 and H12) expressed reduced levels of endoglin in situ. Lung and spinal cord vasculature of newborn H9 were considered normal because they showed no morphological abnormalities. However these normal vessels expressed relative endoglin levels that were reduced to 53%, when compared with age-matched controls. There was no significant difference (95% confidence interval) in the relative endoglin levels on the endothelium of blood vessels of the lung and central nervous system. In the vasculature of lung tissue adjacent to a PAVM in the 78-year-old patient, H12, the relative endoglin levels were reduced to 56% of those in age-matched control lung tissue. There was no significant difference (95% confidence) between the relative endoglin levels of arteries, veins and capillaries in these patients. Our findings raise the possibility that an HHT1-affected individual could be identified by estimating the relative levels of endoglin on vessels in normal skin biopsies, which would not require affected vessels such as telangiectases.
Rupture of CAVMs causes a significant number of cerebral hemorrhages that can be fatal. CAVMs are mostly seen in children 5-9 and are likely congenital, arising during a period critical to development of brain vasculature. 10 PAVMs also occur in children, but the majority present during adolescence or adult life. 13,14 Common to both angiogenesis and vasculogenesis is the process of remodeling, which occurs in CAVMs and PAVMs and involves changes in lumen diameter and vessel wall thickness. In the CAVM of newborn H9, we demonstrated that the feeding artery was highly abnormal but still expressed endoglin on its endothelium, although at reduced levels. In the aneurysmic dilatation, smooth muscle cells were disorganized, leading to a media of variable thickness. No internal elastica lamina was present, and the endothelial layer was dispersed and very thin, due to increased pressure. The aneurysmic dilation of patient H9 was extremely large, with a diameter of 8 to 11 mm, a portion of which is illustrated in Figure 7A ▶ . The PAVM of patient H12 was in fact smaller with 6 to 7 mm in diameter in the dilatation shown in Figure 8 ▶ . It demonstrated similar characteristics, with thin-wall dilatation, interrupted elastica lamina, variable thickness of smooth muscle cells, and dispersed and disorganized adventitia. In both lesions, the mean density values of PECAM-1 and endoglin were both reduced because of the larger surface area of the dilated endothelium (Tables 2 and 3) ▶ ▶ . However the endoglin/PECAM-1 ratio in the AVMs was similar to that observed in normal vessels of the patients. These observations are compatible with an ongoing remodeling process and suggest that CAVMs/PAVMs are not due to a focal loss of endoglin in the lesions.
Other factors/receptors regulating vascular development and integrity, such as vascular endothelial growth factor observed in the subendothelial layer and the perivascular spaces 11,12 and Tie-1 receptor present on the endothelium of CAVMs, 12 must contribute to the formation of CAVMs and PAVMs. These molecules are necessary for vascular development and their presence in focal areas of CAVMs suggests active angiogenesis and vascular remodeling. 11,12 The familial form of venous malformations was shown to be associated with an activating mutation in the kinase domain of the Tie-2, expressed on endothelial cells. 43 Tie-2 is the receptor for angiopoietin-1, 44 a factor that indirectly stimulates the differentiation of smooth muscle cells and plays critical roles during vessel formation. 45 Failure to recruit smooth muscle cells may lead to abnormal proliferation of endothelial cells, characteristic of venous malformations.
TGF-β1 is another important factor implicated in vasculogenesis/angiogenesis, because it regulates interactions between endothelial cells and both mesenchymal and smooth muscle cells of the vessel wall. 46,47 Endoglin is a component of the TGF-β1–β3 receptor complex, 23,24 which can modulate several responses to these ligands, as demonstrated in the U-937 monocytic cell line. 48 TGF-β3 has been implicated in lung development such that an altered response to this ligand, in an endoglin-deficient individual, could contribute to the generation of PAVMs. 49
It has been proposed that CAVMs are caused by a defect in early vascular development and are associated with ongoing abnormal vascular remodeling. 10 Our results in the newborn support this idea and suggest that a mutation in endoglin could perturb the regulatory effects of TGF-β on the early development of brain vessels. However, because most CAVMs are not associated with HHT1, we must conclude that endoglin is only one of several genes regulating brain vasculature. For PAVMs, 70% to 80% are found in HHT1 patients, 14 suggesting a more frequent contribution of endoglin deficiency in their generation. Because TGF-β and other factors are critical for vascular homeostasis, any disruption of their effects by altering their receptors or signaling pathways could lead to abnormal vessel function and subsequent vascular malformations.
The recent observations that endoglin-null mice die of vascular defects in early gestation, 50 similar to those observed for TGF-β1– and TGF-β receptor II–null mice, suggest that, indeed, endoglin is critical for vascular development and that its function is likely related to an altered response to TGF-β1. We also observed that some heterozygous mice on a specific genetic background developed HHT. 51 These novel data confirm that a single copy of endoglin confers susceptibility to the disease but that modifier genes contribute to the development of vascular abnormalities. Furthermore, not all mice of the susceptible strain developed the disease, suggesting that epigenetic factors such as shear stress and environmental conditions are also implicated in the generation of arteriovenous malformations.
Acknowledgments
We gratefully acknowledge the patients who participated in our study. We thank Dr. D. Y. Mason (Oxford, UK.) for providing mAb JC70A to PECAM-1, Dr. E. A. Wayner (Seattle, WA) for mAbs P3D1 and P4A4, and Dr. B. Seon for providing mAb SN6h to endoglin. We thank Dr. C. Smith, Dr. B. Mullen, Ms I. Diplock, and Dr. J. Tessitore, for providing patient samples and paraffin-embedded sections.
Footnotes
Address reprint requests to Dr. Michelle Letarte, Blood and Cancer Research Program, Hospital for Sick Children, 555 University Avenue, Toronto, Canada M5G 1X8. E-mail: mablab@sickkids.on.ca.
Supported by Heart and Stroke Foundation of Ontario grant NA3434 and by Medical Research Council of Canada grant MT6247. A. B. is a recipient of a Studentship from the Medical Research Council of Canada, and M. L. is a Terry Fox Research Scientist of the National Cancer Institute of Canada.
References
- 1.Guttmacher AE, Marchuk DA, White RIJ: Hereditary hemorrhagic telangiectasia. N Engl J Med 1995, 333:918-924 [DOI] [PubMed] [Google Scholar]
- 2.Wirth JA, Pollak JS, White RIJ: Pulmonary arteriovenous malformations. Current Pulmonology and Critical Care Medicine. 1996, :pp 261-298 Mosby-Yearbook, St. Louis [Google Scholar]
- 3.Braverman IM, Keh A, Jacobson BS: Ultrastructure and three-dimensional organization of the telangiectases of hereditary hemorrhagic telangiectasia. J Invest Dermatol 1990, 95:422-427 [DOI] [PubMed] [Google Scholar]
- 4.Fayad PB: Neurologic Manifestations of Hereditary Hemorrhagic Telangiectasia. Edited by S Gilman, LB Goldstein, SG Waxman. La Jolla, CA, Neurobase, 1995
- 5.Kikuchi K, Kowada M, Sasajima H: Vascular malformations of the brain in hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber disease). Surg Neurol 1994, 41:374-380 [DOI] [PubMed] [Google Scholar]
- 6.Kadoya C, Momota Y, Ikegami Y, Urasaki E, Wada S, Yokota A: Central nervous system arteriovenous malformations with hereditary hemorrhagic telangiectasia: report of a family with three cases. Surg Neurol 1994, 42:234-239 [DOI] [PubMed] [Google Scholar]
- 7.Jessurun GA, Kamphuis DJ, van der Zande FH, Nossent JC: Cerebral arteriovenous malformations in the Netherlands Antilles: high prevalence of hereditary hemorrhagic telangiectasia-related single and multiple cerebral arteriovenous malformations. Clin Neurol Neurosurg 1993, 95:193-198 [DOI] [PubMed] [Google Scholar]
- 8.Roy C, Noseda G, Arzimanoglou A, Harpey JP, Binet MH, Vaur C, Caille B: Maladie de Rendu-Osler révélée par la rupture d’un anévrysme artériel cérébral chez un nourrisson. Arch Fr Pediatr 1990, 47:741-742 [PubMed] [Google Scholar]
- 9.Belzic I, Yaseen H, Voirin J, Bonte JB, Laloum D: Malformations artério-veineuses cérébrales dans une probable forme familiale de maladie de Rendu-Osler. Ann Pediatr 1992, 39:301-304 [PubMed] [Google Scholar]
- 10.Challa VR, Moody DM, Brown WR: Vascular malformations of the central nervous system. J Neuropathol Exp Neurol 1995, 54:609-621 [DOI] [PubMed] [Google Scholar]
- 11.Sonstein WJ, Kader A, Michelsen WJ, Llena JF, Hirano A, Casper D: Expression of vascular endothelial growth factor in pediatric and adult cerebral arteriovenous malformations: an immunocytochemical study. J Neurosurg 1996, 85:838-845 [DOI] [PubMed] [Google Scholar]
- 12.Hatva E, Jääskeläinen J, Hirvonen H, Alitalo K, Haltia M: Tie endothelial cell-specific receptor tyrosine kinase is upregulated in the vasculature of arteriovenous malformations. J Neuropathol Exp Neurol 1996, 55:1124-1133 [DOI] [PubMed] [Google Scholar]
- 13.White R, Jr, Lynch-Nyhan A, Terry P, Buescher PC, Farmlett EJ, Charnas L, Shuman K, Kim W, Kinnison M, Mitchell SE: Pulmonary arteriovenous malformations: techniques and long-term outcome of embolotherapy. Radiology 1988, 169:663-669 [DOI] [PubMed] [Google Scholar]
- 14.Shovlin C, Letarte M: Hereditary hemorrhagic telangiectasia and pulmonary arteriovenous malformations: issues in clinical management and review of pathogenic mechanisms. Thorax 1999, 54:714-729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heutink P, Haitjema T, Breedveld GJ, Janssen B, Sandkuijl LA, Bontekoe CJ, Westerman CJ, Oostra BA: Linkage of hereditary haemorrhagic telangiectasia to chromosome 9q34 and evidence for locus heterogeneity. J Med Genet 1994, 31:933-936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McAllister KA, Grogg KM, Johnson DW, Gallione CJ, Baldwin MA, Jackson CE, Helmbold EA, Markel DS, McKinnon WC, Murrell J, McCormick MK, Pericak-Vance MA, Heutink P, Oostra BA, Haitjema T, Westerman CJJ, Porteous ME, Guttmacher AE, Letarte M, Marchuk DA: Endoglin, a TGF-β binding protein of endothelial cells, is the gene for hereditary haemorrhagic telangiectasia type 1. Nat Genet 1994, 8:345-351 [DOI] [PubMed] [Google Scholar]
- 17.Porteous ME, Curtis A, Williams O, Marchuk D, Bhattacharya SS, Burn J: Genetic heterogeneity in hereditary haemorrhagic telangiectasia. J Med Genet 1994, 31:925-926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Berg JN, Guttmacher AE, Marchuk DA, Porteous ME: Clinical heterogeneity in hereditary haemorrhagic telangiectasia: are pulmonary arteriovenous malformations more common in families linked to endoglin? J Med Genet 1996, 33:256-257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shovlin CL, Hughes JM, Tuddenham EG, Temperley I, Perembelon YF, Scott J, Seidman CE, Seidman JG: A gene for hereditary haemorrhagic telangiectasia maps to chromosome 9q3. Nat Genet 1994, 6:205-209 [DOI] [PubMed] [Google Scholar]
- 20.McDonald MT, Papenberg KA, Ghosh S, Glatfelter AA, Biesecker BB, Helmbold EA, Markel DS, Zolotor A, McKinnon WC, Vanderstoep JL: A disease locus for hereditary haemorrhagic telangiectasia maps to chromosome 9q33–34. Nat Genet 1994, 6:197-204 [DOI] [PubMed] [Google Scholar]
- 21.Gougos A, Letarte M: Primary structure of endoglin, an RGD-containing glycoprotein of human endothelial cells. J Biol Chem 1990, 265:8361-8364 [PubMed] [Google Scholar]
- 22.Fernández-Ruiz E, St-Jacques S, Bellón T, Letarte M, Bernabéu C: Assignment of the human endoglin gene (END) to 9q34→qter. Cytogenet Cell Genet 1993, 64:204-207 [DOI] [PubMed] [Google Scholar]
- 23.Cheifetz S, Bellón T, Calés C, Vera S, Bernabeu C, Massagué J, Letarte M: Endoglin is a component of the transforming growth factor-β receptor system in human endothelial cells. J Biol Chem 1992, 267:19027-19030 [PubMed] [Google Scholar]
- 24.Pece Barbara N, Wrana J, Letarte M: Endoglin is an accessory protein that interacts with the signaling receptor complex of multiple members of the transforming growth factor-β superfamily. J Biol Chem 1999, 274:584–594 [DOI] [PubMed]
- 25.McAllister KA, Baldwin MA, Thukkani AK, Gallione CJ, Berg JN, Porteous ME, Guttmacher AE, Marchuk DA: Six novel mutations in the endoglin gene in hereditary hemorrhagic telangiectasia type 1 suggest a dominant-negative effect of receptor function. Hum Mol Genet 1995, 4:1983-1985 [DOI] [PubMed] [Google Scholar]
- 26.Yamaguchi H, Azuma H, Shigekiyo T, Inoue H, Saito S: A novel missense mutation in the endoglin gene in hereditary hemorrhagic telangiectasia. Thromb Haematol 1997, 77:243-247 [PubMed] [Google Scholar]
- 27.Shovlin CL: Molecular defects in rare bleeding disorders: hereditary haemorrhagic telangiectasia. Thromb Haematol 1997, 78:145-150 [PubMed] [Google Scholar]
- 28.Pece N, Vera S, Cymerman U, White RIJ, Wrana JL, Letarte M: Mutant endoglin in hereditary hemorrhagic telangiectasia type 1 is transiently expressed intracellularly and is not a dominant negative. J Clin Invest 1997, 100:2568-2579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gallione CJ, Klaus DJ, Yeh EY, Stenzel TT, Xue Y, Anthony KB, McAllister KA, Baldwin MA, Berg JN, Lux A, Smith JD, Vary CPH, Craigen WJ, Westermann CJJ, Warner ML, Miller YE, Jackson CE, Guttmacher AE, Marchuk DA: Mutation and expression analysis of the endoglin gene in hereditary hemorrhagic telangiectasia reveals null alleles. Hum Mutat 1998, 11:286-294 [DOI] [PubMed] [Google Scholar]
- 30.Cymerman U, Vera S, Pece-Barbara N, Bourdeau A, White RIJ, Dunn J, Letarte M: Identification of hereditary hemorrhagic telangiectasia type 1 in newborns by protein expression and mutation analysis of endoglin. Pediat Res 2000, 47 (in press) [DOI] [PubMed]
- 31.Vincent P, Plauchu H, Hazan J, Faure S, Weissenbach J, Godet J: A third locus for hereditary haemorrhagic telangiectasia maps to chromosome 12q. Hum Mol Genet 1995, 4:945-949 [DOI] [PubMed] [Google Scholar]
- 32.Johnson DW, Berg JN, Gallione CJ, McAllister KA, Warner JP, Helmbold EA, Markel DS, Jackson CE, Porteous ME, Marchuk DA: A second locus for hereditary hemorrhagic telangiectasia maps to chromosome 12. Genome Res 1995, 5:21-28 [DOI] [PubMed] [Google Scholar]
- 33.Johnson DW, Berg JN, Baldwin MA, Gallione CJ, Marondel I, Yoon SJ, Stenzel TT, Speer M, Pericak-Vance MA, Diamond A, Guttmacher AE, Jackson CE, Attisano L, Kucherlapati R, Porteous ME, Marchuk DA: Mutations in the activin receptor-like kinase 1 gene in hereditary haemorrhagic telangiectasia type 2. Nat Genet 1996, 13:189-195 [DOI] [PubMed] [Google Scholar]
- 34.Berg JN, Gallione CJ, Stenzel TT, Allen WP, Schwartz CE, Jackson CE, Porteous ME, Marchuk DA: The activin receptor like kinase 1 gene: genomic structure and mutations in hereditary hemorrhagic telangiectasia type 2. Am J Hum Genet 1997, 61:60-67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Klaus DJ, Gallione CJ, Antony K, Yeh EY, Yu J, Lux A, Johnson DW, Marchuk DA: Novel missense and frameshift mutations in the activin receptor-like kinase-1 gene in hereditary hemorrhagic telangiectasia. Hum Mutat (Mutations in Brief online, number 164), 1998, http://journals.wiley.com/1059-7794/mutbf/htm [DOI] [PubMed]
- 36.Pece Barbara N, Cymerman U, Vera S, Marchuk D, Letarte M: Expression analysis of four endoglin missense mutations suggests haploinsufficiency is the predominant mechanism for hereditary hemorrhagic telangiectasia type 1. Hum Mol Genet 1999, 8:2171–2181 [DOI] [PubMed]
- 37.Pichuantes S, Vera S, Bourdeau A, Pece N, Kumar S, Wayner EA, Letarte M: Mapping epitopes to distinct regions of the extracellular domain of endoglin using bacterially expressed recombinant fragments. Tissue Antigens 1997, 50:265-276 [DOI] [PubMed] [Google Scholar]
- 38.Letarte M, Greaves A, Vera S: CD105 (Endoglin) Cluster Report: Leukocyte Typing V: White Cell Differentiation Antigens. Edited by SF Schlossman, L Boumsell, W Gilks, J Harlan, T Kishimoto, C Morimoto, J Ritz, S Shaw, R Silverstein, T Springer, T Tedder, R Todd. Oxford, UK, Oxford University Press, 1995, pp 1756–1759
- 39.Letarte M, Bourdeau A, Vera S, Pece N, Greaves A, Dignat-Georges F, Mutin M, Kraling B, Linask K, O’Brien E, Labinaz M, Ross J, Parish C, Bernabeu C: EC2 CD105 Workshop panel report: Leukocyte Typing VI: White Cell Differentiation Antigens. Edited by T Kishimoto, H Kikutani, AEGK von dem Borne, SM Goyert, DY Mason, M Miyasaka, L Moretta, K Okumura, S Shaw, TA Springer, K Sugamura, H Zola. New York, Garland Publishing Inc, 1997, pp 703–708
- 40.Haruta Y, Seon BK: Distinct human leukemia-associated cell surface glycoprotein GP160 defined by monoclonal antibody SN6. Proc Natl Acad Sci USA 1986, 83:7898-7902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Seon BK, Matsuno F, Haruta Y, Barcos M: CD105 Workshop: Immunohistochemical detection of CD105 in the vascular endothelium of human malignant and non-malignant tissues. Leukocyte Typing VI: White Cell Differentiation Antigens. Edited by T Kishimoto, H Kikutani, AEGK von dem Borne, SM Goyert, DY Mason, M Miyasaka, L Moretta, K Okumura, S Shaw, TA Springer, K Sugamura, H Zola. New York, Garland Publishing Inc, 1997, pp 709–710
- 42.Parums DV, Cordell JL, Micklem K, Heryet AR, Gatter KC, Mason DY: JC70: a new monoclonal antibody that detects vascular endothelium associated antigen on routinely processed tissue sections. J Clin Pathol 1990, 83:752-757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vikkula M, Boon LM, Carraway KR, Calvert JT, Diamonti AJ, Goumnerov B, Pasyk KA, Marchuk DA, Warman ML, Cantley LC, Mulliken JB, Olsen BR: Vascular dysmorphogenesis caused by an activating mutation in the receptor tyrosine kinase TIE2. Cell 1996, 87:1181-1190 [DOI] [PubMed] [Google Scholar]
- 44.Davis S, Aldrich TH, Jones PF, Acheson A, Compton DL, Jain V, Ryan TE, Bruno J, Radziejewski C, Maisonpierre PC, Yancopoulos GD: Isolation of angiopoietin-1, a ligand for the TIE2 receptor, by secretion-trap expression cloning. Cell 1996, 87:1161-1169 [DOI] [PubMed] [Google Scholar]
- 45.Suri C, Jones PF, Patan S, Bartunkova S, Maisonpierre PC, Davis S, Sato TN, Yancopoulos GD: Requisite role of angiopoietin-1, a ligand for the TIE2 receptor, during embryonic angiogenesis. Cell 1996, 87:1171-1180 [DOI] [PubMed] [Google Scholar]
- 46.Dickson MC, Martin JS, Cousins FM, Kulkarni AB, Karlsson S, Akhurst RJ: Defective haematopoiesis and vasculogenesis in transforming growth factor-β1 knock out mice. Development 1995, 121:1845-1854 [DOI] [PubMed] [Google Scholar]
- 47.Folkman J, D’Amore PA: Blood vessel formation: what is its molecular basis? Cell 1996, 87:1153-1155 [DOI] [PubMed] [Google Scholar]
- 48.Lastres P, Letamendía A, Zhang H, Rius C, Almendro N, Raab U, López LA, Langa C, Fabra A, Letarte M, Bernabéu C: Endoglin modulates cellular responses to TGF-β1. J Cell Biol 1996, 133:1109-1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kaartinen V, Voncken JW, Shuler C, Warburton D, Bu D, Heisterkamp N, Groffen J: Abnormal lung development and cleft palate in mice lacking TGF-β3 indicates defects of epithelial-mesenchymal interaction. Nat Genet 1995, 11:415-421 [DOI] [PubMed] [Google Scholar]
- 50.Li DY, Sorensen LK, Brooke BS, Urness LD, Davis EC, Taylor DG, Boak BB, Wendel DP: Defective angiogenesis in mice lacking endoglin. Science 1999, 284:1534-1537 [DOI] [PubMed] [Google Scholar]
- 51.Bourdeau A, Dumont DJ, Letarte M: A murine model of hereditary hemorrhagic telangiectasia. J Clin Invest. 1999, 104:1343-1351 [DOI] [PMC free article] [PubMed] [Google Scholar]