

Abstract
Microglial activation and redistribution toward blood vessels are some of the earliest observable events occurring within the central nervous system (CNS) during fatal murine cerebral malaria (FMCM). To investigate stimuli that might modulate microglial reactivity during FMCM we have performed two experimental manipulations and observed microglial responses in retinal whole mounts. First, to determine whether increased blood-brain barrier (BBB) permeability in the absence of the malaria parasite initiates the microglial changes, BBB function was compromised experimentally by intracarotid injection of arabinose and retinae were examined 12, 24, or 36 hours later. Second, to determine whether the immune response against the malaria parasite modulates microglial reactivity, infected mice were treated with dexamethasone before day 4 postinoculation. This treatment regime ameliorates cerebral complications without affecting parasite growth. We observed that increased BBB permeability was sufficient to elicit thickening of microglial processes and redistribution of microglia toward the vasculature, characteristic of the early stages of FMCM. However, despite the presence of plasma constituents in the CNS for up to 36 hours, microglia with amoeboid and vacuolated morphology were not observed. Dexamethasone treatment inhibited the up-regulation of α-D-galactose expression and reactive morphological changes in microglia during FMCM. These results suggest that disruption of the CNS milieu by entry of plasma constituents, or circulating malaria parasites in the absence of an immune response, by themselves are insufficient to induce the reactive microglial changes that are characteristic of FMCM. In addition, dexamethasone-sensitive event(s), presumably associated with immune system activation, occurring within the first few days of malaria infection are essential for the development of reactive microglia and subsequent fatal neurological complications.
Fatal murine cerebral malaria (FMCM) is an immunopathological process. In particular, T cells, 1-3 monocytes, 4 and their secretory products appear to be essential for the development of the disease in the murine model. Recently, we have shown that the host response to FMCM is not restricted to the periphery or the cerebral vascular endothelium. Within the central nervous system (CNS) parenchyma, astrogliosis and degeneration of astrocytes, 5 activation of microglia, 6 and increased c-fos and cytokine expression 7,8 are observed and may be important in the initiation and perpetuation of the cerebral complications associated with FMCM.
Of the events so far observed within the CNS parenchyma, changes in microglia are the earliest, occurring within 2 to 3 days postinoculation (p.i.), that is, at least 3 days before the onset of cerebral symptoms and 4 days before death. 6 Morphological changes included retraction of ramified processes, soma enlargement, an increasingly amoeboid appearance, and vacuolation. There was also redistribution of activated microglia toward retinal vessels, in particular toward the venous side of the vascular endothelium. Another striking feature was the up-regulation of α-D-galactose residues on microglia, made evident by an increase in Griffonia simplicifolia (GS) lectin staining. Consistent with previous findings, 9,10 only a small population of microglia in the uninfected adult mouse retina were GS lectin-positive. 6 However, there was a striking increase in the focal density of GS-positive microglia, indicative of their activation, 11 during the progression of the disease. In a “resolving” model of murine cerebral malaria, 4 the microglial changes were transient and much less pronounced. 12
Our previous studies using the retinal whole mount technique have shown a strong relationship between microglial activation, by morphological criteria, and events occurring at the blood-brain barrier (BBB) during FMCM. 6 Microglia redistribute toward the retinal vessels with compromised barrier properties, changes in microglial morphology are initiated in those microglia juxtaposed with the retinal vasculature, and tumor necrosis factor-α (TNF-α) production by microglia is most commonly found in areas close to the cerebral vessels. 8 Comparable microglial changes, including evidence of activation, in human cerebral malaria have been reported. 13,14
A possible initiating event for microglial changes is the early increase in BBB permeability seen in FMCM. 4,15,16 To determine whether the breakdown in barrier properties alone is sufficient to initiate microglial morphological and distributional changes in the absence of parasite inoculation, we now have experimentally induced an increase in BBB permeability through intracarotid injection of arabinose. 17 This procedure produces vasodilatation and endothelial cell dehydration, stretching endothelial cell membranes, and increasing the permeability of intercellular tight junctions. 18
Considering protein concentration in the CNS extracellular space is about 200-fold less than that of the plasma, 19 it has been hypothesized that exposure to plasma constituents as a result of increased BBB permeability may play a role in the modulation of microglia function and phenotype. 20 For example, the expression by microglia of CD4 and sialic acid binding receptor are regulated by exposure to an inducing agent in serum. 20 Microglia may alter their morphology in response to increased BBB permeability because they phagocytose foreign protein and thereby act as sinks to preserve the CNS microenvironment. 21 Furthermore, circulating factors in the blood, including kinins and amino acids such as glutamine and glycine, may cause stress or injury to brain cells and subsequently modulate microglial activity. 22,23
Microglia are also responsive to products of the immune system. Inflammatory mediators and/or high concentrations of potassium from damaged cells provide signals necessary to promote microglial activation and release of reactive oxygen species. 24 In addition, microglia can be stimulated by substances such as lipopolysaccharide (LPS) and interferon-γ (IFN-γ) to secrete cytokines such as interleukin (IL)-1, 25 IL-6, 26 and TNF-α. 27,28 Therefore, in a second series of experiments we have compared the microglial changes seen in the FMCM model with those in the same model after treatment with dexamethasone, an anti-inflammatory and immunosuppressive agent. 29 Previously, we have shown that dexamethasone can protect FMCM mice from death due to cerebral complications without affecting the growth of the parasite. 30 However, to be effective the dexamethasone must be administered 2 to 3 days before the expression of cerebral symptoms.
Using a combination of dexamethasone intervention, experimental permeabilization of the BBB, and the retinal whole mount technique we have carried out a detailed study of potential events leading to the early activation of microglia during FMCM.
Materials and Methods
Mice
CBA/T6 mice (6–8 weeks old) weighing 20–25 g were obtained from the Blackburn Animal House, University of Sydney. A minimum of 6 mice from each experimental group was sacrificed at the times indicated and their retinae were dissected as previously described, 31 then prepared for GS lectin histochemistry and nucleoside diphosphatase (NDPase) histochemistry as detailed below. For all parameters and treatments studied, two independent observers examined the retinal preparations.
FMCM Inoculation Procedure
Mice were given i.p. injections of 10 6 erythrocytes parasitized with Plasmodium berghei ANKA, suspended in 200 μl of phosphate buffered saline (PBS). Before sacrifice, blood smears were taken and the parasitemias were determined. 4,32 To ensure that the mouse/parasite combination followed the usual course of the disease, an inoculated group of mice from each series was allowed to progress through the course of infection until death, which invariably occurred between days 6 and 8 p.i.
Visualization of Microglia with GS Lectin and NDPase Histochemistry
As described previously, microglia and the retinal vasculature were visualized using the GS isolectin B4 (GS lectin), which binds to α-D-galactose. 6,10,33 Microglia also were visualized using NDPase histochemistry. 6,8
Dexamethasone Treatment
FMCM mice were given 300-μl injections of Dexadreson V (75 mg/kg; Intervet International, Boxmeer, The Netherlands) subcutaneously on day 0 and day 1 p.i., a procedure which totally protects them against cerebral complications, or on day 3 and day 4 p.i., which greatly ameliorates, but does not completely abolish, those symptoms. 30 Mice were sacrificed at day 7 p.i. (the terminal stage of the disease in untreated mice, which die with cerebral complications), day 13 p.i. (a time point between the terminal stage of the disease in untreated mice and the time of death from hemolysis in dexamethasone-treated mice), or the time of death in treated mice (day 15–22 p.i.). Retinal whole mounts were prepared from these mice. Before sacrifice, parasitemias were determined and behavioral observations were made and these were compared with the untreated FMCM group.
Intracarotid Injection of Arabinose
CBA/T6 mice were anesthetized with an i.p. injection of Avertin (12 μl/g body weight) and their necks were shaved with electric clippers. An incision was made centrally over the trachea and the carotid artery was exposed by separating the sternohyoid, sternomastoid, and omohyoid muscles. The tissue fascia in the area was removed, displaying the carotid sheath which invests the common carotid artery, internal jugular vein, vagus nerve, and ansa cervicalis. Arabinose (200 μl of 1.6 mol/L solution in isotonic saline; Sigma, St. Louis, MO) with 2% (w/v) Evans blue solution was injected slowly into the carotid, the needle was removed and a small piece of Gelfoam (Upjohn, Rydalmere, Australia) was applied to allow clotting. Polymyxin B (1 μg/ml; Calbiochem, La Jolla, CA) was included in the injection mixture to counter any possible effects of endotoxin contamination. Once the bleeding had stopped the wound was sutured.
At 12, 24, or 36 hours after co-injection of arabinose and Evans blue, 6 mice were sacrificed by CO2 asphyxiation and the contralateral and ipsilateral brain segments and retinae were examined macroscopically for leakage of Evans blue-labeled albumin. Successful intracarotid injections showed Evans blue-albumin staining of the cerebral hemisphere and retina ipsilateral to the injection site. Several controls were performed to be certain that the observations were due to osmotic barrier opening only and not to other effects, such as damage to the carotid artery or excessive blood loss. First, coronal brain sections were stained for hematoxylin and eosin. Mice showing ischemic damage (most often seen as the appearance of shrunken neurons) were eliminated from the study. Second, mice showing neurological symptoms (loss of balance or paralysis) after they regained consciousness were also excluded from the study.
Evaluation of Microglial Density
Outlines of retinae were drawn using a graticule in the 10× eyepeice (Olympus, Tokyo, Japan). The density of GS lectin-positive or NDPase-positive microglia within each 1-mm 2 region was counted, at a magnification of 40×. These values were then multiplied by a magnification correction factor (×16) to give cells/mm 2 and converted into a grading scale. A representative retina was mapped for each time point. This sampling technique provides a general estimate of changes in microglial density within the retina during the progression of the disease and illustrates whether this microglial response occurs over the whole retina or in focal regions.
Analysis of Microglial Distribution
Photomicrographs of retinae were taken at 800× total magnification using an Olympus Vanox microscope equipped with Normarski optics. Photographic montages were assembled and drawings of the retinal microglia and vasculature were made on transparencies. This technique illustrates whether there is a change in the association of microglia with each other or with vessels at a particular level of the vascular tree within the retina during the progression of the disease.
Results
Dexamethasone Treatment Ameliorates Morphological Changes in Microglia and Prevents Death Due to Cerebral Complications in FMCM
Consistent with earlier observations, FMCM mice died 7 days p.i. with P. berghei ANKA, exhibiting cerebral symptoms including hemiplegia, convulsions, and coma. 4,34 We previously have shown that morphological changes in microglia begin very early in FMCM, at days 2–3 p.i., which is 3 to 4 days before the onset of cerebral symptoms. 6 These changes include retraction of ramified processes, soma enlargement, and the adoption of an amoeboid morphology with vacuoles (arrows in Figure 1, A and B ▶ ). Such changes were at maximum frequency and severity at the terminal stage (day 7 p.i.) of FMCM (Figure 1, A ▶ -C).
Figure 1.
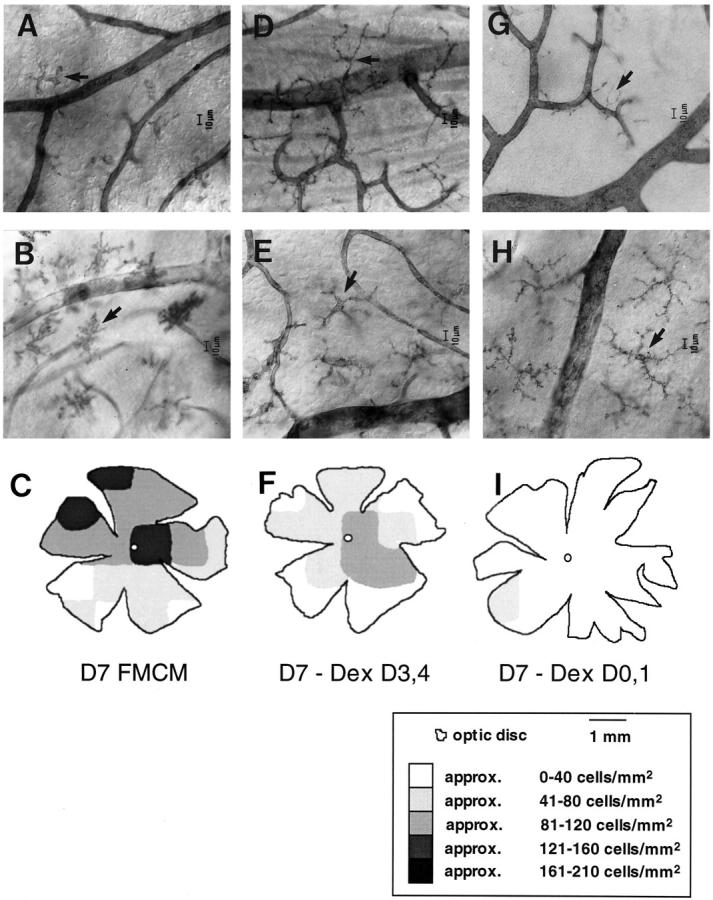
Micrographs show the amelioration of morphological changes of microglia in dexamethasone-treated FMCM mice at day 7 p.i. A−C: Microglia visualized with the GS lectin (A) and NDPase histochemistry (B), and the density of GS lectin-labeled microglia (C) at the terminal stage of FMCM. D−F: FMCM mice treated with dexamethasone on days 3 and 4 p.i. There was a marked reduction in the average density of microglia visualized using the GS lectin (compare C with F). This reduction in density was concomitant with a reduction in the extent of microglial morphological changes visualized with the GS lectin (D) and NDPase (E) histochemistry. Intensely stained GS lectin- and NDPase-labeled microglia with retracted processes and enlarged somas were still evident. However, these changes were mild compared with those in FMCM mice not treated with dexamethasone. G−I: FMCM mice treated with dexamethasone at days 0 and 1 p.i. The average density of GS lectin-labeled microglia (I) was similar to uninfected mice (21 versus 17 cells/mm2). 6 Similarly, microglia labeled with the GS lectin had a morphology typical of resting microglia (G). NDPase-labeled microglia also showed a typical morphology of resting microglia (H). However, the number of microglia visualized was greater than the number of GS lectin-labeled microglia (compare arrows in G and H), because NDPase labels the total microglial population. C, F, and I: Retinal maps showing the density of GS lectin-labeled microglia from FMCM mice treated with dexamethasone. The number of labeled microglia was determined within a 1-mm 2 region, and the cells/mm 2 were converted to a gray scale. Each map is of a single retina representative of the changes observed. Each micrograph is representative of 6 mice.
FMCM mice treated with dexamethasone on days 3 and 4 p.i. showed mild cerebral symptoms, including disturbed gait and movements on day 7 p.i., but then recovered to die of severe hemolysis between days 15–22 p.i., showing no further cerebral involvement. 30 At day 7 p.i., microglia were more intensely stained and displayed shorter and thicker processes and an enlarged cell body, compared with those in uninfected mice. These morphological changes could be found using both GS lectin histochemistry (arrow, Figure 1D ▶ ) and NDPase histochemistry (arrow, Figure 1E ▶ ). However, the changes were mild compared with those found at day 7 p.i. in FMCM mice not treated with dexamethasone (compare Figure 1A ▶ with 1D, and Figure 1B ▶ with 1E).
Mice treated with dexamethasone on days 0 and 1 p.i. displayed no cerebral symptoms at any stage, including on day 7 p.i., but died between days 15 and 22 p.i. At day 7 p.i., microglia displayed the ramified morphology typical of resting microglia. 6 This amelioration of microglial morphological changes could be found using both GS lectin histochemistry (arrow, Figure 1G ▶ ) and NDPase histochemistry (arrow, Figure 1H ▶ ). These observations are summarized in Table 1 ▶ .
Table 1.
Microglial Response in Dexamethasone-Treated Mice during FMCM
| Dexamethasone treatment (day p.i.) | Blood-brain barrier permeability | Increased process complexity* | Retraction of processes and soma enlargement | Amoeboid morphology with vacuoles |
|---|---|---|---|---|
| No treatment | +++ | +++ | +++ | +++ |
| 3 and 4 | + | ++ | + | − |
| 0 and 1 | − | + | + | − |
Mice were sacrificed at day 7 p.i., the terminal stage of the disease in untreated mice. Microglial changes are given on a subjective rating scale: −, no change; +, small degree of change found in all preparations; ++, intermediate change found in all preparations; +++, maximum change equivalent to that seen in FMCM.
*Increased process complexity refers to the appearance of small distensions or an increase in tortuosity of processes. Amoeboid microglia include the GS lectin-labeled population without processes but with a cell body >15 μm. n = 6 mice per group.
Dexamethasone Prevents Microglial Activation Indicated by Increased α-D-Galactose Expression
Focal increases in the density of GS lectin-labeled microglia occur during the progression of FMCM, probably indicating a change in the activation status of these cells. 6,11,35 In dexamethasone-treated mice there was a substantial decrease in the number of GS lectin-labeled microglia at day 7 p.i. compared with untreated FMCM mice (86 cells/mm2; compare Figure 1C ▶ with 1F and 1I). The earlier the mice were treated with dexamethasone, the fewer microglia were labeled with the lectin. In mice treated with dexamethasone on days 3 and 4 p.i. with P. berghei ANKA and sacrificed at day 7 p.i., there was an increase in density of microglia in some focal regions (48 cells/mm 2 compared with uninfected mice, 17 cells/mm2). However, the density in the dexamethasone-treated mice was much less than that found in FMCM mice not treated with this agent (compare Figure 1C ▶ with 1F). A low density of GS lectin-labeled microglia was seen in mice treated with dexamethasone on days 0 and 1 p.i. (21 cells/mm2). This extent of labeling was similar to that of uninfected mice (17 cells/mm2). 6 It was evident in mice treated with dexamethasone that there was an amelioration of morphological changes concomitant with the reduction in up-regulation of α-D-galactose residues on microglia (Figure 1) ▶ . This finding is in agreement with the suggestion that changes in microglial morphology are correlated with changes in phenotype. 36
Morphological Changes in Microglia in Response to an Experimentally Induced Increase in BRB Permeability
In agreement with previous observations, 5 opening of the blood-retinal barrier (BRB) in retinae ipsilateral to the injection site was evident by the gross leakage of Evans blue-albumin complex from the retinal vessels, resulting in poor vessel delineation and high background fluorescence in the surrounding parenchyma. In contrast, minimal staining was noted in the contralateral retina. Infusion of vehicle (isotonic saline plus polymyxin B) did not result in an increase in BRB permeability in either ipsilateral retinae or contralateral retinae. 5 Mice showing any neurological symptoms after they regained consciousness or with cerebral tissue showing histological damage were excluded from the study (see Materials and Methods for details). Microglia from mice that had suffered ischemic damage as a result of damage to the carotid artery were characterized by their lack of processes and a cell diameter >15 μm (Figure 2A) ▶ .
Figure 2.
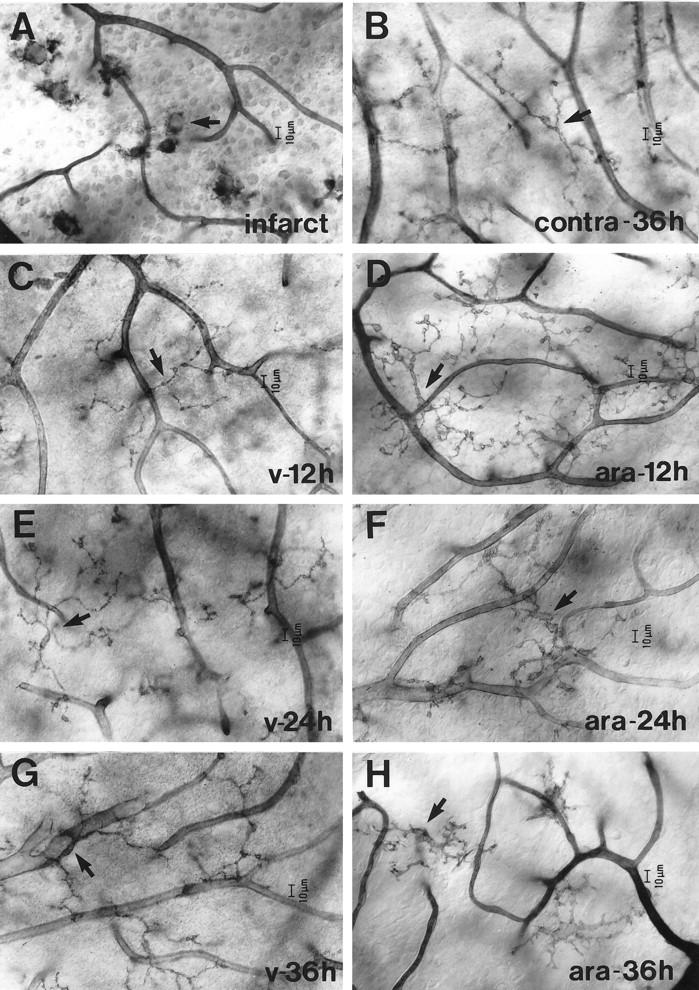
Microglial response to an infarct (A), a single injection of saline containing 1.6 mol/L arabinose, 2% (w/v) Evans blue, and 1 μg/ml polymyxin B (D, F, and H) and saline control containing 2% (w/v) Evans blue and 1 μg/ml polymyxin B (C, E, and G). Retinae were prepared for nucleoside diphosphatase (NDPase) histochemistry at 12 (C and D), 24 (E and F), or 36 (G and H) hours p.i. A: NDPase-labeled retinal microglia devoid of processes in mice with an experimentally-induced infarct (arrow). B: NDPase-labeled microglia in the contralateral retina at 36 hours post-i.c. injection of arabinose (arrow). As with contralateral retinae at 12 and 24 hours, there were no major changes in microglia morphology or distribution. C, E, and G: NDPase labeling of microglia in the retina, ipsilateral to the injection site of vehicle (arrow). No major morphological or spatial distributional changes in microglia were seen in mice injected with vehicle. D, F, and H: An equivalent region in the ipsilateral retina from mice treated with the arabinose mixture. D: At 12 hours post-i.c. injection of arabinose, microglia displayed tortuous processes with prominent distensions and a hypertrophied cell body (arrow). F and H: From 24 hours post-i.c. injection of arabinose, microglia displayed shorter processes and further enlargement of the cell body (arrow). Each micrograph is representative of 6 mice.
Microglia from the contralateral retina of mice given an intracarotid injection of arabinose displayed a ramified morphology, typical of resting microglia, 12, 24, and 36 hours (Figure 2B) ▶ later. In response to an intracarotid injection of vehicle, retinal microglia on the ipsilateral side displayed a ramified morphology, typical of resting microglia, at 12 (Figure 2C) ▶ , 24 (Figure 2E) ▶ , and 36 hours after i.c. injection (Figure 2G) ▶ , suggesting that the injection procedure itself did not cause major changes in microglial morphology.
In contrast, at 12 hours post-i.c. injection of arabinose, microglia from the ipsilateral retina displayed tortuous processes with prominent distensions and a hypertrophied cell body (Figure 2D) ▶ . From 24 hours post-i.c. injection of arabinose, numerous microglia displayed shorter processes and further enlargement of the cell body (Figure 2F) ▶ in addition to those morphological changes found at 12 hours. These morphological changes persisted until the last observed time point (36 hours post-i.c. injection of arabinose, Figure 2H ▶ ). These observations are summarized in Table 2 ▶ .
Table 2.
Microglial Response in the Ipsilateral Retina after an Increase in Blood-Brain Barrier Permeability Induced by Intracarotid Injection of Arabinose
| Time post-injection (h) | Increased process complexity* | Retraction of processes and soma enlargement | Amoeboid morphology with vacuoles |
|---|---|---|---|
| 12 | +++ | − | − |
| 24 | +++ | + | − |
| 36 | +++ | +++ | − |
| Infarct 36 | no processes | +++ | +++ |
Microglial changes are given on the scale: −, no change; +, small degree of change found in all preparations; ++, intermediate change found in all preparations; +++, maximum change equivalent to that seen in FMCM. Ipsilateral retinae showed maximal increase in BBB permeability to albumin that persisted for approximately 4 hours after intracarotid injection of arabinose.
*Increased process complexity refers to the appearance of small distensions or an increase in tortuosity of processes. Amoeboid microglia include the GS lectin-labeled population without processes but with a cell body >15 μm. n = 6 mice per group.
No Change in the Total Number of NDPase-Labeled Microglia after an Experimentally Induced Increase in BRB Permeability
Because NDPase labels the total population of retinal microglia, NDPase histochemistry was used to investigate whether an experimentally-induced increase in BRB permeability causes an increase in total numbers of microglia. There was no consistent increase in the numbers of NDPase-labeled microglia at each time point. The average density of microglia in different areas of the retina in arabinose-treated mice, 12 to 36 hours after injection, varied between 90 and 94 cells/mm 2 (Figure 3, A ▶ -C). Furthermore, there was no obvious proliferative response in microglia that were redistributed toward the retinal vessels at 12 (Figure 3D) ▶ , 24 (Figure 3E) ▶ , or 36 hours (Figure 3F) ▶ after injection of arabinose. The average density of NDPase-labeled microglia during FMCM ranged between 99 and 118 cells/mm2. 6 Microglia could not be visualized with the GS lectin in mice given an intracarotid injection of arabinose, because arabinose interferes with the binding of the lectin to the galactose residues.
Figure 3.
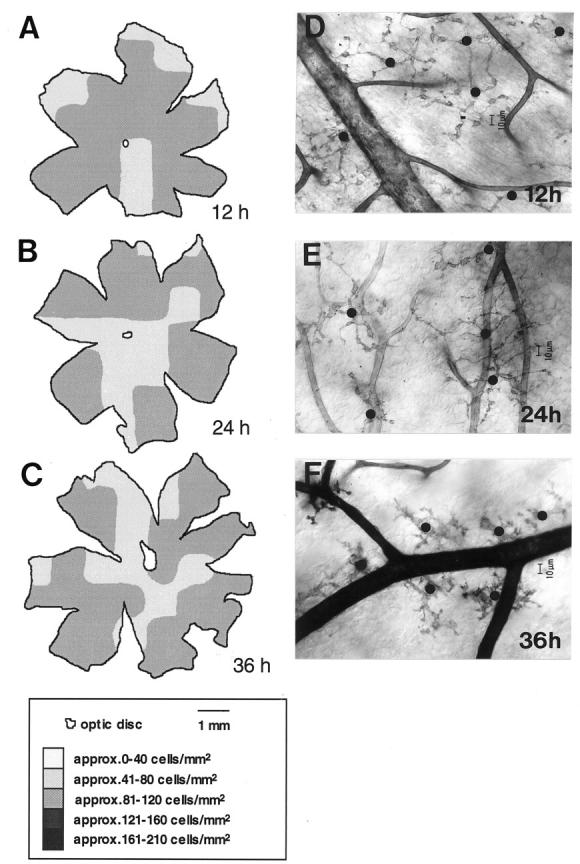
Maps showing representative densities of NDPase-labeled microglia from mice given an intracarotid injection of 1.6 mol/L arabinose + 2% (w/v) Evans blue and polymyxin B. The number of labeled microglia was determined within a 1-mm 2 region, and the cells/mm 2 were converted to a gray scale. Retinal maps show that there was a change in microglial density in response to osmotic barrier opening at 12 (A), 24 (B), and 36 (C) hours after i.c. arabinose injection. D−F: Despite the progressive morphological changes of microglia from 12 to 24 hours after arabinose administration, there was no increase in the total number of microglia visualized using NDPase histochemistry. The black dots over the cell bodies of microglia in D, E, and F illustrate that although the microglia with their long and tortuous processes cover larger areas of the retina at 12 (D) and 24 (E) hours after arabinose compared with those at 36 hours (F), the total number of microglia does not change dramatically. Each map is a single mouse retina. Each micrograph is representative of 6 mice.
Redistribution of Microglia Toward Retinal Vessels in Response to an Experimentally Induced Increase in BRB Permeability
Figure 4 ▶ shows diagrammatically the microglial distributional changes in the ipsilateral retina after an intracarotid injection of arabinose. Microglia found in the retina ipsilateral to the i.c. injection of vehicle and in control animals were seen in a regular array over the entire tissue (Figure 4A) ▶ . However, from 12 hours post-i.c. administration of arabinose, it was evident that there was a closer association of microglia with each other and with vessels at all levels of the vascular tree. Microglia were similarly distributed at 24 hours post-i.c. arabinose. In contrast, at 36 hours post-i.c. arabinose, there appeared to be many more microglia in close association with the arterial side of the vascular tree (Figure 4B) ▶ . This finding contrasts with the redistribution of microglia during FMCM, where microglia were found most closely associated with the venous side of the vascular tree. 8
Figure 4.
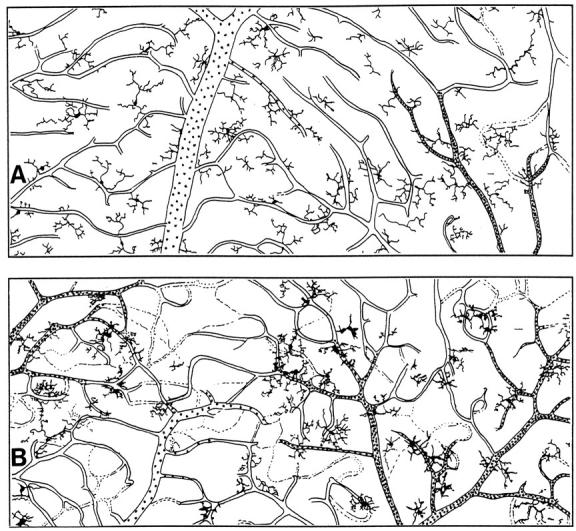
Tracings of photographic montages showing microglial distributional changes in response to intracarotid injection of 1.6 mol/L arabinose, 2% (w/v) Evans blue, and 1 μg/ml polymyxin B or vehicle alone. A: In the ipsilateral retina from mice given an intracarotid injection of vehicle, microglia were regularly distributed like those found in untreated animals. 5 B: At 36 hours after injection of arabinose there was a marked redistribution of microglia toward the arterial side of the vascular tree. The arteries are the narrower caliber vessels with frequent stippling; the veins are the wider caliber vessels with sparse stippling.
Discussion
Microglial changes are evident very early in FMCM, well before the onset of cerebral symptoms. 6,8 As early as day 2 to 3 p.i., some microglia showed an increase in GS lectin staining intensity and distensions on their processes, progressing to a decrease in process length and an increase in soma size. These less ramified microglia, with an activated morphology, 36,37 increased in numbers until the terminal stage of the disease. Similarly, redistribution of microglia toward the retinal vessels began around day 3 p.i. Because these microglial responses are among the earliest observable events in FMCM, they are unlikely to be a consequence of tissue damage, but rather a potential cause of, or contributor to, the cerebral complications. To better understand the contribution of microglia to the neuropathological process, we investigated two potential stimuli that might modulate microglial activation during the course of infection to see whether these factors alone could account for the microglial changes that are observed during FMCM.
Microglia Are Not Activated by Circulating Malaria Parasites in the Absence of an Immune Response
Observations from the present study lead us to suggest an association between the immune response to the malaria parasite, the activation status of microglia, and the fatal neurological complications of FMCM. From Figure 1 ▶ it is evident that the earlier the infected mice were treated with dexamethasone, the less activated the microglia became, and this coincided with less severe neurological symptoms. Microglial activation and neurological symptoms were totally abrogated in mice treated with dexamethasone at the time of parasite inoculation. Neither treatment of FMCM mice with dexamethasone at days 0 and 1 p.i., nor treatment at days 3 and 4 p.i., affected the growth of the parasite. These results suggest that parasite antigens and soluble factors released into the circulation after parasite growth, in the absence of an immune response, are insufficient to active microglia.
Site of Dexamethasone Action
Because the dexamethasone-sensitive event(s) occur early in the pathogenic process, it has been suggested that the drug acts at the stage of T cell activation. 30 Given the key role played by microglia in the pathogenesis of FMCM, 6 (and these results), an alternative mechanism is that dexamethasone ameliorates cerebral complications by preventing the early involvement of microglia. Dexamethasone has been shown to inhibit TNF-α production by LPS-stimulated murine and human microglia in vitro by reducing translation of this cytokine, 38,39 TNF-α plays an essential role in the pathogenesis of FMCM, 40 and microglia produce this cytokine during the course of the disease. 8 Castano and colleagues, 41 however, found that proliferation and activation of microglia during Wallerian degeneration were not affected by dexamethasone treatment. From these observations they hypothesized that microglia do not respond to glucocorticoids in vivo. Thus, a direct effect of dexamethasone on microglia in vivo in FMCM remains to be unequivocally established.
Microglia Display Shorter and Thicker Processes and Redistribute toward the Vasculature in Response to Plasma Constituents
As suggested by Perry and colleagues, 42,43 activation of microglia may be a consequence of increased permeability of the CNS barrier to macromolecules. In regions of the CNS where the BBB is absent, or after a breakdown in the BBB, microglia express molecules that are normally either absent or present at a low level. 20 For example, the expression of CD4 and sialoadhesin, a macrophage receptor for sialic acid, on rat microglia is in part regulated by exposure to constituents of the plasma. 20
Previous studies 15,16 have shown a general increase in permeability of the BRB in FMCM as early as day 2 to 3 p.i. This coincides with the observed microglial responses, so it is possible that movement of plasma molecules across the CNS barrier initiates the morphological changes and accumulation of microglia toward the retinal vessels. This hypothesis is supported by the coincidence between the decrease in BRB permeability and the decrease in GS lectin labeling and microglial morphological changes in malaria-infected, dexamethasone-treated animals compared with their counterparts not treated with the drug (Figure 1) ▶ . Microglia from mice treated with dexamethasone after the initial increase in BBB/BRB permeability (days 2–3 p.i.) displayed both an up-regulation of α-D-galactose residues on their surface and changes in morphology compared with their counterparts in uninfected mice. However, these changes were mild compared with FMCM mice not treated with dexamethasone and, likewise, the neurological symptoms were less severe. Furthermore, dexamethasone treatment (days 0 and 1 p.i.) before the permeability changes completely ameliorated the increase in BBB permeability, up-regulation of α-D-galactose residues and microglial morphological changes, and the development of any cerebral symptoms.
The role of plasma proteins in the initiation of the microglial response is further supported by the changes induced by an intracarotid injection of arabinose (Figures 2–4) ▶ ▶ ▶ , because an experimentally induced increase in BBB/BRB permeability resulted in changes in morphology and distribution of microglia. It should be noted, however, that there was a major difference between microglial changes that resulted from intracarotid injection of arabinose and those observed during FMCM. In the former case, there was a preferential redistribution of microglia toward the arterial side of the vascular tree (Figure 4B) ▶ , whereas microglia from FMCM mice showed preferential redistribution toward the venous side. 6 This contrast is most likely due to the differences in the agent that modifies permeability in these different situations. Because the arabinose solution is injected into the carotid artery, the arabinose passes through the arterial side of the vascular tree before the venous side. Therefore, the arterial vasculature will be subjected to a higher concentration of arabinose. As a result, microglia may redistribute toward the area with the highest extravascular concentration of arabinose or plasma constituents. In contrast, inflammatory agents that cause an increase in vascular permeability are more likely to affect the venous side of the vascular tree. Indeed, observations in retinal whole mounts have shown a more pronounced leakage of plasma proteins into the parenchyma on the venous side of the vascular tree in FMCM 15 and experimental allergic encephalomyelitis. 44 Furthermore, there is a loss of astrocyte ensheathment of vessel segments, predominantly on the venous side of the circulation, at the terminal stage of FMCM. 5 This loss of astrocytes is not observed following an experimentally induced increase in BBB permeability. 5 During FMCM, microglia may be recruited to the site of astrocyte damage to phagocytose astrocytic debris and foreign protein that has entered through the damaged BBB. A phagocytic role for microglia is consistent with the adoption of the amoeboid/vacuolated morphology that is routinely observed during FMCM.
In summary, we have investigated the response of microglia to circulating malaria parasites in the absence of an immune response and to disruption of the CNS microenvironment with plasma constituents. These are the stimuli most likely to modulate microglial activity during FMCM. Changes in microglial morphology, such as thickening and distension of microglial processes and redistribution of microglia toward the vascular endothelium, can be initiated by an increase in permeability of the CNS barrier to macromolecules. However, an increase in BBB permeability is not sufficient to induce reactive microglia with an amoeboid and/or vacuolated microglia like those seen during FMCM. In contrast, dexamethasone-sensitive event(s), probably related to immune activation, occurring within the first few days of infection are necessary for the development of reactive microglial changes and subsequent fatal neurological complications.
From the current study we conclude that a likely course of events in FMCM is as follows: Malaria parasites produce a vasoactive factor that leads to increased BBB permeability. This in turn results in the movement of plasma constituents into the CNS, which initiates the redistribution of microglia and astrocytes 5 toward blood vessels. Modest morphological changes of microglia, denoting a departure from their resting state, would result from exposure to, and phagocytosis of, extravasated plasma constituents and/or cell debris. These mild microglial changes are necessary, but not sufficient, for the development of cerebral pathology. Rather, concurrent dexamethasone-sensitive immunopathological events are the most crucial, because once they are initiated the disease becomes irreversible and invariably fatal. Further reactive microglial changes, unique to animals that go on to develop irreversible cerebral complications, might be caused by malarial exoantigens entering the CNS through the compromised BBB. These reactive changes also could be induced, or reinforced, by cytokines derived from circulating leukocytes, or monocytes adhering to the cerebral microvascular endothelium. The secretion of various neuroactive and neurotoxic factors by peripheral or local CNS immunocompetent cells, such as monocytes and microglia, respectively, may interfere with CNS functions, contributing to coma and death. Tumor necrosis factor 8,40 and metabolites of the kynurenine pathway of tryptophan metabolism 45 may be of particular importance in causing derangement of CNS functions.
Acknowledgments
We thank Clive Jeffery for photographic assistance and Dr. Weiyun Yu for assistance with development of the intracarotid injection procedure.
Footnotes
Address reprint requests to Professor Nicholas Hunt, Department of Pathology (D06), University of Sydney, Sydney, N.S.W. 2006, Australia. E-mail: nhunt@pathology.usyd.edu.au.
Supported by grants to N. H. and T. C.-L. from the National Health and Medical Research Council and Sydney University Research Grants Scheme. I. M. was supported by an Australian Postgraduate Award.
References
- 1.Finley RW, Mackey LJ, Lambert PH: Virulent P. berghei malaria: prolonged survival and decreased cerebral pathology in T cell deficient nude mice. J Immunol 1982, 129:2213-2218 [PubMed] [Google Scholar]
- 2.Finley R, Weintraub J, Lewis JA, Engers HD, Zubler R, Lambert P-H: Prevention of cerebral malaria by adoptive transfer of malaria specific cultured T cells into mice infected with Plasmodium berghei. J Immunol 1983, 131:1522-1526 [PubMed] [Google Scholar]
- 3.Grau GE, Fajardo LF, Piguet P-F, Allet B, Lambert P-H, Vassalli P: Tumor necrosis factor (cachectin) as an essential mediator in murine cerebral malaria. Science 1987, 237:1210-1212 [DOI] [PubMed] [Google Scholar]
- 4.Neill AL, Hunt NH: Pathology of fatal and resolving cerebral malaria. Parasitology 1992, 105:165-175 [DOI] [PubMed] [Google Scholar]
- 5.Medana IM, Chan-Ling T, Hunt NH: Redistribution and degeneration of retinal astrocytes in experimental murine cerebral malaria: relationship to disruption of the blood-retinal barrier. Glia 1996, 16:51-64 [DOI] [PubMed] [Google Scholar]
- 6.Medana IM, Hunt NH, Chan-Ling T: Early activation of microglia during fatal murine cerebral malaria. Glia 1997, 19:91-103 [DOI] [PubMed] [Google Scholar]
- 7.Ma N, Harding AJ, Pamphlett R, Chaudhri G, Hunt NH: Increased c-fos expression in the brain during experimental murine cerebral malaria: possible association with neurological complications. J Infect Dis 1997, 175:1480-1489 [DOI] [PubMed] [Google Scholar]
- 8.Medana IM, Hunt NH, Chaudhri G: Tumor necrosis factor-α expression in the brain during fatal murine cerebral malaria: evidence for production by microglia and astrocytes. Am J Pathol 1997, 150:1473-1486 [PMC free article] [PubMed] [Google Scholar]
- 9.Htain W-W, Leong S-K, Ling E-A: A comparative Mac-1 immunocytochemical and lectin histochemical study of microglial cells in the normal and athymic mice. Glia 1994, 12:44-51 [DOI] [PubMed] [Google Scholar]
- 10.Streit WJ, Kreutzberg GW: Lectin binding by resting and reactive microglia. J Neurocytol 1987, 16:249-260 [DOI] [PubMed] [Google Scholar]
- 11.Maddox DE, Shibata S, Goldstein IJ: Stimulated macrophages express a new glycoprotein receptor reactive with Griffonia simplicifolia I-B4 isolectin. Proc Natl Acad Sci USA 1982, 79:166-170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Medana IM: The involvement of astrocytes and microglia in the immunopathology of fatal murine cerebral malaria. PhD Thesis, University of Sydney, Australia, 1996, 192 pp
- 13.Janota I, Doshi B: Cerebral malaria in the United Kingdom. J Clin Pathol 1979, 32:769-772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brown HC, Hien TT, Day NPJ, Mai NTH, Chuong LV, Chau TTH, Loc PP, Phu NH, Bethell D, Farra J, Gatter KC, White NJ, Turner GDH: Evidence of blood brain barrier dysfunction in human cerebral malaria. Neuropathol App Neurobiol 1999, 25:331-340 [DOI] [PubMed] [Google Scholar]
- 15.Chan-Ling T, Neill AL, Hunt NH: Early microvascular changes in murine cerebral malaria detected in retinal whole mounts. Am J Pathol 1992, 140:1121-1130 [PMC free article] [PubMed] [Google Scholar]
- 16.Neill AL, Chan-Ling T, Hunt NH: Comparisons between microvascular changes in cerebral and non-cerebral malaria mice, using the retinal whole mount technique. Parasitology 1993, 107:477-487 [DOI] [PubMed] [Google Scholar]
- 17.Fredericks WR, Rapoport SI: Reversible osmotic opening of the blood-brain barrier in mice. Stroke 1987, 19:266-268 [DOI] [PubMed] [Google Scholar]
- 18.Rapoport SI, Fredericks WR, Ohno K, Pettigrew KD: Quantitative aspects of reversible osmotic opening of the blood-brain barrier. Am J Physiol 1980, 238:R421-R431 [DOI] [PubMed] [Google Scholar]
- 19.Tibbling G, Link H, Ohman S: Principles of albumin and IgG analyses in neurological disorders. I. Establishment of reference values. Scand J Clin Lab Invest 1977, 37:385-390 [DOI] [PubMed] [Google Scholar]
- 20.Perry VH, Crocker PR, Gordon S: The blood-brain barrier regulates the expression of a macrophage sialic acid-binding recpetor on microglia. J Cell Sci 1992, 101:201-207 [DOI] [PubMed] [Google Scholar]
- 21.Mucke L, Eddleston M: Astrocytes in infectious and immune-mediated diseases of the central nervous system. FASEB J 1993, 7:1226-1232 [DOI] [PubMed] [Google Scholar]
- 22.Richmon JD, Fukuda K, Sharp FR, Noble LJ: Induction of HSP-70 after hyperosmotic opening of the blood-brain barrier in the rat. Neurosci Lett 1995, 202:1-4 [DOI] [PubMed] [Google Scholar]
- 23.Richmon JD, Fukuda K, Mauda N, Sato M, Bergeron M, Sharp FR, Panter SS, Noble LJ: Induction of hemoxygenase-1 after hyperosmotic opening of the blood-brain barrier. Brain Res 1988, 780:108-118 [DOI] [PubMed] [Google Scholar]
- 24.Colton CA, Gilbert DL: Production of superoxide anions by a CNS macrophage, the microglia. FEBS Lett 1987, 223:284-288 [DOI] [PubMed] [Google Scholar]
- 25.Giulian D, Baker TJ, Shih L-C, Lachman LB: Interleukin-1 of the central nervous system is produced by amoeboid microglia. J Exp Med 1986, 164:594-604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Frei K, Malipiero UV, Leist TP, Zinkernagel M, Schwab ME, Fontana A: On the cellular source and function of interleukin 6 produced in the central nervous system in viral diseases. Eur J Immunol 1989, 19:689-694 [DOI] [PubMed] [Google Scholar]
- 27.Lavi E, Suzumura A, Murasko DM, Murray EM, Silberberg DH, Weiss SR: Tumor necrosis factor induces expression of MHC class I antigens on mouse astrocytes. J Neuroimmunol 1988, 18:245-253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sawada M, Kondo N, Suzumura A, Marunouchi T: Production of tumor necrosis factor-α by microglia and astrocytes in culture. Brain Res 1989, 491:394-397 [DOI] [PubMed] [Google Scholar]
- 29.Issekutz TB: Effects of anti-inflammatory agents on lymphocyte migration stimulated by the interferons, tumor necrosis factor and cutaneous inflammation. Int J Immunopharmacol 1989, 11:725-732 [DOI] [PubMed] [Google Scholar]
- 30.Neill AL, Hunt NH: Effects of endotoxin and dexamethasone on cerebral malaria in mice. Parasitology 1995, 111:443-454 [DOI] [PubMed] [Google Scholar]
- 31.Chan-Ling T: Glial, neuronal and vascular cytogenesis in wholemounted cat retina. Microscopy Res Tech 1997, 36:1-16 [DOI] [PubMed] [Google Scholar]
- 32.Rest JR: Cerebral malaria in inbred mice. I. A new model and its pathology. Trans Roy Soc Trop Med Hyg 1982, 76:410-415 [DOI] [PubMed] [Google Scholar]
- 33.Chan-Ling T, Halasz P, Stone J: Development of retinal vasculature in the cat: processes and mechanisms. J Comp Neurol 1990, 9:459-478 [DOI] [PubMed] [Google Scholar]
- 34.Thumwood CM, Hunt NH, Clark IA, Cowden WB: Breakdown of the blood-brain barrier in murine cerebral malaria. Parasitology 1988, 96:579-589 [DOI] [PubMed] [Google Scholar]
- 35.Warfel AH, Zucker-Franklin D: Specific ligation of surface α-D-galactosyl epitopes markedly affects the quantity of four major proteins secreted by macrophages. J Leukocyte Biol 1992, 52:80-83 [DOI] [PubMed] [Google Scholar]
- 36.Thomas WE: Brain macrophages: evaluation of microglia and their functions. Brain Res Rev 1992, 17:61-74 [DOI] [PubMed] [Google Scholar]
- 37.Vrabec F: Activated human microglia under pathological conditions. Archiv Klin Experiment Ophthal 1975, 196:49-60 [DOI] [PubMed] [Google Scholar]
- 38.Chao CC, Hu S, Close K, Choi CS, Molitor TW, Novick WJ, Peterson PK: Cytokine release from microglia: differential inhibition by pentoxifylline and dexamethasone. J Infect Dis 1992, 166:847-853 [DOI] [PubMed] [Google Scholar]
- 39.Peterson PK, Hu S, Sheng WS, Kravitz FH, Molitor TW, Chatterjee D, Chao CC: Thalidomide inhibits tumor necrosis factor-alpha production by lipopolysaccharide- and lipoarabinomannan-stimulated human microglial cells. J Infect Dis 1995, 172:1137-1140 [DOI] [PubMed] [Google Scholar]
- 40.Lucas R, Lou JN, Juillard P, Moore M, Bluethmann H, Grau GE: Respective roles of TNF receptors in the development of experimental cerebral malaria. J Neuroimmunol 1997, 72:143-148 [DOI] [PubMed] [Google Scholar]
- 41.Castano A, Lawson LJ, Fearn LJ, Perry VH: Activation and proliferation of microglia are insensitive to glucocorticoids in Wallerian degeneration. Eur J Neurosci 1996, 8:581-588 [DOI] [PubMed] [Google Scholar]
- 42.Perry VH, Gordon S: Macrophages and microglia in the nervous system. Trends Neurosci 1988, 11:273-277 [DOI] [PubMed] [Google Scholar]
- 43.Perry VH, Gordon S: Macrophages and the nervous system. Int Rev Cytol 1991, 125:203-244 [DOI] [PubMed] [Google Scholar]
- 44.Hu P, Pollard J, Hunt N, Chan-Ling T: Microvascular and cellular responses in the retina of rats with acute experimental allergic encephalomyelitis (EAE). Brain Pathol 1998, 8:487-498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sanni LA, Tattam B, Thomas SR, Moore D, Chaudhri G, Stocker R, Hunt NH: Dramatic changes in oxidative tryptophan metabolism along the kynurenine pathway in cerebral and non-cerebral malaria. Am J Pathol 1998 152:611–619 [PMC free article] [PubMed]