

Abstract
Hurler disease resulting from a deficiency in α-l-iduronidase, which causes an accumulation of dermatan sulfate and heparan sulfate glycosaminoglycans, is characterized by connective tissue and skeletal deformations, cardiomyopathy, cardiac valve defects, and progressive coronary artery stenosis. In this report, we present evidence that accumulation of dermatan sulfate but not heparan sulfate moieties is linked to impaired elastic fiber assembly that, in turn, contributes substantially to the development of the clinical phenotype in Hurler disease. Our data suggest that dermatan sulfate-bearing moieties bind to and cause functional inactivation of the 67-kd elastin-binding protein, a molecular chaperone for tropoelastin, which normally facilitates its secretion and assembly into elastic fibers. We demonstrate that, in contrast to normal skin fibroblasts and cells from Sanfilippo disease, which accumulate heparan sulfate, Hurler fibroblasts show reduced expression of elastin-binding protein and do not assemble elastic fibers, despite an adequate synthesis of tropoelastin and sufficient production of a microfibrillar scaffold of elastic fibers. Because cultured Hurler fibroblasts proliferate more quickly than their normal counterparts and the addition of exogenous insoluble elastin reduces their proliferation, we suggest that cell contacts with insoluble elastin play an important role in controlling their proliferation.
Hurler disease belongs to a group of inherited metabolic storage diseases and is caused by a primary deficiency in lysosomal α-l-iduronidase, 1-10 which causes an accumulation of dermatan sulfate (DS) and heparan sulfate (HS) glycosaminoglycans. 11,12 It has also been established that Hurler disease patients demonstrate variable levels of deficiency in β-galactosidase. 13,14 In addition to dwarfism and mental retardation, patients with Hurler disease are characterized by skeletal malformations, hernias, and multiorgan lesions of connective tissue. 15-20 Most patients with Hurler disease demonstrate symptoms of systemic hypertension, cardiac valvular insufficiency, cardiomyopathy, and a striking coronary artery stenosis that causes death before the end of the second decade. 21-30 Angiographic studies, as well as quantitative evaluation at necropsy, indicate that coronary artery lesions in children with Hurler disease are usually diffuse, develop in all four coronary arteries, and cause severe (75–100%) luminal narrowings. 27,31-33 In addition to the presence of vacuolated Hurler cells, the affected tissues, including coronary arteries, are characterized by an intracellular and pericellular accumulation of DS- and HS-containing glycosaminoglycans and glycolipids, 22,30 as well as high deposition of collagen and poorly developed elastic fibers. The basis for impaired elastic fiber deposition in Hurler disease has not been previously addressed, nor even recognized as a part of the pathogenic mechanism responsible for the clinical features of this disease.
The mature elastic fibers and laminae present in connective tissues and blood vessel walls are complex structures made of polymeric (insoluble) elastin in which polypeptide chains of tropoelastin are covalently cross-linked and placed on a scaffold of 12-nm microfibrils that consists of several glycoproteins, eg, fibrillins and microfibril-associated glycoproteins (MAGPs). 34-38 Tropoelastin is a soluble 70-kd precursor of extracellular elastin, 39,40 synthesized by such cells as fibroblasts, chondrocytes, and smooth muscle cells (SMCs), which has to be secreted and properly positioned on the microfibrillar scaffold 41-44 before being cross-linked by lysyl oxidase. 45 Several other molecules are also colocalized with elastic fibers. 46-49
Our group has previously shown that the early stages of elastogenesis are controlled by the 67-kd elastin-binding protein (EBP) that is identical to the enzymatically inactive spliced variant of β-galactosidase, 50,51 which also has a galactolectin domain that can bind free galactosugars and galactosugar-bearing moieties. 52-54 The EBP acts as a recycling molecular chaperone that protects the highly hydrophobic tropoelastin molecules from intracellular self-aggregation and premature degradation. 55,56 The EBP also facilitates the orderly assembly of tropoelastin on the microfibrillar scaffold of growing elastic fibers. 57 We have established that the orderly release of tropoelastin from its transportation complex with the EBP occurs at the cell surface, just after the galactolectin domain of the EBP binds to the highly glycosylated microfibrillar scaffold of elastic fibers. 56,57 The binding of galactosamine-containing residues, protruding from polyglycosylated fibrillin molecules and/or from fibrillin-associated chondroitin sulfate proteoglycans 58 to the galactolectin domain of EBP, causes such a conformational change of this protein that it dissociates from tropoelastin. The released tropoelastin molecules then anchor to the MAGPs, either via a positively charged domain located at its C-terminal or via a region encoded by exon 30. 59 We have established that such a coordinated elastogenesis can be disrupted by a pericellular accumulation of galactosugar-bearing moieties, such as chondroitin sulfate or DS, which induces premature shedding of the EBP from the cell surface and the release of tropoelastin far away from microfibrilar acceptors. 49,54 In contrast, the glucosugar-bearing glycosaminoglycan, HS, does not bind to the EBP nor disrupt elastogenesis in cultures of arterial SMCs.
Our in vitro studies aimed at elucidation of the pathomechanism of impaired elastogenesis in Hurler disease were encouraged by analysis of autopsy material that demonstrated a striking lack of elastic fibers in cardiac valve tissue and in the neointimal coronary lesions of Hurler patients (see Figure 1 ▶ ). In the current study, we tested the hypothesis that an excessive intracellular and pericellular accumulation of DS, but not HS, induces shedding of the EBP and subsequent impaired elastogenesis in Hurler disease. Immunohistochemical and biochemical analyses of cultured human skin fibroblasts indicate that cells derived from children with Hurler disease (demonstrating excessive urinary excretion of DS and HS) lose their newly synthesized EBP. In contrast to fibroblasts from healthy individuals and from patients with Sanfilippo disease, which accumulate HS only, 60 cultured Hurler fibroblasts do not assemble elastic fibers despite an adequate synthesis of tropoelastin and a sufficient production of the components of the microfibrillar scaffold (MAGPs and fibrillin). At the same time, cultured Hurler fibroblasts synthesize more fibronectin and demonstrate a higher proliferation rate than normal and Sanfilippo fibroblasts. Therefore, we suggest that these features are linked to the impaired elastogenesis in the Hurler phenotype.
Figure 1.
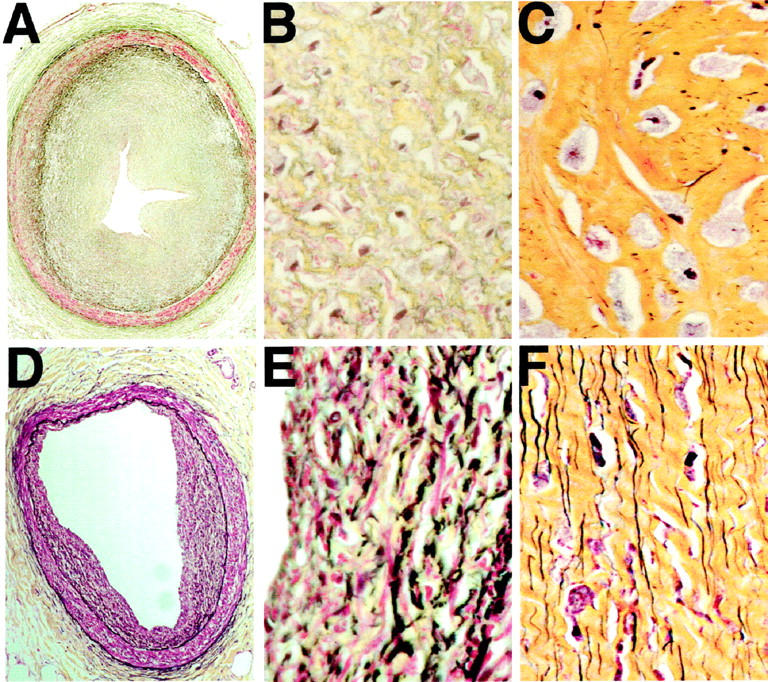
A: Micrograph of extramural coronary artery from a 9-year-old child with Hurler disease, showing intimal thickening with 90% narrowing (original magnification, ×20). B: The high-power image (×400) of the same neointima, demonstrating extensive glycosaminoglycan deposition (green) and only minimal amounts of elastin (black). Note that the dark blue staining objects are nuclei of Hurler cells, not elastin fibrils. C: Micrograph of mitral valve chordal tissue from a 9-year-old child with Hurler disease, showing that the extracellular matrix contains abundant collagen (yellow) but only minimal amounts of elastin (black); original magnification, ×400. D: Micrograph of the extramural coronary artery from a 10-year-old patient with Sanfilippo disease showing focal intimal thickening (original magnification, ×20). E: The high-power image (×400) of the same artery indicates that, in marked contrast to the Hurler disease lesion (B), neointimal cells in the Sanfilippo patient deposited matrix material containing abundant elastic fibers (black). F: Micrograph of the mitral-valve chordal tissue from the Sanfilippo patient, showing abundant deposition of numerous and well-formed elastin fibers (black). All histological sections were stained with Movat’s pentachrome, which shows elastin as black, glycosaminoglycans as green, collagen as yellow, smooth muscle as red, and nuclei as dark blue.
Although we were not able to obtain and test living SMCs derived from Hurler disease coronaries, we believe that our results with available Hurler disease fibroblasts, indicating links between DS deposition, impaired elastogenesis, and increased cellular proliferation, are relevant and contribute to an understanding of the pathomechanism responsible for the development of the peculiar occlusive lesions in the coronary arteries of Hurler disease patients.
Materials and Methods
Materials
All chemical grade reagents were from Sigma Chemical Co. (St. Louis, MO). α-Minimum essential medium, fetal bovine serum (FBS), and other cell culture products were obtained from GIBCO Life Technologies (Burlington, ON, Canada). 4B-sepharose was from Pharmacia (Uppsala, Sweden). Powdered bovine ligamentum nuchae insoluble elastin and the polyclonal antibody to tropoelastin were purchased from Elastin Products Co., Inc. (Owensville, MI). The anti-S-Gal polyclonal antibody raised to the elastin/laminin-binding domain of the alternatively spliced variant of β-galactosidase 50 and the bovine ciliary zone (BCZ) monoclonal antibody 61 were used to detect the 67-kd EBP. Microfibrillar proteins were detected with a polyclonal antibody to MAGP and to human fibrillin 1 from Elastin Products Co., Inc. Monoclonal antibody to fibronectin (mAB1940) was obtained from Chemicon (Temecula, CA), and polyclonal anti-fibronectin antibody was obtained from ICN (Costa Mesa, CA). Polyclonal antibody to human collagen type I was a generous gift of L. W. Fischer (The National Institutes of Health, Bethesda, MD). Secondary antibodies, including fluorescein-conjugated goat anti-rabbit and goat anti-mouse antibodies, were purchased from Sigma. The horseradish peroxidase-conjugated goat anti-rabbit antibody used for Western blotting was from Biorad (Hercules, CA). The chemiluminescence detection kit and radiolabeled reagents, 3H-valine, 3H-serine, 35S-methionine, and 3H-thymidine, were purchased from Amersham Canada Ltd. (Oakville, ON, Canada).
Histopathology
To justify the clinical relevance of our in vitro studies aimed at the pathomechanism of impaired elastogenesis, we examined the distribution of extracellular-matrix components in 4-μm-thick histological sections of extramural coronary arteries and the mitral valve chordae tendineae obtained at the autopsies of a 9-year-old male with Hurler disease, a 10-year-old female with Sanfilippo disease, and a 15-year-old normal male who died suddenly but did not suffer from any systemic disease. All histological sections were stained with Movat’s pentachrome, 62 which shows elastin as black, glycosaminoglycans as green, collagen as yellow, smooth muscle as red, and nuclei as dark blue. Previous studies have confirmed that the distribution of black-stained material with Movat’s method entirely overlaps with immunodetectable elastin. 63,64
Fibroblast Cultures
Skin biopsies of three Hurler disease patients diagnosed at The Hospital for Sick Children in Toronto included an 18-week-old fetus (case 7131), a 9-month-old female (case 8180), a 21-month-old male (case 8339); three patients diagnosed with the Sanfilippo disease included a 21-month-old female (case 7995), a 9-month-old male (case 8849), and a 9-month-old male (case 7825); skin biopsies from three normal children of 4, 7, and 36 months of age (cases 3858, 4212, and 4184, respectively) were used as the source of tested fibroblasts. The rationale for using cells from Sanfilippo disease was that their storage of glycosaminoglycans is limited to HS-bearing moieties and does not include DS. 2-4,60,65 All fibroblasts were originally isolated by collagenase digestion of the biopsies and then were passaged by trypsinization and maintained in α-minimum essential medium supplemented with 20 mmol/L 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, 1% antibiotics/antimycotics, 10% FBS, and 1% l-glutamate. Expressions of the EBP, as well as the deposition of extracellular elastin, collagen type I, fibronectin, fibrillin I, and MAGP, were then compared by immunohistochemistry at passages 2–5 of cultured fibroblasts. Fibroblasts derived from the skin of normal children and from Sanfilippo patients were also cultured in the presence or absence of 400 μg/ml of exogenous DS or HS. Cell proliferation was then assessed by the incorporation of 3H-thymidine, and the production of their insoluble elastin and fibronectin was assessed biochemically and by immunohistochemistry.
In a separate series of experiments, we tested whether partial digestion of the cell surface-associated DS moieties by β-galactosidase or chondroitinase ABC would restore normal deposition of elastic fibers by the Hurler disease fibroblasts. The confluent cultures of Hurler fibroblasts were, therefore, maintained in a normal medium in the presence or absence of 0.2 U/day of chondroitinase ABC for 3 and 10 days or plated on glass coverslips and placed (face up) on the top of subconfluent cultures of β-galactosidase-complementary DNA-transfected Chinese hamster ovary (CHO) cells secreting the active enzyme into the conditioned medium. 68 The parallel cocultures of Hurler fibroblasts with a comparable number of nontransfected CHO cells were also used for comparison. Both types of cocultures were terminated after 3 and 10 days, and the production of elastic fibers was assessed by immunohistochemistry and by insoluble elastin assay as described below.
Immunostaining
Subconfluent 48-hour-old cultures of normal, Hurler, and Sanfilippo fibroblasts demonstrating nonoverlapping cellular edges, as well as 10-day-old dense cultures that produce abundant extracellular matrix, were used. Forty eight-hour cultures fixed in cold 100% methanol at −20°C for 30 minutes were incubated with monospecific anti-S-Gal antibody, 50 which recognizes the EBP (2 mg/ml diluted 1:100); with BCZ antibody (5 mg/ml, diluted 1:200), which recognizes a different epitope on the EBP 61 ; or with a monoclonal antibody recognizing DS. 69 For comparison, the parallel cultures of normal fibroblasts exposed to 1 mg/ml of DS were also analyzed.
Ten-day-old confluent cultures of the normal, Hurler, and Sanfilippo fibroblasts were fixed in cold 100% methanol and then incubated with polyclonal antibody to tropoelastin (2 mg/ml diluted 1:100), 66 with monoclonal antibody to fibronectin (1 mg/ml diluted 1:1000) and with polyclonal antibody to collagen type I (1 mg/ml diluted 1:1000) as previously described. 50,51 The parallel cultures scheduled for immunohistochemical assessement of microfibrillar components were fixed in 0.5% paraformaldehyde at room temperature for 15 minutes, blocked in phosphate-buffered saline containing 0.1 mmol/L ammonium chloride, then washed in phosphate-buffered saline and treated with specific polyclonal antibody to fibrillin (2 mg/ml diluted 1:100), or additionally pretreated for 10 minutes with 3 mol/L guanidine HCl containing 50 mmol/L dithiothreitol, alkylated with 100 mmol/L iodoacetamide for 15 minutes, washed in phosphate-buffered saline, and then immunostained with the specific polyclonal antibody to microfibril-associated glycoprotein at the same concentration. 67 All cultures were incubated with the appropriate fluorescein-conjungated secondary antibodies (fluorescein-conjugated goat anti-rabbit or goat anti-mouse antibodies) for an additional hour. Nuclei were counterstained with propidium iodide. Morphometric analysis of 10-day-old cultures immunostained with antibodies recognizing extracellular-matrix components was performed using an Olympus AH-3 microscope attached to a CCD camera (Optronix) and a computer-generated video analysis system (Image-Pro Plus software, Media Cybernetics, Silver Spring, MD). In each analyzed group, 50 low-power fields (×20) from three separate cultures (derived from different patients) were analyzed, and the areas occupied by the particular immunodetectable components were quantified. The abundance of each immunodetectable component was then expressed as a percentage of the entire analyzed field.
Tropoelastin and Insoluble Elastin Assays
Hurler, Sanfilippo, and control fibroblasts were grown to confluency in 10-cm cell culture dishes in quadruplicates. 20 μCi of 3H-valine were added to each dish along with fresh media. Cultures were then incubated for 72 hours, and soluble and insoluble elastin were assessed separately in each culture. First, media were collected and immunoprecipitated with a polyclonal antibody to tropoelastin, and then the soluble proteins present in the intracellular compartments were extracted by an overnight incubation with 0.1 mol/L acetic acid, and the intracellular tropoelastin was immunoprecipitated from such extracts as previously described, 55 and then quantitatively assessed after scintillation counting. The remaining cultures, containing cell remnants and deposited insoluble extracellular matrix, were then scraped in 0.1 N NaOH, sedimented by centrifugation, and boiled in 0.5 ml of 0.1 N NaOH for 45 minutes to solubilize all matrix components except elastin. The resulting pellets containing the insoluble elastin were then solubilized by boiling in 200 μl of 5.7 N HCl for 1 hour, and the aliquots were mixed with scintillation fluid and counted. 70
Isolation of EBP
To compare patterns of EBP expression by the control and the Hurler fibroblasts, we carried out pulse-chase experiments. Fibroblasts at passage 4 were initially plated at 1 × 106cells/dish to form a subconfluent culture and then incubated in triplicate in serum-free Medium 199 for 6 hours and pulsed with 15 μCi/ml 14C-serine in serine-free medium for 1 hour. The cultures were then rinsed well and chased in fresh Medium 199 for 5, 15, 30, and 45 minutes. At the end of each chase period, the cell layers and the media were processed separately. To isolate the EBP from the cell layers of Hurler and control fibroblasts, the standard elastin-affinity chromatography technique was used. Fibroblasts were scraped from each culture dish, suspended in 1 ml of 0.1 mol/L bicarbonate buffer, pH 8, and extracted with 0.1 mol/L lactose, 0.1 mol/L dithiothreitol, and 0.25% octyl-β-glucoside in the presence of proteinase inhibitors in the following final concentrations: 2 mmol/L benzamidine, 2 mmol/L EACA, 2 mmol/L phenylmethylsulfonyl fluoride,1 mmol/L ethylenedinitrilo tetraacetic acid, and 1 mg/ml trasylol. Extraction took place over 3 hours at 4°C with constant stirring, and the remaining insoluble material was removed by centrifugation. The supernatants were dialyzed exhaustively (12,000–14,000 molecular weight cut-off) at 4°C against 0.1 mol/L sodium bicarbonate, pH 8, containing proteinase inhibitors. The EBP present in extracts of each culture was then purified by insoluble elastin affinity chromatography, as described previously. 50,51 Briefly, samples of fibroblast extracts were mixed with 1 mg of insoluble elastin and rotary shaken for 1 hour at 4°C. At the end of the incubation, the unbound material was removed by washing the insoluble elastin affinity slurries with 0.1 mol/L sodium bicarbonate buffer, pH 8, until the absorption A280 of the eluant returned to background level. The elastin slurries were pelleted by centrifugation, suspended in 62.5 mmol/L Tris-HCl buffer, pH 6.8, containing 2% sodium dodecyl sulfate, 10% glycerol, 5% mercaptoethanol, and 0.001% bromophenol blue with dithiothreitol and boiled for 5 minutes. The EBPs were resolved by 7.5% to 12% polyacrylamide gel electrophoresis followed by autoradiography. The identity of the 67-kd EBP was additionally confirmed by immunoblotting with the affinity-purified polyclonal antibody raised to human tropoelastin (5 mg/ml diluted 1:10 000), or with anti S-Gal antibody recognizing EBP (2 mg/ml diluted 1:5000) followed by horseradish peroxidase-conjugated goat anti-rabbit antibody-conjugated secondary antibodies diluted 1:5000, and signals were developed with the chemiluminescence detection system. The medium from each culture was also mixed with a cocktail of protease inhibitors and then subjected to elastin affinity chromatography, sodium dodecyl sulfate-polyacrylamide electrophoresis, and autoradiography as described above.
Production of Fibronectin Assays
Normal skin fibroblasts, as well as fibroblasts from the Hurler and Sanfilippo patients (1 × 10 6 cells/dish), were maintained in Medium 199 containing 5% FBS. After the first 24 hours of incubation, 20 μCi/ml of 35S-methionine were added to each sample, and cultures were kept for another 48 hours. After this period, the culture medium was removed and stored in the presence of a protease inhibitors cocktail. The cell layers were extracted overnight at 4°C with 3 mol/L guanidine-HCl, 10 mmol/L 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, 0.1 mol/L dithiothreitol, and 0.5% octyl-β-glucoside, in the presence of protease inhibitors. After centrifugation of insoluble material, the supernatant was dialyzed exhaustively at 4°C against 0.1 mol/L sodium bicarbonate, pH 8, containing protease inhibitors. Fibronectin was then extracted separately from the samples of media and cell layers using 1-ml gelatin 4B-sepharose columns, eluted, and resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis. The 220-kd bands were dissected from the gels and counted by liquid scintillation spectometry as previously described. 71 The relatively large volume of 4B-sepharose present in each column excluded the possibility that fibronectin present in the FBS that was added to the medium would compete for binding with radiolabeled fibronectin produced by SMCs. The amount of radiolabeled fibronectin was normalized for DNA content determined from each culture, using Hoescht Reagent H3313, as previously described. 72 Immunoblots with polyclonal anti-fibronectin antibody of the flow-through and washes from the gelatin 4B-sepharose columns did not detect any 220-kd fibronectin or significant amounts of its degradation products, thus indicating that fibronectin was quantitatively recovered from media and cell extracts.
The parallel cultures of normal and Sanfilippo fibroblasts were also kept for 72 hours in the presence or absence of DS or HS (both in concentration 400 μg/ml), and Hurler fibroblasts were exposed to chondroitinase ABC (0.2 U/day). Their production of secreted and unassembled fibronectin, as well as cell-and matrix-associated fibronectin, were assessed by the incorporation of 35S-methionine as described above.
Assessment of Cell Proliferation
Fibroblasts from normal skin, as well as Hurler disease fibroblasts, were suspended in α-MEM containing 5% FBS and initially plated in six-well dishes at a density of 50,000 cells per well. The medium was changed 24 hours later, and parallel cultures were maintained for the next 48 hours, either in normal medium or in the presence of 1 mg/well of powdered insoluble elastin. The cell density was then roughly estimated in each culture under the inverted microscope with Nomarski optics and then cells were trypsynized and counted in a hemocytometer. Parallel sextoplicate cultures incubated as above were also exposed to 3H-thymidine (2 μCi/well) for the last 24 hours. These cultures were then washed in PBS and treated with cold trichloroacetic acid twice for 10 minutes at 4°C. Then, 0.5 ml of 0.3 N NaOH was added to all dishes for 30 minutes, and subsequently 200-μl aliquots of each culture were mixed with scintillation fluid and counted.
In a separate experiment the influence of 48 hours of exposure to DS, and HS (both in concentrations of 400 μg/ml) on incorporation of 3H-thymidine was also tested in cultures of normal human fibroblasts and in cultures of SMCs isolated (by collagenase digestion) from pig coronary artery (CA SMC). Pig intestinal SMCs (INT SMC), which do not produce elastin, were also used as an additional control.
Incorporation of 3H-thymidine to Hurler fibroblasts was additionally tested after their exposure to 0.2 U/day of chondroitinase ABC.
Analysis of Data
In all biochemical studies, mean and standard deviations were calculated and statistical analyses were carried out by an analysis of variance.
Results
The assessment of histological sections from the coronary artery intimal lesions and from mitral valve chordae tendineae additionally confirmed that the lack of well assembled elastic fibers is the most striking feature distinguishing certain connective tissue structures in Hurler and Sanfilippo patients. Sections of the extramural coronary arteries from the 9-year-old Hurler patient stained with Movat’s pentachrome showed extensive concentric intimal hyperplasia with a reduction in lumen area of ≤90% (Figure 1A) ▶ . These nearly occlusive intimal lesions were composed of numerous SMCs and were particularly rich in glycosaminoglycans and collagen, but contained very little elastin (Figure 1B) ▶ . Besides size and multifocal distribution, the lack of elastin was the most peculiar feature distinguishing the coronary artery intimal lesions of this Hurler patient from the single lesion detected in the coronary artery of the age-matching patient with San-filippo disease, which demonstrated abundant, well-formed elastic fibers (Figure 1, D and E) ▶ . A similar focal intimal lesion with well-formed elastin was also seen in the coronary artery of the 15-year-old boy who died suddenly, but did not suffer of any systemic disease (not shown). In sections of the mitral valve chordae tendineae from the Hurler patient, the characteristic Hurler cells filled with a storage product (known to be DS and HS) were surrounded by matrix made up mostly of abundant collagen and containing only a few short elastin fibers (Figure 1C) ▶ . In contrast, sections from the mitral valve chordae of the Sanfilippo patient (Figure 1F) ▶ showed numerous, well-formed, long elastin fibers adjacent to Sanfilippo cells filled with the storage product (known to be HS). In fact, the abundance and distribution of elastic fibers in Sanfilippo mitral valve chordae did not differ from these detected in normal valve chordae (not shown).
Immunostaining with the specific antibodies to extracellular-matrix components revealed that collagen type I, and major components of the microfibrillar scaffold (MAGP and fibrillin I) were deposited by Hurler fibroblasts in amounts similar to those in cultures of normal skin fibroblasts and cultures of Sanfilippo fibroblasts. Morphometric analysis revealed, however, that amounts of fibronectin produced by Hurler fibroblasts significantly exceeded those present in cultures of normal and Sanfilippo fibroblasts and that they did not deposit immunodetectable elastic fibers (Figure 2) ▶ . Lack of elastin deposition in Hurler cell cultures was striking indeed. As depicted in Figure 3 ▶ , normal (A) and Sanfilippo (C) fibroblasts produced long, branching elastic fibers, whereas Hurler fibroblasts (B) did not deposit any extracellular elastin. Immunohistochemistry also revealed that both normal and Sanfilippo fibroblasts lose their ability for elastic fiber assembly when cultured in the presence of exogenous DS (Figure 3, D and F) ▶ . It is interesting that Hurler fibroblasts restored deposition of the elastic fibers when cultured in the presence of 0.2 U/day of chondroitinase ABC or when cocultured with CHO cells secreting active β-galactosidase that is capable of partial degradation of extracellular DS moieties (Figure 3E) ▶ . Immunostaining with anti-S-Gal antibodies recognizing 67-kd EBP indicated that Hurler fibroblasts show greatly diminished levels of this protein as compared with control and Sanfilippo fibroblasts. Fibroblasts derived from normal skin (Figure 3G) ▶ and Sanfilippo fibroblasts (Figure 3I) ▶ demonstrated the EBP antigen present along the entire cell surface. In Hurler fibroblasts (Figure 3H) ▶ , the cell surface EBP was substantially reduced. Moreover, normal fibroblasts cultured in the presence of 400 μg/ml of DS (Figure 3J) ▶ showed very weak stainability with anti-S-Gal antibody. Immunostaining with BCZ antibody, which recognizes a different epitope on the EBP, confirmed these results (data not shown).
Figure 2.

Morphometric analysis of 10-day-old cultures of normal, Hurler, and Sanfilippo fibroblasts immunostained with the specific antibodies to extracellular-matrix (ECM) components. Hurler fibroblasts deposit only negligible amounts of immunodetectable extracellular elastin. Whereas the amounts of fibronectin produced by Hurler fibroblasts significantly exceed those present in cultures of normal and Sanfilippo fibroblasts, their deposition of collagen type I, fibrillin I, and MAGP does not differ from normal and Sanfilippo fibroblasts. In each analyzed group, 50 low-power fields (×20) from three separate cultures (derived from different patients) were analyzed, and the area occupied by the particular immunodetectable component was quantified. The abundance of each component was then expressed as a percentage of the entire analyzed field (mean ± SD), and results from cultures of Hurler and Sanfilippo fibroblasts were statistically compared with those in cultures of normal skin fibroblasts (*P < 0.001; **P < 0.005).
Figure 3.
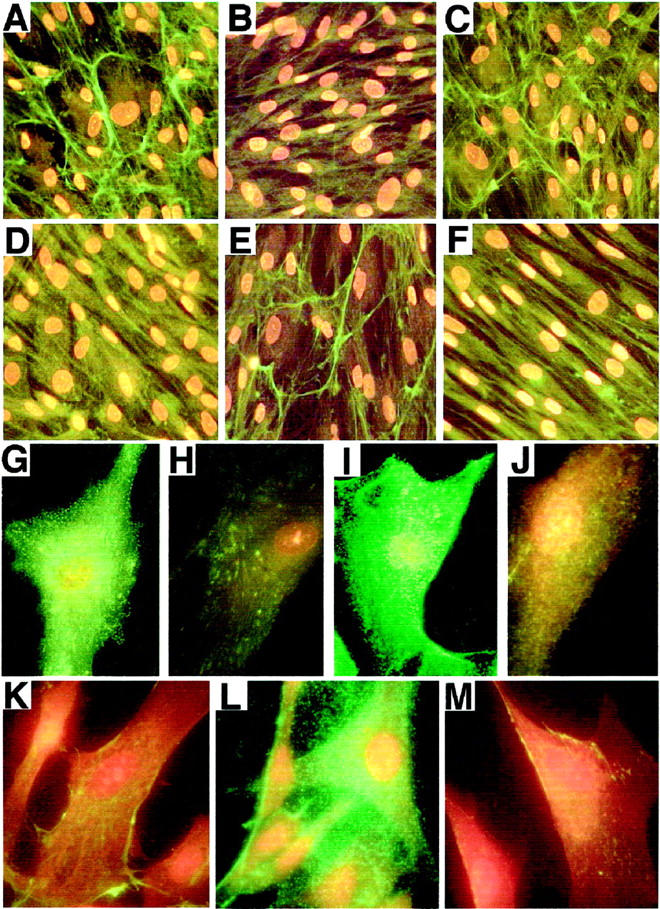
A–F: Representative photomicrographs of 10-day-old cultures immunostained with antitropoelastin antibody. A and C: Normal and Sanfilippo fibroblasts, respectively, produced long, branching elastic fibers, whereas Hurler fibroblasts (B) did not deposit any extracellular elastin. D and F: Both normal and Sanfilippo fibroblasts, respectively, lose their ability for elastic fiber deposition when cultured in the presence of exogenous DS. E: Hurler fibroblasts restored deposition of the elastic fibers when cocultured with CHO cells secreting active β-galactosidase. G–J: Representative photomicrographs of 48-hour-old cultures immunostained with anti-S-Gal antibody recognizing EBP. H: Hurler fibroblasts show greatly diminished levels of EBP, as compared with control (G) and Sanfilippo (I) cells. J: Normal fibroblasts cultured in the presence of 400 μg/ml of DS display a very low level of EBP. K–M: Representative photomicrographs of cultures immunostained with anti-DS antibody. Cultured Hurler fibroblasts (L) express substantially more immunodetectable DS then normal fibroblasts (K). M: Hurler fibroblasts lose their extensive cell surface deposition of DS when cocultured for 24 hours with CHO cells secreting active β-galactosidase.
The metabolic labeling with 3H-valine confirmed immunochemical observations indicating that secretion of tropoelastin by Hurler fibroblasts was selectively impaired. As showed in Figure 4 ▶ (upper panel), most of the metabolically labeled tropoelastin produced by fibroblasts from normal children (cases 3858, 4184, 4212) and from Sanfilippo patients (cases 7825, 7995, 8849) could be immuoprecipitated from the culture medium, and only a relatively small fraction of this 3H-valine-labeled protein was recovered from intracellular extracts. This was in contrast to Hurler fibroblasts (cases 7131, 8180, 8339) which retained the majority of their metabolically labeled tropoelastin intracellularly. Consistently, Hurler fibroblasts incorporated much less 3H-valine into extracellular insoluble elastin than cells taken from normal or from Sanfilippo skin biopsies (Figure 4 ▶ , middle panel). Normal and Sanfilippo fibroblasts cultured in the presence of DS, but not HS, demonstrated significantly reduced levels of insoluble elastin, which resembled those detected in untreated cultures of Hurler fibroblasts (Figure 4 ▶ , lower panel). Moreover, Hurler fibroblasts maintained in the presence of chondroitinase ABC or cocultured with the β-Gal-transfected CHO cells, which secrete active β-galactosidase (enzymes capable of partial degradation of DS), restored their deposition of insoluble elastin.
Figure 4.

Upper panel: Quantitative analysis of immunoprecipitable 3H-valine-labeled tropoelastin from the conditioned media (striated bars) and from the cell extracts (white bars) of 3-day-old cultures. In contrast to normal (cases 3858, 4212, 4184) and Sanfilippo (cases 7825, 7995, 8849) fibroblasts, Hurler fibroblasts (cases 7131, 8180, 8339) retain the majority of their metabolically labeled tropoelastin intracellularly. Middle panel: Consistently, Hurler fibroblasts incorporate much less 3H-valine into extracellular insoluble elastin than cells taken from normal or from Sanfilippo skin biopsies. Lower panel: Normal and Sanfilippo fibroblasts cultured in the presence of 400 μg of DS, but not HS, demonstrate significantly reduced levels of metabolically labeled insoluble elastin, compared with untreated controls. Hurler fibroblasts cocultured for 3 days with the β-Gal-transfected CHO cells (β-Gal) secreting active β-galactosidase or exposed to 0.2 U/day of chondroitinase ABC (Chnase-ABC) restore their deposition of insoluble elastin. Cocultures with untransfected CHO cells (CHO) did not improve production of insoluble elastin by cultured Hurler fibroblasts, as compared with the untreated controls (Contr.). Values of mean ± SD from five different experiments were collected, and respective results from cultures of Hurler and Sanfilippo fibroblasts were statistically compared with those in cultures of normal skin fibroblasts. Results from normal, Sanfilippo, and Hurler fibroblasts treated with different additions were statistically compared with respective untreated controls (*P < 0.001).
Immunohistochemistry with monoclonal antibody recognizing DS additionally confirmed that Hurler fibroblasts cocultured for 3 days with the β-Gal-transfected CHO cells lose their extensive expression of DS (see Figure 3, K–M ▶ ). Consistently, immunostaining with anti-S-Gal and BCZ antibodies indicated that Hurler cells, which were maintained in the presence of chondroitinase ABC or β-galactosidase and lose their extensive DS, regained their cell surface expression of the EBP (data not shown).
Because Hurler fibroblasts demonstrated only weak immunostainability with the EBP-recognizing antibodies, we also tested whether those DS-storing cells are primarily deficient in the EBP or whether they lose this recyclable tropoelastin chaperone, due to the DS-dependent shedding. The metabolic pulse-labeling with radioactive serine and the further chase of this tracer to the 67-kd EBP indicated that both normal and Hurler fibroblasts initially synthesize comparable amounts of this EBP. In contrast to normal cells, which retain the majority of the labeled EBP, Hurler fibroblasts quickly lose this newly produced protein, which can be detected in increased amounts in the respective conditioned media during the chase (Figure 5) ▶ .
Figure 5.

Representative autoradiographs showing levels of the 67-kd EBP during the chase after metabolic pulse-labeling with radioactive serine. Results indicate that both normal and Hurler fibroblasts initially synthesize comparable amounts of EBP, isolated by elastin affinity columns. In contrast to normal cells, which retain the majority of the labeled EBP, Hurler fibroblasts steadily lose this newly produced protein, which can be detected in the increased amounts in the respective conditioned media during the chase.
Biochemical assays confirmed results obtained by immunohistochemistry and demonstrated that Hurler disease fibroblasts showed increased synthesis of fibronectin as compared with normal or Sanfilippo fibroblasts. It is interesting that incubation of Hurler fibroblasts in the presence of 0.2 U/day of chondroitinase ABC caused significant reduction of their fibronectin production (Figure 6A) ▶ . Moreover, assays of metabolically labeled, soluble fibronectin in the conditioned medium, as well as cell- and matrix-associated fibronectin, indicated that exogenous DS, but not HS, significantly increased the synthesis of fibronectin in cultures of normal and Sanfilippo fibroblasts (Figure 6B) ▶ . In fact, the addition of DS, a galactosugar-bearing GAG (causing shedding of the EBP) elevated fibronectin synthesis by normal fibroblasts to the levels observed in cultures of untreated Hurler fibroblasts. The Northern blots detecting steady-state levels of fibronectin messenger RNA showed no significant differences when control and GAG-treated cultures were compared after 24 hours (Figure 6C) ▶ , suggesting that the mechanism of increased fibronectin production is likely due to post-transcriptional and even post-translational regulation.
Figure 6.

A: Three-day-old cultures of Hurler fibroblasts demonstrate higher levels of secreted and unassembled, and cell- and matrix-associated fibronectin, as compared with normal and Sanfilippo fibroblasts. Incubation of Hurler fibroblasts in the presence of 0.2 U/day of chondroitinase ABC (Chnase ABC) causes reduction of their fibronectin levels. B: Addition of DS, but not HS (both in concentrations of 400 μg/ml), significantly increased net production of fibronectin in 48-hour cultures of normal fibroblasts. Production of fibronectin was assessed by incorporation of 35S-methionine into the 220-kd protein isolated by gelatin-sepharose affinity and identified as fibronectin by immunoblotting. Results from normal, Sanfilippo, and Hurler fibroblasts treated with different additions were statistically compared with respective untreated controls. Values of mean ± SD from three different experiments were statistically compared (*P < 0.001). C: Northern blot analysis of confluent normal fibroblasts cultures incubated for 24 hours in the presence or absence of 400 μmol/L DS or HS. Levels of fibronectin mRNA detected in cultures treated with both glycosaminoglycans did not differ from levels observed in untreated control culture. Levels of ethidium bromide-stained 28S and 18S ribosomal RNA indicate equal sample loading.
Comparison of cultured fibroblasts, using phase-contrast microscopy, additionally revealed that the growth rate of cultured Hurler fibroblasts was considerably higher than of those taken from normal children (Figure 7A) ▶ . The results of experiments aimed at quantification of this phenomenon showed that, whereas normal or Sanfilippo fibroblasts initially plated at concentrations of 50,000 cells/well grew to an average 90,000 cells/well in 3 days, the Hurler fibroblasts plated with the same initial density reached an average of 250,000 cells/well in 3 days. Similar results indicating a higher proliferation of Hurler fibroblasts were obtained when the incorporation of 3H-thymidine was assessed (Figure 7B) ▶ . Phase-contrast microscopy, direct cell counting, and 3H-thymidine incorporation all indicated that the high proliferation rate of Hurler fibroblasts could be significantly reduced in cultures treated with exogenous insoluble elastin (Figure 7, A and B) ▶ . Moreover, we found that the addition of DS, but not HS, significantly increased incorporation of 3H-thymidine to normal and Sanfilippo fibroblasts in 3-day-old cultures. It is important that Hurler fibroblasts cultured in the presence of chondroitinase ABC decreased their 3H-thymidine incorporation (Figure 7C) ▶ .
Figure 7.
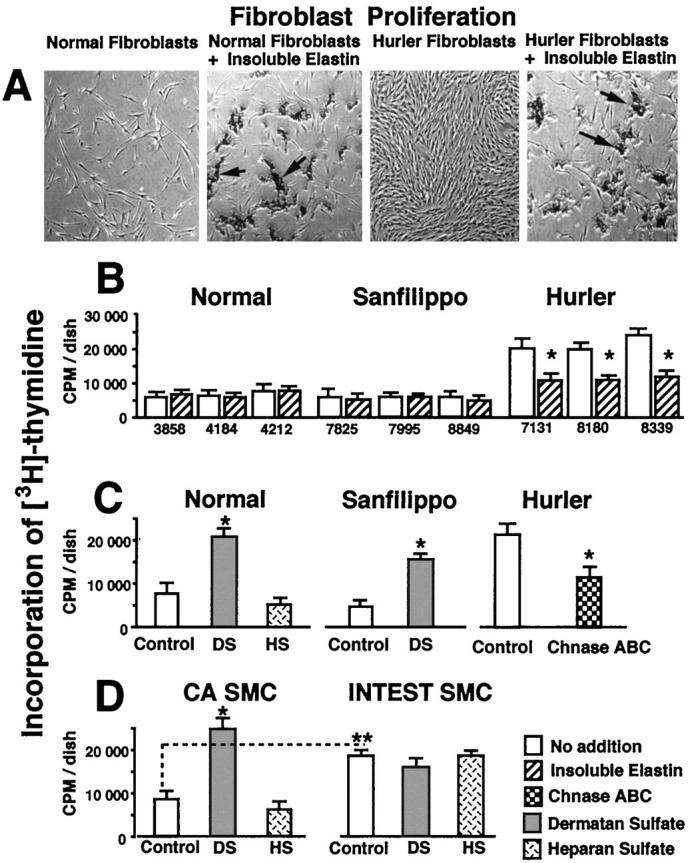
A: Phase-contrast micrographs of 3-day-old cultures, illustrating that the growth rate of Hurler fibroblasts was considerably higher than those taken from normal children. Whereas the addition of insoluble elastin (arrows) to cultured normal fibroblasts did not affect their final density, addition of insoluble elastin (arrows) to cultures of Hurler fibroblasts substantially reduced their number in 3-day-old culture. B: Incorporation of 3H-thymidine also indicates higher growth of Hurler fibroblasts as compared with normal and Sanfilippo fibroblasts in 3-day-old cultures. These assays also illustrate that high proliferation of Hurler fibroblasts was significantly reduced in cultures treated with exogenous insoluble elastin (striated bars). C: Addition of DS, but not HS (both in concentrations of 400 μg/ml) significantly increased incorporation of 3H-thymidine to normal and Sanfilippo fibroblasts. Hurler fibroblasts cultured for 3 days in the presence of chondroitinase ABC (0.2 U/day) (Chnase ABC) decrease their 3H-thymidine incorporation. D: Pig intestinal SMCs (INTEST SMC), which do not produce elastin, incorporate more radioactive thymidine then the elastin-producing pig coronary artery SMCs (CA SMC). CA SMCs incubated for 3 days in the presence of 400 μg/ml of DS, up-regulate their incorporation of labeled thymidine, as compared with the untreated control and cells incubated with HS. In contrast, neither DS nor HS affected proliferation of pig intestinal SMCs. In all experiments, cells were plated with the same initial density 50,000 cells/well. Values of mean ± SD from 3 different experiments were statistically compared with untreated controls within the same cell type (*P < 0.002). Proliferation rates of different cell types tested were also assessed (**P < 0.05).
Additional assessment of cellular proliferation indicated that, similarly to normal human fibroblasts, the elastin-producing coronary artery SMCs selectively upregulated their incorporation of labeled thymidine when incubated in the presence of DS. This was in contrast to intestinal SMCs (which do not produce elastin), which did not change their rate of 3H-thymidine incorporation when treated with DS or HS. (Figure 7D) ▶ .
Discussion
Among all inherited diseases that affect the structure, distribution, and abundance of elastic fibers, only supravalvular aortic stenosis, Williams syndrome, and cutis laxa have been directly connected with primary alterations in the elastin gene. 73-76 Other genetic diseases that severely affect the elastic fiber integrity (Marfan syndrome, pseudoxanthoma elasticum, Buschke-Ollendorff syndrome, Menkes disease) have been linked to primary defects in microfibrillar proteins or to errors in copper metabolism and lysyl oxidase. 34-39,42,77,78
In this paper we have presented data indicating that abnormal tissue accumulation of DS in Hurler disease leads to a secondary deficiency in the 67-kd EBP and to impaired elastic fiber assembly.
Elastic fiber assembly takes place in the infoldings of the plasma membrane 34,55,79,80 in which microfibrils are found grouped in small bundles and serve to align the tropoelastin molecules in precise register so that cross-linking regions are juxtaposed before oxidation by lysyl oxidase. 34-39,45 We have previously established that the 67-kd EBP (identical to the catalytically inactive spliced variant of β-galactosidase) forms an intracellular complex with tropoelastin and serves as its molecular chaperone, which escorts this very hydrophobic and nonglycosylated elastin precursor through the secretory pathways and protects it from intracellular aggregation and enzymatic degradation. 51,55 We have demonstrated that the majority of the EBP, after dissociation from its ligand at the assembly sites, recycles back to the endosomal compartment of the cell to reassociate with the newly synthesized tropoelastin. 56 We have also shown that incubation of elastin-producing cells (eg, ear cartilage chondrocytes and vascular SMCs) with galactose, lactose, and agarose or with N-acetyl-galactosamine-containing glycosaminoglycans, chondroitin sulfate, and DS causes depletion of the EBP. 51-54 These galactosugar-bearing moieties accumulating in the endosomal or pericellular compartments bind to the lectin domain of the EBP and cause a premature release of tropoelastin from its transporter and shedding of the EBP from the cell surface. 81 Histopathological observations of Hurler disease tissues indicate that the affected sites are characterized by the presence of vacuolated Hurler cells, demonstrating intracellular and pericellular accumulations of DS and HS glycosaminoglycans. These affected sites are often rich in collagen fibers, but contain a poorly developed system of elastic fibers (Figure 1) ▶ . Because the Hurler fibroblasts were capable of normal synthesis of tropoelastin (Figure 4) ▶ and did not demonstrate any higher elastolytic activity than normal fibroblasts (data not shown), it follows that impaired formation of extracellular fibers, but not their accelerated degradation, is responsible for the low net content of insoluble elastin in the tissues of Hurler patients. In fact, increased elastolysis would not be expected, because DS can sequestrate and inactivate leukocytic elastase. 82,83
Because the HS-accumulating fibroblasts taken from Sanfilippo patients demonstrate strong immunostaining with antibodies recognizing the EBP and good production of elastic fibers, the excessive accumulation of DS and not HS should account for the disruption of elastogenesis in Hurler fibroblasts. This conclusion is further supported by the observation that exogenous DS, but not HS, added to the culture of normal or Sanfilippo fibroblasts caused impaired elastogenesis and that degradation of DS by treatment of Hurler fibroblasts with β-galactosidase and chondroitinase ABC restored their deposition of the extracellular elastic fibers. Our results are also consistent with the observation of McGowan and colleagues, 84 who reported that galactosamine-containing glycosaminoglycans, chondroitin sulfate, and DS increase the quantity of soluble elastin in the conditioned medium and decrease the deposition of insoluble elastin in the extracellular matrix formed by cultured rat lung fibroblasts.
Our data indicate that the impaired elastogenesis in Hurler disease can be directly linked with the functional deficiency in the EBP. The metabolic pulse-labeling of cultured fibroblasts clearly demonstrated that, whereas the initial synthesis of the EBP by Hurler fibroblasts did not differ from the normal fibroblasts, Hurler cells steadily lose this protein during the 30- to 45-minute chase. These results are consistent with previous data demonstrating that an excess of galactosugar-bearing moieties in the pericellular space triggers shedding of the EBP from the cell surface of arterial SMCs, which in turn eliminates its recycling and subsequently inhibits tropoelastin secretion and its extracellular assembly into the elastic fibers. 54-56
Immunohistochemistry and results of our metabolic studies also suggest that a constant shedding of the EBP from cell surfaces of Hurler fibroblasts may be additionally linked to the up-regulation of fibronectin production. This claim is additionally supported by the fact that normal fibroblasts exposed to DS, but not to HS, also demonstrated increased net deposition of fibronectin. Present findings are also consistent with our previous data demonstrating that chondroitin sulfate- and DS-dependent accumulation of metabolically labeled fibronectin in arterial SMCs is directly related to shedding of the EBP 85 and a subsequent unmasking of an adjacent interleukin-1 (IL-1) receptor. 71 Because we have previously demonstrated that removal of the cell surface EBP from SMCs allows access of IL-1β to its receptor, with induction of intracellular signals leading to a net increase in the production of fibronectin, 71 we suggest that the loss of the EBP may be also responsible for triggering a similar mechanism in Hurler fibroblasts that have been cultured in the presence of 10% FBS, which contains IL-1β and other growth factors.
Our studies of cultured Hurler fibroblasts that are characterized by impaired elastogenesis also show that these cells proliferate much faster than their normal counterparts. This increased rate of Hurler fibroblast proliferation resembles the situation in which restenotic lesions narrow arteries after angioplasty, when the increased SMC proliferation coincides with an abnormal accumulation of DS proteoglycans and with a substantial decrease in the insoluble elastin content. 86-88 It is interesting that this increased rate of cellular proliferation in Hurler disease occurs despite accumulation of HS compounds, which have been shown as a inhibitors of vascular SMC proliferation and neointimal formation. 89-90
In Hurler disease, DS moieties, in addition to lysosomes, also accumulate on the cell surface (Figure 3L) ▶ . It is therefore conceivable that they may successfully compete with small HS glycosaminoglycans and prevent their binding and sequestration of certain growth factors, as in basic fibroblast growth factor (bFGF) and polyamines (eg, spermine) that would normally induce an antiproliferative effect. 89,90 On the other hand, we should not ignore that HS has been shown to be an agent that can up-regulate platelet-derived growth factor receptors on human lung fibroblasts, 91 and that the interaction of bFGF with heparin and HS can potentiate or antagonize bFGF activity, depending on the size of the saccharide used. Oligosaccharides based on HS structures as small as six sugar residues have been demonstrated to bind to bFGF and block its activity, whereas structures larger than 10 sugar residues potentiate mitogenic effects of bFGF. 92 Thus, nondegraded long chains of HS accumulating in Hurler disease cells may potentially contribute to the net effect of higher than normal proliferation, if released from the lysososmal compartment. Alternatively, the lack of the antiproliferative effect of HS may be because nondegraded HS moieties are stored in lysosomes and do not interfere with cell surface-derived mitogenic signals that are facilitated by DS. This idea is strengthened by the finding that Sanfilippo fibroblasts, which store HS alone, do not show any slower proliferation rate than normal fibroblasts (Figure 7C) ▶ . Moreover, accumulation of nondegraded HS moieties, both in Hurler patients (Figure 1, A ▶ and B), and in Sanfilippo patients (Figure 1, D and E) ▶ did not arrest excessive SMC proliferation nor inhibit the development of intimal thickening observed in both diseases. Therefore, the presence of DS compounds on the surface of Hurler cells, which triggers EBP shedding and leads to impaired elastogenesis, may be a crucial factor that is responsible for the dramatic difference in the magnitude of intimal lesions observed in both diseases.
Because the extensive proliferation of cultured Hurler fibroblasts could be reduced to a normal level after the administration of exogenous insoluble elastin (Figure 7, A and B) ▶ or substantially decreased after exposure to chondroitinase ABC (Figure 7C) ▶ that removes DS and causes the restoration of normal elastogenesis, we suggest that interactions between cells and the insoluble elastin may either directly initiate antimitogenic signals or block the mitogenic signals transduced by the common growth factors present in the FBS-containing medium. Results indicating that intestinal SMCs, which do not deposit elastin, incorporate more radioactive thymidine than the elastin-depositing cells (normal human skin fibroblasts and pig coronary artery SMCs) and the fact that only those elastoblasts up-regulate their proliferation when exposed to DS (Figure 7G) ▶ additionally implicate involvement of elastin and its receptor in the process controlling cellular proliferation. Our observation is consistent with the recent report of Li and colleagues, 93 who showed that newborn transgenic mice lacking elastin protein die as a result of arterial blockage with abnormal intimal thickenings, which consist of proliferating SMCs. Studies of normal lung development also indicate that the relatively late initiation of elastin deposition (last trimester of embryonic life) coincides with a steady decrease in proliferation rates of SMCs in the rat pulmonary vasculature after birth. 94
The progressive intimal thickenings developed in the coronary arteries of Hurler disease patients (Figure 1) ▶ are characterized by the peculiar lack of elastin, and, in this respect, closely resemble the proliferative lesions developed in transgenic mice lacking elastin. 93 Our data additionally indicate that the DS-induced shedding of the EBP, causing impaired elastogenesis, can be directly linked to the increased cellular proliferation (Figure 7C) ▶ and up-regulation of fibronectin deposition (Figure 6B) ▶ , which in turn facilitates cellular migration. In this respect, this shedding may play an important part in the pathomechanism leading to development of occlusive lesions in coronary arteries, which can cause death of Hurler disease patients. We cannot conclude, however, that a high proliferation of Hurler fibroblasts may be solely induced by the lack of the insoluble elastin. This proliferation may also be promoted by the antiadhesive properties of DS-rich proteoglycans, 95 by the presence of nonassembled or partially degraded tropoelastin-derived peptides that promote progression of the cell cycle, 96,97 or by a DS-facilitated mitogenic response to such mitogens as FGF-2 98 or hepatocyte growth factor/scatter factor 99 present in the serum.
We propose that the accumulation of DS moieties by Hurler fibroblasts, which induces the functional deficiency in the EBP and, consequently, leads to disruption of normal elastogenesis, is directly linked to a deficiency in elastic fibers in the skin, tendons, and cardiac valvular apparatus of Hurler disease patients. Furthermore, because the presence of elastic fibers in the limb buds and their primitive perichondrial tissue has been suggested as a crucial factor in maintaining the proper shape of the normal embryonal skeleton, 100 our results may also be relevant to skeletal deformations found in Hurler disease. Further studies, using fetal autopsy tissues, would be required, however, to confirm this possibility. We also believe that the peculiar lack of elastic fibers in the occlusive coronary lesions in children with Hurler disease (Figure 1, A and B) ▶ is also initiated by the proposed DS-dependent mechanism, leading to functional EBP deficiency.
Our data not only indicate that Hurler disease should be added to the list of inherited diseases that are characterized by impaired elastogenesis but also encourage further studies exploring whether storage of galactosugar-containing glycosaminoglycans (DS, chondroitin sulfate, and keratan sulfate) also cause faulty elastic fiber assembly that might contribute to the development of the characteristic clinical phenotypes that are observed in other types of mucopoolysacharidoses.
Acknowledgments
The authors are very grateful to Dr. Gregory J. Wilson from the Division of Pathology at The Hospital for Sick Children for the histological assessment of the autopsy material and to Dr. John W. Callahan from the Division of Structural Biology and Biochemistry for providing the CHO cells transfected with β-galactosidase cDNA. We are also grateful to Haren Treasurer for the excellent technical assistance.
Footnotes
Address reprint requests to Dr. Aleksander Hinek, Cardiovascular Research Program, The Hospital for Sick Children, 555 University Avenue, Toronto, ON M5G 1X8, Canada. E-mail: alek.hinek@sickkids.on.ca.
Supported by grant PG 13920 from the Medical Research Council of Canada and by a Career Investigator Award from the Heart and Stroke Foundation of Ontario (to A. H.).
References
- 1.Dorfman A, Matalon R: The Hurler and Hunter syndromes. Am J Med 1969, 47:691-707 [DOI] [PubMed] [Google Scholar]
- 2.Dorfman A, Matalon R: The mucopolysaccharidoses. Stanbury JB Wyngaarden JB Frederickson DS eds. The Metabolic Basis of Inherited Diseases. 1972, :pp 1218-1272 McGraw-Hill, New York [Google Scholar]
- 3.Matalon R, Dorfman A: Hurler’s syndrome, an α-L-iduronidase deficiency. Biochem Biophys Res Commun 1972, 47:959-964 [DOI] [PubMed] [Google Scholar]
- 4.McKusick VA: Heritable Disorders of Connective Tissue. 1972:pp 325-346 CV Mosby, St Louis
- 5.Hopwood JJ, Morris CP: The mucopolysaccharidoses: diagnosis, molecular genetics and treatment. Mol Biol Med 1990, 7:38-41 [PubMed] [Google Scholar]
- 6.Taylor JA, Gibson GJ, Brooks DA, Hopwood JJ: α-L-iduronidase in normal and mucopolysaccharidosis type-I human skin fibroblasts. Biochem J 1991, 274:263-267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Scott HS, Litjens T, Nelson PV, Thompson PR, Brooks DA, Hopwood JJ, Morris CP: Identification of mutations in the α-L-iduronidase gene (IDUA) that cause Hurler and Scheie syndromes. Am J Hum Genet 1993, 53:973-986 [PMC free article] [PubMed] [Google Scholar]
- 8.Scott HS, Bunge S, Gal A, Clarke LA, Morris CP, Hopwood JJ: Molecular genetics of mucopolysaccharidosis type I: diagnostic, clinical, and biological implications. Hum Mutat 1995, 6:288-302 [DOI] [PubMed] [Google Scholar]
- 9.Neufeld E, Muenzer J: The mucopolysacharidoses. Scriver CR Beaudet AL Sly WS Valle D eds. The Metabolic Basis of Inherited Diseases. 1997, :pp 2465-2494 McGraw-Hill, New York [Google Scholar]
- 10.Peters C, Shapiro EG, Krivit W: Hurler syndrome: past, present, and future. J Pediatr 1998, 133:7-9 [DOI] [PubMed] [Google Scholar]
- 11.Whitley CB, Ridnour MD, Draper KA, Dutton CM, NegIia JP: Diagnostic test for mucopolysaccharidosis. 1. Direct method for quantifying excessive urinary glycosaminoglycan excretion. Clin Chem 1989, 35:374-379 [PubMed] [Google Scholar]
- 12.Byers S, Rozaklis T, Brumfield LK, Ranieri E, Hopwood JJ: Glycosaminoglycan accumulation and excretion in the mucopolysaccharidoses: characterization and basis of a diagnostic test for MPS. Mol Genet Metab 1998, 65:282-290 [DOI] [PubMed] [Google Scholar]
- 13.MacBrinn M, Okada S, Woollacott M, Patel V, Ho MW, Tappel AL, O’Brien JS: β-Galactosidase deficiency in the Hurler syndrome. N Engl J Med 1969, 281:338-343 [DOI] [PubMed] [Google Scholar]
- 14.Ho MW, O’Brien JS: Hurler’s syndrome: deficiency of a specific β-galactosidase isoenzyme. Science 1969, 165:611-613 [DOI] [PubMed] [Google Scholar]
- 15.Haust MD, Landing BH: Histochemical studies in Hurler’s disease. J Histochem Cytochem 1961, 9:79-86 [DOI] [PubMed] [Google Scholar]
- 16.Lagundoff D, Ross R, Benditt EP: Histochemical and electron microscopic study in a case of Hurler’s disease. Am J Pathol 1962, 41:273-286 [PMC free article] [PubMed] [Google Scholar]
- 17.Shapiro LJ: The mucopolysacharidoses, glycoproteinoses and mycolipidoses. Pediatrics. Edited by AM Rudolph. Norwalk, CT, Appleton & Lange, Appleton-Century-Crofts, 1991, pp 375–382
- 18.Roubicek M, Gehler J, Spranger J: The clinical spectrum of α-L-iduronidase deficiency. Am J Med Genet 1985, 20:471-481 [DOI] [PubMed] [Google Scholar]
- 19.Bunge S, Clements PR, Byers S, Kleijer WJ, Brooks DA, Hopwood JJ: Genotype-phenotype correlations in mucopolysaccharidosis type I using enzyme kinetics, immunoquantification and in vitro turnover studies. Biochim Biophys Acta 1998, 1407:249-256 [DOI] [PubMed] [Google Scholar]
- 20.Belcher RW: Utrastructure of the skin in the genetic mucopolysaccharidoses. Arch Pathol 1972, 94:511-518 [PubMed] [Google Scholar]
- 21.Cawdery JE, Leonard JV: Hurler syndrome with cardiomyopathy in infancy. J Pediatr 1989, 114:430-432 [DOI] [PubMed] [Google Scholar]
- 22.Schieken RM, Kerber RE, Ionasescu VV, Zellweger H: Cardiac manifestations of the mucopolysaccharidoses. Circulation 1975, 52:700-705 [DOI] [PubMed] [Google Scholar]
- 23.Renteria VG, Ferrans VJ, Jones M, Roberts WC: Intracellular collagen fibrils in prolapsed (“floppy”) human atrioventricular valves. Lab Invest 1976, 35:439-443 [PubMed] [Google Scholar]
- 24.Renteria VG, Ferrans VJ, Roberts WC: The heart in the Hurler syndrome: gross, histologic and ultrastructural observations in five necropsy cases. Am J Cardiol 1976, 38:487-501 [DOI] [PubMed] [Google Scholar]
- 25.Johnson GL, Vine DL, Cottrill CM, Noonan JA: Echocardiographic mitral valve deformity in the mucopolysaccharidoses. Pediatrics 1981, 67:401-406 [PubMed] [Google Scholar]
- 26.Perkins DG, Haust MD: Ultrastructure of myocardium in the Hurler syndrome. Possible relation to cardiac function. Virchows Arch A Pathol Anat Histol 1982, 394:195-205 [DOI] [PubMed] [Google Scholar]
- 27.Haust MD: Arterial involvement in genetic diseases. Am J Cardiovasc Pathol 1987, 1:231-285 [PubMed] [Google Scholar]
- 28.Vinallonga X, Sanz N, Balaguer A, Miro L, Ortega JJ, Casaldaliga J: Hypertrophic cardiomyopathy in mucopolysaccharidoses: regression after bone marrow transplantation. Pediatr Cardiol 1992, 13:107-109 [DOI] [PubMed] [Google Scholar]
- 29.Nelson J, Shields MD, Mulholland HC: Cardiovascular studies in the mucopolysaccharidoses. J Med Genet 1990, 27:94-100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dangel JH: Cardiovascular changes in children with mucopolysaccharide storage diseases and related disorders—clinical and echocardiographic findings in 64 patients. Eur J Pediatr 1998, 157:534-538 [DOI] [PubMed] [Google Scholar]
- 31.Brosius FC, III, Roberts WC: Coronary artery disease in the Hurler syndrome: qualitative and quantitative analysis of the extent of coronary narrowing at necropsy in six children. Am J Cardiol 1981, 47:649-653 [DOI] [PubMed] [Google Scholar]
- 32.Braunlin EA, Hunter DW, Krivit W, Burke BA, Hesslein PS, Porter PT, Whitley CB: Evaluation of coronary artery disease in the Hurler syndrome by angiography. Am J Cardiol 1992, 69:1487-1489 [DOI] [PubMed] [Google Scholar]
- 33.du Cret RP, Weinberg EJ, Jackson CA, Braunlin EA, Boudreau RJ, Kuni CC, Carpenter BM, Hunter DW, Krivit W, Bodeau G: Resting Tl-201 scintigraphy in the evaluation of coronary artery disease in children with Hurler syndrome. Clin Nucl Med 1994, 19:975-978 [DOI] [PubMed] [Google Scholar]
- 34.Mecham RP, Heuser J: The elastic fiber. Cell Biology of the Extracellular Matrix, 2nd ed. Edited by ED Hay. New York, Plenum Press, pp 79–109
- 35.Parks WC, Pierce RA, Lee KA, Mecham RP: Elastin. Adv Mol Cell Biol 1993, 6:131-181 [Google Scholar]
- 36.Rosenbloom J, Abrams WR, Mecham RP: Extracellular matrix 4: the elastic fiber. FASEB J 1993, 7:1208-1218 [PubMed] [Google Scholar]
- 37.Christiano AM, Uito J: Molecular pathology of elastic fibers. J Invest Derm 1994, 103:53-57 [DOI] [PubMed] [Google Scholar]
- 38.Pasquali-Ronchetti I, Baccarani-Contri M: Elastic fiber during development and aging. Microsc Res Tech 1997, 38:428-435 [DOI] [PubMed] [Google Scholar]
- 39.Vrhovski B, Weiss AS: Biochemistry of elastin. Eur J Biochem 1998, 258:1-18 [DOI] [PubMed] [Google Scholar]
- 40.Debell L, Tamburro AM: Elastin: molecular description and function. Int J Biochem Cell Biol 1999, 31:261-272 [DOI] [PubMed] [Google Scholar]
- 41.Sakai LY, Keene DR, Engvall E: Fibrillin, a new 350-kD glycoprotein is a component of extracellular microfibrils. J Cell Biol 1986, 103:2499-2509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kielty CM, Shuttleworth CA: Fibrillin-containing microfibrils: structure and function in health and disease. Int J Biochem Cell Biol 1995, 27:747-760 [DOI] [PubMed] [Google Scholar]
- 43.Zhang H, Apfelroth SD, Hu W, Davis EC, Sanguinieti C, Bonadio J, Mecham RR, Ramirez F: Structure and expression of fibrillin-2, a novel microfibrillar component preferentially located in elastic matrices. J Cell Biol 1994, 124:855-863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gibson MA, Hatzinikolas G, Kumaratilake JS, Sandberg LB, Nicholl JK, Sutherland GR, Cleary EG: Further characterization of proteins associated with elastic fiber microfibrils including the molecular cloning of MAGP-2 (MP25). J Biol Chem 1996, 271:1096-1103 [DOI] [PubMed] [Google Scholar]
- 45.Kagan HM, Vaccaro CA, Bronson RE, Tang SS, Brody JS: Ultrastructural immunolocalization of lysyl oxidase in vascular connective tissue. J Cell Biol 1986, 103:1121-1128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Baccarani-Contri M, Vincenzi D, Cicchetti F, Mori G, Pasquali-Ronchetti I: Immunocytochemical localization of proteoglycans within normal elastic fibers. J Cell Biol 1990, 53:305-312 [PubMed] [Google Scholar]
- 47.Bressan GM, Daga-Gordini D, Colornbatti A, Castellani I, Mariga V, Volpin D: Emilin, a component of elastic fibers preferentially located at the elastin-microfibrils interface. J Cell Biol 1993, 12:201-212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Roark EF, Keene DR, Haundenschild CC, Godyna S, Little CD, Argraves WS: The association of human fibulin-1 with elastic fibers: an immunohistological, ultrastructural, and RNA study. J Histochem Cytochem 1995, 43:401-411 [DOI] [PubMed] [Google Scholar]
- 49.Hinek A, Wrenn DS, Mecham RP, Barondes SH: The elastin receptor: a galactoside-binding protein. Science 1988, 239:1539-1541 [DOI] [PubMed] [Google Scholar]
- 50.Hinek A, Rabinovitch M, Keeley F, Okamura-Oho Y, Callahan J: The 67-kD elastin/laminin-binding protein is related to an enzymatically inactive, alternatively spliced form of β-galactosidase. J Clin Invest 1993, 9:1198-1205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Privitera S, Prody CA, Callahan JW, Hinek A: The 67-kd enzymatically inactive alternatively spliced variant of β-galactosidase is identical to the elastin/laminin-binding protein. J Biol Chem 1998, 273:6319-6321 [DOI] [PubMed] [Google Scholar]
- 52.Mecham RP, Hinek A, Entwistle R, Wrenn DS, Griffin GL, Senior RM: Elastin binds to a multifunctional 67-kilodalton peripheral membrane protein. Biochemistry 1989, 283:3716-3722 [DOI] [PubMed] [Google Scholar]
- 53.Mecham RP, Whitehouse L, Hay M, Hinek A, Sheetz MP: Ligand affinity of the 67 kd elastin/laminin binding protein is modulated by the protein’s lectin domain: visualization of elastin/laminin-receptor complexes with gold-tagged ligands. J Cell Biol 1991, 113:187-194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hinek A, Mecham RP, Keeley F, Rabinovitch M: Impaired elastin fiber assembly related to reduced 67-kD elastin binding protein in fetal lamb ductus arteriosus and in cultured aortic smooth muscle cells treated with chondroitin sulfate. J Clin Invest 1991, 88:2083-2094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hinek A, Rabinovitch M: 67-kD elastin binding protein is a protective companion of extracellular insoluble elastin, and intracellular tropoelastin. J Cell Biol 1994, 126:563-574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hinek A, Keeley F, Callahan J: Recycling of the 67-kd elastin binding protein in arterial monocytes is imperative for secretion of tropoelastin. Exp Cell Res 1995, 220:312-324 [DOI] [PubMed] [Google Scholar]
- 57.Hinek A: Nature and multiple functions of the 67 kD elastin/laminin binding protein. Cell Adhes Commun 1994, 2:185-193 [DOI] [PubMed] [Google Scholar]
- 58.Kielty CM, Whittaker SP, Shuttleworth CA: Fibrillin: evidence that chondroitin sulphate proteoglycans are components of microfibrils and associate with newly synthesised monomers. FEBS Lett 1996, 386:169-173 [DOI] [PubMed] [Google Scholar]
- 59.Brown P, Mecham L, Tisdale C, Mecham RP: The cysteine residues in the carboxy terminal domain of tropoelastin form an intrachain disulfide bond that stabilizes a loop structure and positively charged pocket. Biochem Biophys Res Commun 1992, 186:549-555 [DOI] [PubMed] [Google Scholar]
- 60.Kaplan P, Wolfe LS: Sanfilippo syndrome type D. J Pediatr 1987, 110:267-271 [DOI] [PubMed] [Google Scholar]
- 61.Mecham RP, Hinek A, Cleary EG, Kucich U, Rosenbloom J: Development of immunoreagents to ciliary zonules that react with protein components of elastic fibers. Biochem Biophys Res Commun 1988, 151:822-826 [DOI] [PubMed] [Google Scholar]
- 62.Musto L: Modified Movat pentachrome stain. J Histotechnol 1996, 9:173-174 [Google Scholar]
- 63.deChalain T, Phyllips JH, Hinek A: Bio-engineering of porcine and human elastic cartilage in transplants of aggregated auricular chondrocytes embedded in hydrogel containing alginate, collagen and κ-elastin. J Biomed Mat Res 1999, 44:280-288 [DOI] [PubMed] [Google Scholar]
- 64.Starcher B, Pierce R, Hinek A: UVB irradiation selectively stimulates deposition of new elastic fibers by dermal fibroblasts surrounding the hair follicles and sebaceous glands in mice. J Invest Dermatol 1999, 112:450-455 [DOI] [PubMed] [Google Scholar]
- 65.Hopwood JJ, Elliott H: Isolation and characterization of N-acetylglucosamine 6-sulfate from the urine of a patient with Sanfilippo type D syndrome and its occurrence in normal urine. Biochem Int 1983, 6:831-838 [PubMed] [Google Scholar]
- 66.Prosser J, Whitehouse LA, Parks WC, Hinek A, Park PW, Mecham RP: Polyclonal antibodies to tropoelastin and specific detection and measurement of tropoelastin in vitro. Connect Tissue Res 1991, 265:265-279 [DOI] [PubMed] [Google Scholar]
- 67.Gibson MA, Kumaratilake JS, Cleary EG: The protein components of the 12-nanometer microfibrils of elastic and nonelastic tissues. J Biol Chem 1989, 264:4590-4598 [PubMed] [Google Scholar]
- 68.Zhang S, McCarter JD, Okamura-Oho Y, Yaghi F, Hinek A, Withers SG, Callahan JW: Kinetic mechanism and characterization of human β-galactosidase precursor secreted by permanently-transfected CHO cells. Biochem J 1994, 304:281-288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Takagi M, Ono Y, Maeno M, Miyashita K, Omiya K: Immunohistochemical and biochemical characterization of sulphated proteoglycans in embryonic chick bone. J Nihon Univ Sch Dent 1997, 39:156-163 [DOI] [PubMed] [Google Scholar]
- 70.Hinek A, Rabinovitch M: Ductus arteriosus smooth muscle cells produce a 52 kD tropoelastin associated with impaired assembly of elastic laminae. J Biol Chem 1993, 268:1405-1413 [PubMed] [Google Scholar]
- 71.Hinek A, Molossi S, Rabinovitch M: Functional interplay between interleukin-1 receptor and elastin binding protein regulates fibronectin production in coronary artery smooth muscle cells. Exp Cell Res 1996, 225:122-131 [DOI] [PubMed] [Google Scholar]
- 72.West D, Sattar A, Kumar S: A simplified in situ solubilization procedure for the determination of DNA, and cell number in tissue cultured mammalian cells. Anal Biochem 1985, 147:289-295 [DOI] [PubMed] [Google Scholar]
- 73.Ewart AK, Jin W, Atkinson D, Morris CA, Keating MT: Supravalvular aortic stenosis associated with a deletion disrupting the elastin gen. J Clin Invest 1994, 93:1071-1077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ewart AK, Morris CA, Atkinson D, Jin W, Sternes K, Spallone P, Stock AD, Leppert M, Keating MT: Hernizygosity at the elastin locus in a developmental disorder, Williams syndrome. Nat Genet 1993, 5:11-16 [DOI] [PubMed] [Google Scholar]
- 75.Curran ME, Atkinson DL, Ewart AK, Morris CA, Leppert MF, Keating MT: The elastin gene is disrupted by a translocation associated with supravalvular aortic stenosis. Cell 1993, 73:159-168 [DOI] [PubMed] [Google Scholar]
- 76.Tassabehji M, Metcalfe K, Donnai D, Hurst J, Reardon W, Burch M, Readm AP: Elastin: genomic structure and point mutations in patients with supravalvular aortic stenosis. Hum Mol Genet 1997, 6:1029-1036 [DOI] [PubMed] [Google Scholar]
- 77.Lee B, Godfrey M, Vitale E, Hori H, Mattei MG, Sarfarazi M, Tsipouras P, Ramirez F, Hollister DW: Linkage of Marfan syndrome and a phenotypically related disorder to two different fibrillin genes. Nature 1991, 352:330-334 [DOI] [PubMed] [Google Scholar]
- 78.Sandberg LB, Soskel NT, Leslie JG: Elastin structure, biosynthesis, and relation to disease states. N Engl J Med 1981, 304:566-579 [DOI] [PubMed] [Google Scholar]
- 79.Hinek A, Thyberg J, Friberg U: Heterotopic transplantation of isolated aortic cells, an electron microscopical study. Cell Tissue Res 1976, 172:59-79 [DOI] [PubMed] [Google Scholar]
- 80.Hinek A, Thyberg J: Electron microscopic observations on the formation of elastic fibers in primary cultures of aortic smooth muscle cells. J Ultrastruct Res 1977, 60:12-20 [DOI] [PubMed] [Google Scholar]
- 81.Hinek A: Biological functions of the non-integrin elastin/laminin receptor. Biol Chem 1996, 377:471-480 [PubMed] [Google Scholar]
- 82.Volpi N: Inhibition of human leukocyte elastase activity by chondroitin sulfates. Chem Biol Interact 1997, 105:157-167 [DOI] [PubMed] [Google Scholar]
- 83.Ying QL, Kemme M, Saunders D, Simon SR: Glycosaminoglycans regulate elastase inhibition by oxidized secretory leukoprotease inhibitor. Am J Physiol 1997, 272:533-541 [DOI] [PubMed] [Google Scholar]
- 84.McGowan SE, Liu R, Harvey CS: Effects of heparin and other glycosaminoglycans on elastin production by cultured neonatal rat lung fibroblasts. Arch Biochem Biophys 1993, 302:322-331 [DOI] [PubMed] [Google Scholar]
- 85.Hinek A, Boyle J, Rabinovitch M: Vascular smooth muscle cells detachment from elastin and migration through elastic laminae is promoted by chondroitin sulfate-induced shedding of the 67-kD cell surface elastin-binding protein. Exp Cell Res 1992, 203:344-353 [DOI] [PubMed] [Google Scholar]
- 86.Tao Z, Smart FW, Figueroa JE, Glancy DL, Vijayagopal P: Elevated expression of proteoglycans in proliferating vascular smooth muscle cells. Atherosclerosis 1997, 135:171-179 [DOI] [PubMed] [Google Scholar]
- 87.Wight TN, Lara S, Riessen R, Le Baron R, Isner J: Selective deposits of versican in the extracellular matrix of restenotic lesions from human peripheral arteries. Am J Pathol 1997, 151:963-973 [PMC free article] [PubMed] [Google Scholar]
- 88.Radhakrishnamurthy B, Tracy RE, Dalferes ER, Jr, Berenson GS: Proteoglycans in human coronary arteriosclerotic lesions. Exp Mol Pathol 1998, 65:1-8 [DOI] [PubMed] [Google Scholar]
- 89.Belting M, Havsmark B, Jonsson M, Persson S, Fransson LA: Heparan sulphate/heparin glycosaminoglycans with strong affinity for the growth-promoter spermine have high antiproliferative activity. Glycobiology 1996, 6:121-129 [DOI] [PubMed] [Google Scholar]
- 90.Bingley JA, Hayward IP, Campbell JH, Campbell GR: Arterial heparan sulfate proteoglycans inhibit vascular smooth muscle cell proliferation and phenotype change in vitro and neointimal formation in vivo. J Vasc Surg 1998, 28:308-318 [DOI] [PubMed] [Google Scholar]
- 91.Malmstrom J, Westergren-Thorsson G: Heparan sulfate upregulates platelet-derived growth factor receptors on human lung fibroblasts. Glycobiology 1998, 8:1149-1155 [DOI] [PubMed] [Google Scholar]
- 92.Foxall C, Wei Z, Schaefer ME, Casabonne M, Fugedi P, Peto C, Castellot JJ Jr, Brandley BK: Sulfated malto-oligosaccharides bind to basic FGF, inhibit endothelial cell proliferation, and disrupt endothelial cell tube formation. J Cell Physiol 1996 168:657–667 [DOI] [PubMed]
- 93.Li DY, Brooke B, Davis EC, Mecham RP, Sorensen LK, Boak BB, Eichwald E, Keating MT: Elastin is an essential determinant of arterial morphogenesis. Nature 1998, 393:276-280 [DOI] [PubMed] [Google Scholar]
- 94.Belknap JK, Weiser-Evans MC, Grieshaber SS, Majack RA, Stenmark KR: Relationship between perlecan and tropoelastin gene expression and cell replication in the developing rat pulmonary vasculature. Am J Respir Cell Mol Biol 1999, 20:24-34 [DOI] [PubMed] [Google Scholar]
- 95.Merle B, Malaval L, Lawler J, Delmas P, Clezardin P: Decorin inhibits cell attachment to thrombospondin-1 by binding to a KKTR-dependent cell adhesive site present within the N-terminal domain of thrombospondin-1. J Cell Biochem 1997, 67:75-83 [PubMed] [Google Scholar]
- 96.Jung S, Rutka JT, Hinek A: Tropoelastin and elastin degradation products induce proliferation of human astrocytoma cell lines. J Neuropathol Exp Neurol 1998, 57:429-448 [DOI] [PubMed] [Google Scholar]
- 97.Hinek A, Jung S, Rutka JT: Suramin causes cell surface aggregation of the elastin receptor molecules on human astrocytoma cells and amplifies their elastin-dependent proliferation. Acta Neuropathol 1999, 97:399-407 [DOI] [PubMed] [Google Scholar]
- 98.Penc SF, Pomahac B, Winkler T, Dorschner RA, Eriksson E, Herndon M, Gallo RL: Dermatan sulfate released after injury is a potent promoter of fibroblast growth factor-2 function. J Biol Chem 1998, 273:28116-28121 [DOI] [PubMed] [Google Scholar]
- 99.Lyon M, Deakin JA, Rahmoune H, Fernig DG, Nakamura T, Gallagher JT: Hepatocyte growth factor/scatter factor binds with high affinity to dermatan sulfate. J Biol Chem 1998, 273:271-278 [DOI] [PubMed] [Google Scholar]
- 100.Hurlel JM, Corson G, Daniels K, Reiter RS, Sakai LY, Solursh M: Elastin exhibits a distinctive temporal and spatial pattern of distribution in the developing chick limb in association with the establishment of the cartilaginous skeleton. J Cell Sci 1994, 107:2623-2634 [DOI] [PubMed] [Google Scholar]