

Abstract
Growth hormone (GH) modulates the hypothalamic release of somatostatin and GH-releasing hormone; however, there has been no evidence of GH autoregulation on the pituitary somatotroph. To determine the effects of GH on its own regulation, we examined the pituitaries of giant transgenic mice expressing a GH agonist (E117L), dwarf transgenic mice expressing a GH antagonist (G119K), and dwarf mice devoid of the GH receptor/binding protein (GHR/BP). In the E117L transgenic mice, the number and distribution of pituitary GH-immunoreactive cells were unchanged from nontransgenic littermate controls; an ultrastructural examination revealed typical, densely granulated somatotrophs. In contrast, the pituitaries of the G119K mice contained both moderately granulated somatotrophs and a sparsely granulated (SG) population with well-developed synthetic organelles and a distinct juxtanuclear globular GH-staining pattern. GHR/BP-deficient mice exhibited a marked reduction in the intensity of cytoplasmic GH immunoreactivity; however, prominent GH staining in the juxtanuclear Golgi was seen. GH-immunoreactive cells were increased in number, and the reticulin network pattern was distorted; stains for proliferating cell nuclear antigen confirmed mild hyperplasia. Electron microscopy showed that the somatotrophs were hyperactive SG cells with prominent endoplasmic reticulum membranes, large Golgi complexes, and numerous mitochondria. These findings are consistent with synthetic and secretory hyperactivity in pituitary somatotrophs due to the reduced GH feedback regulation. The changes are most striking in animals that are devoid of GHR/BP and less marked in animals expressing a GH antagonist; both models had reduced insulin-like growth factor-I levels, but the more dramatic change in the GHR/BP animals can be explained by abrogated GH signaling. This represents the first evidence of direct GH feedback inhibition on pituitary somatotrophs, which may have implications for the use of GH analogs in different clinical settings.
Growth hormone (GH) secretion is under the complex control of the hypothalamus with predominant stimulation by GH-releasing hormone (GHRH) and GH-related peptide (GHRP) and inhibition by somatostatin (SRIH). 1 These effects are modulated by peripheral negative feedback signals, including the target growth factor of GH insulin-like growth factor-I (IGF-I), certain amino acids and nutrient metabolites, and other hormones, including glucocorticoids, that act at the level of the adenohypophysis and the hypothalamus. 1 GH itself can alter its own regulation at the level of the hypothalamus, where it modulates the release of GHRH and SRIH. 1 Thus far, however, there has been little evidence for a more direct role for GH in the autoregulation of the pituitary somatotroph. 2
To determine whether GH participates in an autofeedback mechanism at the level of the pituitary somatotroph, we studied the pituitaries of giant transgenic mice expressing a GH agonist (E117L), dwarf transgenic mice expressing a GH antagonist (G119K), and dwarf mice that were devoid of the GH receptor/binding protein (GHR/BP).
Materials and Methods
Transgenic and GH Receptor-Deficient Mice
Production and characterization of transgenic mice expressing either bGH-E117L (GH agonist) or bGH-G119K (GH antagonist) genes have been described in detail. 3,4 The serum from the G119K mice contained approximately 2 μg/ml of the GH antagonist, whereas bGH levels in the E117L mice were approximately 0.55 μg/ml. The IGF-1 levels of the E117L animals were approximately 800 ng/ml, whereas the G119K animals possessed levels of approximately 150 μg/ml. The control animals had IGF-1 levels of 350 ng/ml. 5
The production of mice with a disrupted GHR/BP gene has been described. 6 The GHR/BP−/− mice were genotyped by polymerase chain reaction as reported. 7 The homozygous animals had decreased levels of IGF-1 and elevated serum GH concentrations.
Morphological Methods
The 5-month-old male mice of each group and equal numbers of the age-matched littermate controls were sacrificed by decapitation. At autopsy, the pituitaries were removed and weighed, and the other organs were carefully inspected, weighed, and measured.
For light microscopy, sections of the autopsied tissues were fixed in buffered formalin and embedded in paraffin; 4- to 5-μm-thick sections were stained with hematoxylin and eosin. The pituitaries were also stained with the Gordon-Sweet silver method to demonstrate the reticulin fiber network.
Immunocytochemical stains to localize adenohypophysial hormones were performed using the streptavidin-biotin-peroxidase complex technique. Primary polyclonal antisera directed against rat pituitary hormones were used at the specified dilutions: GH, 1:2500; prolactin, 1:2500; β-thyroid-stimulating hormone (β-TSH), 1:3000; β-follicle-stimulating hormone (β-FSH), 1:600; β-luteinizing hormone (β-LH), 1:2500 (National Hormone and Pituitary Program, Rockville, MD); and adrenocorticotropin prediluted preparation, which was further diluted 1:20 (Dako, Carpinteria, CA). To evaluate cell proliferation, a monoclonal antibody directed against proliferating cell nuclear antigen (PCNA) from Novocastra (Newcastle-On-Tyne, UK) was applied at 1:4000. The entire adenohypophysial area of the immunostained sections was analyzed, and the percentage of immunoreactive cells was determined for each hormone and for PCNA staining.
Double staining was performed to colocalize PCNA and the various pituitary hormones as described previously. 8 For these stains, the cytoplasmic hormone reactivity identified by polyclonal antisera was detected with streptavidin-biotin-peroxidase (Autoprobe III detection system; Biomeda, Foster City, CA) and visualized with the chromogen NovaRED Substrate (Vector Laboratories, Burlingame, CA). The nuclear-PCNA reactivity identified by the monoclonal antibody was localized with the Vectastain elite detection system (Vector Laboratories) and revealed using cobalt 3,3′-diaminobenzidine.
For electron microscopy, small pieces of adenohypophysial tissue were fixed in 2.5% glutaraldehyde, postfixed in 1% osmium tetroxide, dehydrated in graded ethanols, processed through propylene oxide, and embedded in epoxy resin. Semithin sections were stained with toluidine blue, and appropriate areas were selected for the fine structural study. Ultrathin sections were stained with uranyl acetate and lead citrate and investigated with a Philips CM 100 electron microscope.
Results
Gross Appearance
The transgenic mice expressing the E117L agonist were large and resembled animals that overexpress wild-type GH, as described previously. 3 The animals weighed 150% of controls. The pituitaries were proportionately larger than those of the controls (145% of control) but anatomically normal.
The transgenic mice expressing the G119K antagonist exhibited visible somatic-growth suppression (weights 60% of controls) as previously documented. 9 These pituitaries were proportionately smaller than those of the controls (58% of controls) but again were anatomically normal for the size of the animal.
The mice that were deficient in the GH-receptor also exhibited visible somatic-growth suppression (weights 45% of controls) as previously documented. 6,9 Their pituitaries were smaller than those of the controls (58% of controls), but in this model the reduction of the pituitary was less than that of the overall weight.
The physiological features of these mice, including size, GH, IGF-1, and GHR/BP levels, are detailed in Table 1 ▶ .
Table 1.
Physiological Features of Animals Studied
| GH Agonist-Transgenic Mice | GH-Antagonist Transgenic Mice | GHR/BP−/− Mice |
|---|---|---|
| Giant phenotype; body weight 150% of control | Proportionate dwarf phenotype; body weight 60% of control | Proportionate dwarf phenotype; body weight 45% of control |
| Pituitary weight 145% of control | Pituitary weight 58% of control | Pituitary weight 58% of control |
| GH ↑ (agonist bGH) | GH ↑ (antagonist bGH) | GH ↑ (endogenous 30× control)6 |
| IGF-1 ↑ (226% of control)5 | IGF-1 ↓ (42% of control)5 | IGF-1 ↓ (10% of control)6 |
Morphological Findings
In the E117L transgenic mice, all organs were proportionately large but exhibited a relatively normal morphology. In the adenohypophysis, the architecture was normal with an intact reticulin-fiber network delineating acini of a normal and consistent size (Figure 1a) ▶ . The distribution of pituitary GH-immunoreactive cells (64%–68%) was unchanged from the controls (62%–69%). GH immunoreactivity was found as diffuse cytoplasmic staining in the somatotrophs of the controls (Figure 2a) ▶ and the transgenic mice (Figure 2b) ▶ . Electron microscopy demonstrated the presence of somatotrophs with relatively scant profiles of the rough endoplasmic reticulum, small Golgi complexes, and moderate numbers of large, round, evenly electron-dense secretory granules (Figure 3a) ▶ . Lactotrophs, corticotrophs, thyrotrophs, and gonadotrophs were all identified in normal proportions: lactotrophs, 15% to 20%; corticotrophs, 5% to 7%; thyrotrophs, approximately 3%; and gonadotrophs, 10% to 15% of the adenohypophysial cell population. These cells exhibited normal morphology at the light and electron microscopic level (not shown). In general, gonadotrophs were relatively inactive.
Figure 1.
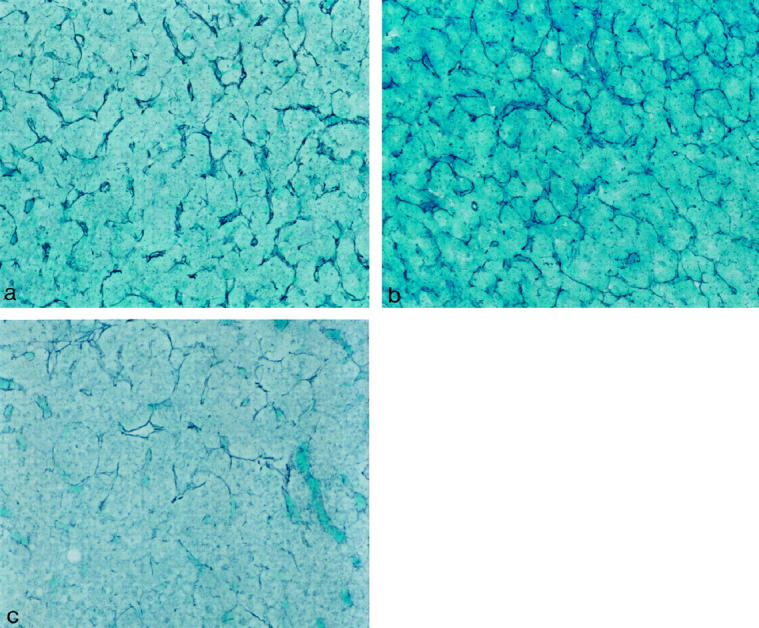
Histology of pituitaries of GH agonist (E117L) mice, GH antagonist (G119K) mice, and GH receptor-deficient mice. a: In the pituitary of a GH agonist (E117L) mouse, the reticulin stain shows an intact pattern of pituitary acinar structure. There is no evidence of hyperplasia or adenoma. b: In the pituitary of a GH antagonist (G119K) mouse, the reticulin stain shows intact pituitary acinar structure. There is no evidence of hyperplasia or adenoma. c: In the pituitary of a GH receptor-deficient mouse, the reticulin stain shows enlargement of pituitary acini with focal loss of reticulin. This is consistent with mild hyperplasia. Gordon Sweet silver stain; original magnification, ×100.
Figure 2.
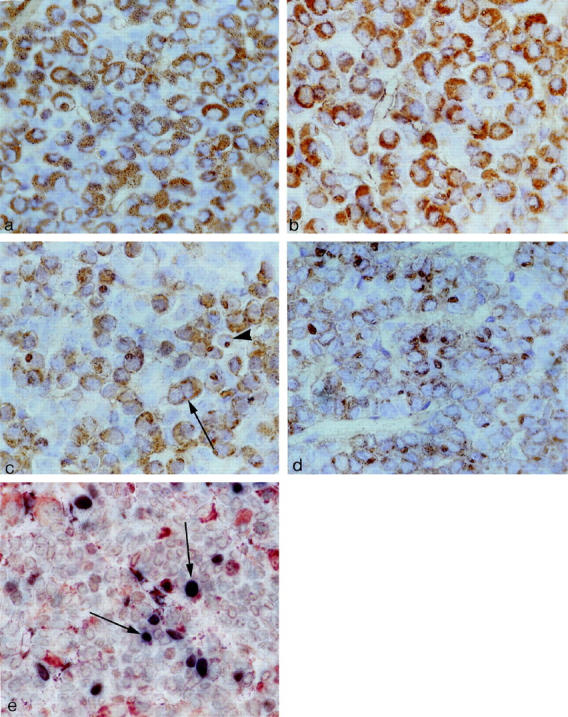
Immunohistochemistry for GH in pituitaries of GH agonist (E117L) mice, GH antagonist (G119K) mice, and GH-receptor-deficient mice. a: Somatotrophs in a control animal are numerous and have homogeneous strong, diffuse cytoplasmic positivity for GH. b: In the pituitary of a GH agonist (E117L) mouse, GH immunoreactivity is identified in somatotrophs that are normal in number and distribution. c: In the pituitary of a GH antagonist (G119K) mouse, GH immunoreactivity is identified in somatotrophs that have diffuse cytoplasmic positivity (arrow), but there are also cells that have a juxtanuclear focal globular-staining pattern consistent with reactivity in the Golgi region of SG cells (arrowhead). d: In the pituitary of a GH receptor-deficient mouse, GH immunoreactivity is identified in numerous somatotrophs, but the intensity of reactivity is weak, and most cells exhibit a prominent juxtanuclear globular staining pattern, consistent with reactivity in the Golgi region of SG cells. Avidin-biotin-peroxidase complex technique; original magnification, ×250. e: Double immunostaining for GH and PCNA identifies scattered cells that contain both markers (arrows), indicating proliferation of somatotrophs; the majority of PCNA-positive cells exhibited no cytoplasmic reactivity for pituitary hormones. Double immunostaining technique; original magnification, ×250.
Figure 3.

Ultrastructure of somatotrophs in pituitaries of GH agonist (E117L) mice, GH antagonist (G119K) mice, and GH receptor-deficient mice. a: In the pituitary of a GH agonist (E117L) mouse, electron microscopy identifies somatotrophs that have moderate numbers of dense secretory granules. The endoplasmic reticulum (arrow) is scant and Golgi complexes (G) are not conspicuous. b: In the pituitary of a GH antagonist (G119K) mouse, electron microscopy identifies densely granulated somatotrophs with numerous secretory granules throughout the cytoplasm (periphery of photograph), but the large central cell has a different morphology. This cell has very well-developed rough endoplasmic reticulum (arrow), and the secretory granules are accumulated within the large juxtanuclear Golgi complex (G) that displaces and disfigures the nucleus. c: In the pituitary of a GH receptor-deficient mouse, the ultrastructural features of somatotrophs are highly atypical; the SG cells have prominent short scattered rough endoplasmic reticulum profiles and a large juxtanuclear Golgi complex (G) but few secretory granules. Original magnifications, ×4000.
The G119K transgenic mice had proportionately small organs; however, their microscopic appearance was not detectably different from the controls with the exception of the adenohypophysis. The histological appearance of the gland was not different from that of the normal control animals, and the reticulin pattern indicated preservation of the acinar structure (Figure 1b) ▶ . The pituitaries contained immunoreactive GH cells that were not detectably increased in number (65–68% of cells) and were normally distributed, but they exhibited unusual immunohistochemical features. Unlike the control tissues with numerous diffusely and strongly immunoreactive, densely granulated cells (cf Figure 2a ▶ ), the somatotrophs comprised a mixed population of diffusely immunoreactive, moderately granulated cells and sparsely granulated (SG) cells with a distinctly juxtanuclear globular-staining pattern for GH (Figure 2c) ▶ . Electron microscopy confirmed the presence of moderately granulated somatotrophs with well-developed endoplasmic reticulum and large Golgi regions, as well as an SG population with well-developed synthetic organelles and a distinct juxtanuclear globular Golgi complex (Figure 3b) ▶ . Other adenohypophysial cell types were present in normal numbers (as above) and exhibited a normal ultrastructural morphology.
The GHR/BP-deficient mice exhibited an obvious dwarf phenotype not different from the G119K transgenic mice; they also had smaller organs but with no discrete pathology. In the pituitaries of these animals that lack the GH receptor binding, there was obvious disruption of the reticulin-fiber network with expansion of acini with loss of reticulin (Figure 1c) ▶ ; this change was diffuse throughout the gland, and no focal lesion was identified. There was an increase in the proportion of GH-immunoreactive cells (76% to 79%, compared with controls, 61% to 67%) and a marked reduction in the intensity of cytoplasmic GH immunoreactivity in individual cells; the numerous somatotrophs exhibited prominent, juxtanuclear Golgi positivity (Figure 2d) ▶ . The immunoreactivity for other pituitary hormones was retained in a normal distribution, but the percentages of the immunoreactive cells for each hormone were proportionately reduced: lactotrophs, 10% to 15%; corticotrophs, 3% to 4%; thyrotrophs, approximately 2%; and gonadotrophs, 6% to 8% of the adenohypophysial cell population. The gonadotrophs were characterized by weak hormonal reactivity. These features were consistent with a subtle and diffuse hyperplasia of the hyperactive somatotrophs. By electron microscopy, typical somatotrophs were not recognized; instead, there was a prominent population of hyperactive SG cells with prominent endoplasmic reticulum membranes, large Golgi complexes, and numerous mitochondria (Figure 3c) ▶ . Other adenohypophysial cell types exhibited normal ultrastructural morphology, but gonadotrophs were relatively inactive.
In the pituitaries of the control animals and the G119K animals, PCNA labeled 3% to 6% of the adenohypophysial cells. In the pituitaries of the E117L animals, PCNA labeled 5% to 9% of the adenohypophysial cells, and, in the GHR/BP-deficient mice, there was an increased PCNA index from 10% to 14% of the adenohypophysial cells, consistent with mild hyperplasia. Colocalization studies identified PCNA-nuclear reactivity in scattered GH-positive cells (Figure 2e) ▶ , but the majority of PCNA-positive nuclei were found in cells that harbored no detectable hormone staining.
Discussion
The role of GH in regulating its own expression and secretion is governed by a multilevel system of endocrine, autocrine, and paracrine interactions between GH and its putative regulators. These include central factors such as GHRH, GHRP, and SRIH, as well as peripheral factors, including IGF-I and GH itself. GHRH and GHRP favor GH secretion, whereas SRIH inhibits GH secretion as is evidenced by in vitro and in vivo studies. 1 In vivo GH administration reduces GH responsiveness to GHRH stimulation in humans. 10 This rapid inhibitory effect occurs prior to a corresponding rise in plasma IGF-I. These findings have been cited as evidence that reduces the likelihood that IGF-1 is responsible for the feedback observed. 11 The role of central GHRH, SRIF, and GH is further complicated by the reciprocal interactions between the GHRH and SRIF regulation within the hypothalamus. GHRH and SRIF expression are closely coupled with evidence that SRIF neurons from the periventricular nuclei synapse on GHRH neurons in the arcuate nucleus. 12 These findings are suggestive of hypothalamic control of an inverse relationship between GHRH and SRIF. Moreover, GH appears to also suppress the hypothalamic expression of the GH receptor. 13 The intracerebroventricular administration of an antisense GH receptor oligonucleotide augments GH secretion while reducing the hypothalamic SRIF expression in the rat. 14 These findings suggest a role for the GH receptor, as well as SRIF, in mediating the inhibition of GH-induced negative feedback. Additionally, GH has been suggested to decrease GHRP receptor expression, 15 further adding to the central indirect mechanisms by which GH can modulate itself. Peripheral negative feedback inhibition by IGF-I on the somatotroph has been well characterized. The in vivo as well as in vitro administration of IGF-I has been shown to reduce GH secretion and gene transcription. 1
The third α-helix of GH is a critical domain involved in GH receptor signaling and biological properties. 4,9,16,17 The substitution of Gly-119 with a variety of amino acids results in GH antagonists, ie, GH analogs, that inhibit GH-receptor signaling systems. 3 Indeed, we have shown that animals transgenic for this substitution are dwarfs 3 and that these animals demonstrate diminished liver and kidney GH-binding activity as well as circulating plasma IGF-I. Thus, this or similar analogs may represent a new pharmacologic approach to the manipulation of the GH/IGF-I axis in various medical conditions. For example, acromegaly is a disease that is characterized by GH and IGF-I excess due to a somatotroph adenoma in which peripheral GH antagonism represents a potential targeted treatment strategy. 18 However, in such patients, the loss of negative feedback inhibition due to the GH antagonists may, in concert with the associated reduction in IGF-I, be envisioned to result in somatotroph hyperplasia and pituitary enlargement.
Pituitary somatotrophs are a remarkably stable cell population in the adenohypophysis. Their number, immunoreactivity, and ultrastructure remain unchanged in various pathological conditions. They are characteristically densely granulated cells with large homogeneous secretory granules. Whereas there may be an increase in the amount of rough endoplasmic reticulum and a prominence of the Golgi region in situations of chronic hyperstimulation by GHRH, 19-21 the functionally suppressed somatotroph cannot be distinguished from a somatotroph that is actively synthesizing and secreting hormone in the normal state. This is evident in the somatotrophs of patients with idiopathic GH deficiency; their somatotrophs are clearly in a resting state and can be stimulated by GHRH, 22 but they are morphologically indistinguishable from normal somatotrophs. 23 It is also true of adenomatous somatotrophs that are exposed to somatostatin analogs. 24 The only morphologically altered somatotroph is the SG variant, a cell type that has been identified exclusively in human pituitary adenomas 25 and as a transient phenomenon in the human fetal pituitary during midgestation, when circulating GH levels reach acromegalic proportions. 26 These SG somatotrophs are characterized by a significant reduction in the number and size of secretory granules, a large Golgi complex, and a unique collection of keratin filaments that form a juxtanuclear whorl, the so-called “fibrous body.” The pathogenesis of this cellular alteration has been unclear, because the administration of GHRH is unable to cause a transition of densely-granulated somatotrophs to this phenotype, 21 and somatostatin is unable to reverse the morphology even when GH release is suppressed. 24
Our data indicate that, at least in the mouse model, marked somatotroph hyperplasia and neoplasia are not significant features of GH antagonism because there is no gross enlargement of the pituitaries in these animals. In GHR/BP-deficient animals, reticulin staining, GH, and PCNA labeling provided evidence of mild somatotroph hyperplasia. The lack of somatotroph hyperplasia in GH-antagonist mice and the relatively subtle changes in GHR/BP-deficient animals contrast with the striking proliferative effect that GHRH stimulation has on these cells. 20,27,28 The subcellular changes, however, indicate that endogenous intracellular GH production may be accelerated in the setting of reduced GH signaling. Well-developed rough endoplasmic reticulum and large juxtanuclear Golgi complexes in SG cells are found in GH antagonist-transgenic mice and moreso in GH receptor-deficient mice; these morphological characteristics resemble the features of stimulated lactotrophs in rodents and humans and of SG somatotroph adenomas in humans. The lack of keratin filaments is the only difference between these stimulated somatotrophs and the cells of SG somatotroph adenomas. This is not a surprising or unexpected difference because the accumulation of keratin filaments in adenohypophysial cells is known to be a species-specific phenomenon; Crooke’s hyalinization in glucocorticoid-suppressed corticotrophs is unique to humans and is not seen in rodent models of glucocorticoid excess.
The SG morphology of somatotrophs is most striking and there is acinar enlargement of the reticulin-fiber network of the gland in the animals that lack GHR/BP, where the GH feedback to the somatotroph would be totally reduced. Because both types of dwarf mice examined in this study have reduced IGF-1 levels, the differences between the somatotrophs in the two models can be explained by direct GH autoregulation that would be completely absent in the GHR/BP-deficient mice. A role for IGF-1 and/or GHRH as mediators of this phenomenon cannot be excluded.
The reverse does not appear to lead to major alterations in the somatotroph. The expression of the potent GH agonist E117L did not alter the subcellular morphology, and there was no evidence of involution or reduction of somatotrophs. This lack of evidence of feedback suppression is consistent with the lack of morphological evidence of involution in the nontumorous adenohypophysis of patients with acromegaly. It seems that this cell type provides morphological evidence of hormonal activity only in a hyperstimulated situation.
In this report we have demonstrated synthetic and secretory hyperactivity in pituitary somatotrophs due to reduced GH feedback regulation. These changes are most striking in animals devoid of GHR/BP, in which there is even mild hyperplasia, and these changes are less marked in animals expressing a GH antagonist. Our findings represent the first evidence of direct GH feedback inhibition on pituitary somatotrophs, which may have implications for the use of GH analogs in different clinical settings.
Acknowledgments
The authors acknowledge the technical assistance of Mr. Kelvin So and Ms. Jackie Pittman.
Footnotes
Address reprint requests to Dr. Sylvia L. Asa, Department of Pathology and Laboratory Medicine, Mount Sinai Hospital, 600 University Avenue, Toronto, Ontario, Canada M5G 1X5. E-mail: sasa@mtsinai.on.ca.
References
- 1.Giustina A, Veldhuis JD: Pathophysiology of the neuroregulation of growth hormone secretion in experimental animals and the human. Endocr Rev 1998, 19:717-797 [DOI] [PubMed] [Google Scholar]
- 2.Kraicer J, Lussier B, Moor BC, Cowan JS: Failure of growth hormone (GH) to feed back at the level of the pituitary to alter the response of the somatotrophs to GH-releasing factor. Endocrinology 1988, 122:1511-1514 [DOI] [PubMed] [Google Scholar]
- 3.Chen WY, Wight DC, Mehta BV, Wagner TE, Kopchick JJ: Glycine 119 of bovine growth hormone is critical for growth-promoting activity. Mol Endocrinol 1991, 5:1845-1852 [DOI] [PubMed] [Google Scholar]
- 4.Chen WY, White ME, Wagner TE, Kopchick JJ: Functional antagonism between endogenous mouse growth hormone (GH) and a GH analog results in dwarf transgenic mice. Endocrinology 1991, 129:1402-1408 [DOI] [PubMed] [Google Scholar]
- 5.Chen NY, Chen WY, Striker GE, Kopchick JJ: Co-expression of bovine growth hormone (GH) and human GH antagonist genes in transgenic mice. Endocrinology 1997, 138:851-854 [DOI] [PubMed] [Google Scholar]
- 6.Zhou Y, Maheshwari HG, He L, Reed M, Lozykowski M, Okada S, Cataldo L, Coschigano K, Wagner TE, Baumann G, Kopchick JJ: A mammalian model for Laron syndrome produced by targeted disruption of the mouse growth hormone receptor/binding protein gene (the Laron mouse). Proc Natl Acad Sci USA 1997, 94:13215-13220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chandrashekar V, Bartke A, Coschigano KT, Kopchick JJ: Pituitary and testicular function in growth hormone receptor gene knockout mice. Endocrinology 1999, 140:1082-1088 [DOI] [PubMed] [Google Scholar]
- 8.Puy LA, Asa SL: The ontogeny of pit-1 expression in the human fetal pituitary gland. Neuroendocrinology 1996, 63:349-355 [DOI] [PubMed] [Google Scholar]
- 9.Chen WY, Chen N-Y, Yun J, Wagner TE, Kopchick JJ: In vitro and in vivo studies of antagonistic effects of human growth hormone analogs. J Biol Chem 1994, 269:15892-15897 [PubMed] [Google Scholar]
- 10.Nakamoto JM, Gertner JM, Press CM, Hintz RL, Rosenfeld RG, Genel M: Suppression of the growth hormone (GH) response to clonidine and GH-releasing hormone by exogenous GH. J Clin Endocrinol Metab 1986, 62:822-826 [DOI] [PubMed] [Google Scholar]
- 11.Ross RJ, Borges F, Grossman A: Growth hormone pretreatment in man blocks the response to growth hormone-releasing hormone: evidence for a direct effect of growth hormone. Clin Endocrinol 1987, 26:117-123 [DOI] [PubMed] [Google Scholar]
- 12.Liposits ZS, Merchenthaler I, Paull WK, Flerko S: Synaptic communication between somatostatinergic axons and growth hormone-releasing factor (GRF) synthesizing neurons in the arcuate nucleus of the rat. Histochemistry 1988, 89:247-252 [DOI] [PubMed] [Google Scholar]
- 13.Bennett PA, Levy A, Sophokleous S, Robinson IC, Lightman SL: Hypothalamic GH receptor gene expression in the rat: effects of altered GH status. J Endocrinol 1995, 147:225-234 [DOI] [PubMed] [Google Scholar]
- 14.Pellegrini E, Bluet-Pajot MT, Mounier F, Bennett P, Kordon C, Epelbaum J: Central administration of a growth hormone (GH) receptor mRNA antisense increases GH pulsatility and decreases hypothalamic somatostatin expression in rats. J Neurosci 1996, 16:8140-8148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bennett PA, Thomas GB, Howard AD, Feighner SD, van den Ploeg LH, Smith RG, Robinson IC: Hypothalamic growth hormone secretagogue-receptor (GHS-R) expression is regulated by growth hormone in the rat. Endocrinology 1997, 138:4552-4557 [DOI] [PubMed] [Google Scholar]
- 16.Chen WY, Wight DC, Wagner TE, Kopchick JJ: Expression of a mutated bovine growth hormone gene suppresses growth of transgenic mice. Proc Natl Acad Sci USA 1990, 87:5061-5065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen WY, Wight DC, Chen NY, Coleman TA, Wagner TE, Kopchick JJ: Mutations in the third α-helix of bovine growth hormone dramatically affect its intracellular distribution in vitro and growth enhancement in transgenic mice. J Biol Chem 1991, 266:2252-2258 [PubMed] [Google Scholar]
- 18.Rodvold KA, van der Lely AJ: Pharmacokinetics and pharmacodynamics of B2036-PEG, a novel growth hormone receptor antagonist in acromegalic subjects. Endocrine Society 81st Annual Meeting 1999, (abstract). Washington, DC, Endocrine Society of America, 1999, p 145
- 19.Thorner MO, Perryman RL, Cronin MJ, Rogol AD, Draznin M, Johanson A, Vale W, Horvath E, Kovacs K: Somatotroph hyperplasia: successful treatment of acromegaly by removal of a pancreatic islet tumor secreting a growth hormone-releasing factor. J Clin Invest 1982, 70:965-977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sano T, Asa SL, Kovacs K: Growth hormone-releasing hormone-producing tumors: clinical, biochemical, and morphological manifestations. Endocr Rev 1988, 9:357-373 [DOI] [PubMed] [Google Scholar]
- 21.Kawakita S, Asa SL, Kovacs K: Effects of growth hormone-releasing hormone (GHRH) on densely granulated somatotroph adenomas and sparsely granulated somatotroph adenomas in vitro: a morphological and functional investigation. J Endocrinol Invest 1989, 12:443-448 [DOI] [PubMed] [Google Scholar]
- 22.Borges JLC, Blizzard RM, Evans WS, Furlanetto R, Rogol AD, Kaiser DL, Rivier J, Vale W, Thorner MO: Stimulation of growth hormone (GH) and somatomedin C in idiopathic GH-deficient subjects by intermittent pulsatile administration of synthetic human pancreatic tumor GH-releasing factor. J Clin Endocrinol Metab 1984, 59:1-6 [DOI] [PubMed] [Google Scholar]
- 23.Schechter J, Kovacs K, Rimoin D: Isolated growth hormone deficiency: immunocytochemistry. J Clin Endocrinol Metab 1984, 59:798-800 [DOI] [PubMed] [Google Scholar]
- 24.Asa SL, Felix I, Kovacs K, Ramyar L: Effects of somatostatin on somatotroph adenomas of the human pituitary: an in vitro functional and morphological study. Endocr Pathol 1990, 1:228-235 [DOI] [PubMed] [Google Scholar]
- 25.Asa SL: Tumors of the Pituitary Gland. Washington, DC, Armed Forces Institute of Pathology, 1998
- 26.Asa SL, Kovacs K, Horvath E, Losinski NE, Laszlo FA, Domokos I, Halliday WC: Human fetal adenohypophysis: electron microscopic and ultrastructural immunocytochemical analysis. Neuroendocrinology 1988, 48:423-431 [DOI] [PubMed] [Google Scholar]
- 27.Stefaneanu L, Kovacs K, Horvath E, Asa SL, Losinski NE, Billestrup N, Price J, Vale W: Adenohypophysial changes in mice transgenic for human growth hormone-releasing factor: a histological, immunocytochemical, and electron microscopic investigation. Endocrinology 1989, 125:2710-2718 [DOI] [PubMed] [Google Scholar]
- 28.Asa SL, Kovacs K, Stefaneanu L, Horvath E, Billestrup N, Gonzalez-Manchon C, Vale W: Pituitary adenomas in mice transgenic for growth hormone-releasing hormone. Endocrinology 1992, 131:2083-2089 [DOI] [PubMed] [Google Scholar]