

Abstract
Mice deficient in the nuclear factor κB (NF-κB)-transactivating gene RelA (p65) die at embryonic days 14–15 with massive liver apoptosis. In the adult liver, activation of the NF-κB heterodimer RelA/p50 can cause hepatocyte proliferation, apoptosis, or the induction of acute-phase response genes. We examined, during wild-type fetal liver development, the expression of the Rel family member proteins, as well as other proteins known to be important for NF-κB activation. We found these proteins and active NF-κB complexes in the developing liver from at least 2 days before the onset of lethality observed in RelA knockouts. This suggests that the timing of NF-κB activation is not related to the timing of lethality. We therefore hypothesized that, in the absence of RelA, embryos were sensitized to tumor necrosis factor (TNF) receptor 1 (TNFR-1)-mediated apoptosis. Thus, we generated mice that were deficient in both RelA and TNFR-1 to determine whether apoptotic signaling through TNFR-1 was responsible for the lethal phenotype. RelA/TNFR-1 double knockout mice survived embryonic development and were born with normal livers without evidence of increased hepatocyte apoptosis. These animals became runted shortly after birth and survived an average of 10 days, dying from acute hepatitis with an extensive hepatic infiltration of immature neutrophils. We conclude that neither RelA nor TNFR-1 is required for liver development and that RelA protects the embryonic liver from TNFR-1-mediated apoptotic signals. However, the absence of both TNFR-1 signaling and RelA activity in newborn mice makes these animals susceptible to endogenous hepatic infection.
The NF-κB transcription factor modulates gene expression in many cellular responses, including inflammation, apoptosis, and liver regeneration. 1-3 The NF-κB transactivating subunit, RelA (p65), plays a critical role in mouse liver development because RelA−/− mice die during mid-gestation from massive hepatocyte apoptosis. 4,5 Whereas much work has been done establishing the roles of NF-κB/Rel proteins in adult tissues, (for recent review, see 6 ), little is known about their functions in the developing animal. Except for RelA−/− mice, other Rel knockouts have not demonstrated critical roles for Rel proteins during development. 7-9 Schmidt-Ullrich et al generated transgenic mice expressing a NF-κB-driven LacZ reporter gene construct to provide information on the timing and cellular location of NF-κB activation during murine development. NF-κB activity was detected late in development, with no evidence of LacZ-positive cells until E12.5. Furthermore, NF-κB transactivation was predominantly noted within the developing brain, spinal medulla, and thymus of their transgenic lines. Only one of their transgenic lines showed any expression in the fetal liver, starting at E13. 10 Thus, the finding by Beg et al that RelA−/− embryos die of massive liver apoptosis is particularly intriguing because of the lack of demonstration of NF-κB activation during mouse liver development.
The tumor necrosis factor (TNF) rapidly and potently activates NF-κB in a variety of cell types, including adult hepatocytes. TNF mediates its cellular responses by signaling through two receptors, TNFR-1 and TNFR-2, but most of its biological functions are signaled through TNFR-1. 11,12 TNFR-1 is a death domain-containing receptor that, on ligand binding and trimerization, recruits the adapter protein TRADD. The signals elicited in response to TNF bifurcate at the level of TRADD binding. The overexpression of TRADD stimulates both apoptosis and NF-κB activation through differential binding domains. 13 The C-terminal portion of TRADD binds the death domain-containing protein FADD, resulting in apoptosis. 14 The N-terminal portion of TRADD binds the TNF receptor-associated factor 2 (TRAF-2), and other more recently identified proteins such as RIP, ultimately resulting in the activation of NF-κB. 15,16
NF-κB activation is initiated by its release from the cytoplasmic inhibitory proteins IκBα or IκBβ. 17 Two IκB kinases, IKK-1 and IKK-2, which are responsible for phosphorylating IκB proteins, are components of a large multisubunit complex. 18,19 Phosphorylation of IκB leads to its ubiquitination and subsequent degradation by the proteasome, resulting in nuclear translocation of NF-κB. 20,21 Recent work has shown that IKK-2 knockouts die during development of liver degeneration and apoptosis, similar to RelA knockouts. 22,23 This suggests that IKK-2 is directly responsible for activating NF-κB in the developing liver.
Work from this and other laboratories demonstrated that TNF is required for the initiation of liver regeneration in mice, and that its proliferative effect on hepatocytes is strictly dependent on signaling through TNFR-1. 3,24 Mice lacking TNFR-1 have high mortality during the early stages of liver regeneration and the surviving animals have a deficit in DNA replication. TNFR-1−/− mice fail to activate the transcription factors NF-κB and STAT3 at the start of liver regeneration. However, DNA synthesis can be restored in these animals by interleukin 6 (IL-6) injection, which also restores STAT3 but not NF-κB activation. 3 These and other experiments 25,26 show that, on one hand, TNF, signaling through TNFR-1 and downstream activation of NF-κB, IL-6, and STAT3, is an important proliferative agent for hepatocytes. On the other hand, direct blockage of NF-κB activation, both in vivo and in hepatocyte cultures, switches the biological effect of TNF from proliferative to apoptotic. 27-30 If, as in liver regeneration, TNFR-1−/− mice fail to activate NF-κB during liver development, it is surprising that these mice develop normally and show no obvious signs of hepatic apoptosis.
To examine whether TNFR-1-mediated signaling is responsible for the lethal phenotype of RelA knockouts, we generated mice deficient in both RelA and TNFR-1. We show that double-knockout mice survive embryonic development and that gestation in these animals is apparently unaffected because newborn mice have normal liver morphology with little detectable apoptosis. Thus, RelA−/− mice die from apoptotic signals mediated through TNFR-1 during liver development. RelA and p50, the components necessary for NF-κB activation, are present during fetal liver development in wild-type (WT) mice from at least E12, suggesting that the timing of NF-κB activation is not related to the timing of embryonic lethality in RelA−/− mice. Survival of the RelA/TNFR-1 double knockouts suggests that neither RelA nor TNFR-1 is critical for hepatic development in mice. However, newborn animals with these deficiencies become sensitive to infection and the neutrophilic invasion that cause damage to the liver and other organs, resulting in early postnatal mortality.
Materials and Methods
Animals
The C57BL/6J (WT) mice were obtained from Jackson Laboratories (Bar Harbor, ME).The RelA heterozygous breeder mice were obtained from Amer Beg (Columbia University, New York, NY), and the TNFR-1 knockout mice have been previously described. 3 To obtain fetal samples, timed matings were performed, and the presence of a vaginal plug the following morning was considered embryonic day 0 (E0). To generate mice deficient in both RelA and TNFR-1, RelA+/− females were crossed with TNFR-1−/− males producing double heterozygous RelA+/−/TNFR-1+/− offspring that were subsequently back crossed. The breeding cages from RelA+/−/TNFR-1+/− matings were examined daily, and the first day that pups were found in the cage was considered day 1 of postnatal life. The number of pups born to each female was recorded, and, within 24 hours, a small piece of tail was removed from each newborn for DNA analysis.
Animals were maintained in a specific pathogen-free facility under 12-hour dark/light cycles and given standard diet and water ad libitum. All animal work was in accordance with policies at the University of Washington.
Isolation of Murine Liver Samples
For isolation of fetal livers, pregnant mice were sacrificed between E12 and E19. Fetuses were dissected free of extra embryonic membranes and decidual tissue and placed in phosphate-buffered saline; livers were carefully removed under a dissecting microscope at 30×. For the isolation of neonatal livers, newborn pups were killed at 2, 4, 7, and 16 days of age, and livers were carefully dissected from the animal.
Polymerase Chain Reaction (PCR) Genotyping
For fetal samples, a portion of the brain was isolated from each sample. For newborns, a small piece of tail was removed. All samples were digested in GNTK buffer, (50 mmol/L KCl, 1.5 mmol/L MgCl2, 10 mmol/L Tris-HCl, pH 8.5, 0.01% gelatin, 0.45% Nonidet P-40, 0.45% Tween-20) supplemented with 100 μg/ml proteinase K at 55°C. Proteinase K was heat inactivated, and 1–3 μl of the extract was used in PCR. For RelA PCR, a three-primer reaction amplified a 120-bp fragment of the WT allele and a 160-bp fragment of the RelA mutant allele. For TNFR-1, a four-primer PCR reaction amplified a 120-bp fragment of the WT allele and a 155-bp fragment of the mutant allele. The PCR products were run on 2% agarose (SeaKem, FMC BioProducts, Rockland, ME) gels and visualized with a UVP gel documentation system (Upland, CA).
Protein Extractions
Whole-cell extracts from the fetal livers were isolated by resuspension in a tissue lysis buffer (20 mmol/L 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), pH 7.5, 2 mmol/L ethylene glycol bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA), 10% glycerol, 1% Tween-20, 1 mmol/L dithiothreitol, 0.5 mmol/L phenylmethyl sulfonyl fluoride, 0.5 mmol/L benzamidine-HCl, 10 μg/ml aprotinin, 10 μg/ml leupeptin, 10 μg/ml pepstatin A) and incubated on ice for 30 minutes. Extracts were cleared by centrifugation at 4°C at 16,000 × g for 15 minutes and stored at −80°C. Whole-cell extracts from the newborn-liver samples were homogenized in a glass homogenizer in 0.5–1 ml of the tissue lysis buffer before incubation on ice and centrifugation.
Nuclear extracts from the fetal liver were isolated in a manner similar to the methods described by Han and Brasier. 31 All buffers were supplemented with the following protease inhibitors before use: 240 μg/ml antipain, 2 μg/ml aprotinin, 0.01 mol/L benzamidine-HCl, 0.2 mmol/L dithiothreitol, 10 μg/ml leupeptin, 10 μg/ml pepstatin A, 0.5 mmol/L phenylmethyl sulfonyl fluoride, 0.15 μmol/L spermine, 0.5 μmol/L spermidine. In a brief description of the procedure, livers were resuspended in Buffer A (50 mmol/L HEPES, pH 7.4, 10 mmol/L KCl, 1 mmol/L ethylenediaminetetraacetic acid (EDTA), 1 mmol/L EGTA, 0.5% Nonidet P-40) and kept on ice for at least 10 minutes. The samples were cleared by centrifugation at 4°C at 4000 × g for 5 minutes. Pellets were resuspended in 200–500 μl of buffer B (1.7 mol/L sucrose, 50 mmol/L HEPES, pH 7.4, 10 mmol/L KCl, 1 mmol/L EDTA, 1 mmol/L EGTA) and centrifuged at 4°C at 16,000 × g for 30 minutes. Pellets were resuspended in buffer C (10% glycerol, 50 mmol/L HEPES, pH 7.4, 400 mmol/L KCl, 1 mmol/L EDTA, 1 mmol/L EGTA), incubated on ice for 30 minutes with frequent vortexing, and centrifuged at 4°C at 16,000 × g for 5 minutes. Extracts were stored at −80°C.
Nuclear extracts from newborn livers were isolated as previously described. 3
Western Blot
Protein was quantitated by the Bradford method (Bio-Rad, Hercules, CA) and 25 μg of protein per lane was resolved on a 10% sodium dodecyl sulfate-polyacrylamide gel. The gels were transferred to nitrocellulose membranes (Amersham, Piscataway, NJ) and blocked overnight at 4°C in Tris-buffered saline containing 0.1% Tween-20 and 5% nonfat milk. Antibodies were obtained from Santa Cruz Biotechnologies, Inc. (Santa Cruz, CA). RelA (#SC-372-G), IκBα (#SC-371), and IκBβ (#SC-945) were used at 1:2000 dilutions and TRAF-2 (#SC-876) was used at 1:1000 dilution. The membranes were incubated for one hour at room temperature, washed with Tris buffered saline-T, and bound with the appropriate horseradish peroxidase-conjugated secondary antibody; blots were washed and, subsequently, detected by ECL (Amersham).
Electrophoretic Mobility Shift Assay (EMSA)
Four μg of nuclear protein were preincubated at room temperature for 10 minutes in a binding buffer (20 mmol/L HEPES, pH 7.5, 60 mmol/L KCl, 5 mmol/L MgCl2, 0.2 mmol/L EDTA, 8% glycerol, 1 μg poly dI/dC, 1% Nonidet P-40, 0.1 mmol/L dithiothreitol, 0.1 mmol/L phenylmethyl sulfonyl fluoride). A [32P]-labeled probe (1 × 10 5 cpm) was added per reaction, and incubation was continued for an additional 30 minutes. Double-stranded probes were obtained from Santa Cruz Biotechnologies, Inc. The NF-κB binding sequence probe used is 5′ AGTTGAGGGGACTTTCCCAGGC 3′. The SIE binding sequence probe used for Stat3 is 5′ GTGCATTTCCCGTAAATCTTGTCTACA 3′. Reactions were electrophoresed through a 5% polyacrylamide, 1× Tris-glycine-EDTA gel. For binding specificity, an unlabeled probe was added to one reaction 30 minutes before the addition of the labeled probe. For supershifts, after the 30-minute incubation of the labeled probe, specific antibodies obtained from Santa Cruz Biotechnologies, Inc. (RelA #SC-372X, p50 #SC-114X, p52 #SC-298X, c-Rel #SC-70X, and Stat3 #SC-482X) were added to indicated samples for an additional 30 minutes. The gels were dried and exposed to film for the detection of bands.
Immunohistochemistry
Embryos isolated at each gestational stage were fixed overnight in 10% buffered-formalin (Fisher Scientific, Pittsburgh, PA) and processed for paraffin embedding. The sections were cut onto glass slides and processed for immunohistochemistry by using the VECTASTAIN ABC kit and protocol (Vector Laboratories, Burlingame, CA). Antibodies were used at 1:100 dilution, and the control reactions were performed with an antibody prebound to a control peptide (Santa Cruz) before incubation on the slide (data not shown). The RelA, IκBα and TRAF-2 antibodies were the same as those used in Western blots. TNF- #SC-1348 and TNFR-1 #SC-1069 were also used. Biotinylated secondary antibodies and VECTASTAIN ABC solution were used at 1:100 dilutions. Antigen detection was through diaminobenzidine tetrahydrochloride (Sigma, St. Louis, MO) staining. The slides were counterstained with hematoxylin (Sigma).
Histology
The samples from newborn pups were fixed overnight in 10% buffered formalin (Fischer Scientific) and processed for paraffin embedding. The sections were cut onto glass slides and processed for routine hematoxylin and eosin staining.
Results
Expression of RelA Protein in WT Fetal Livers Does Not Correlate with Timing of Embryonic Lethality in RelA−/− Mice
RelA−/− mice die within a specific time period during development (E14–E15). Therefore, we first investigated in WT (C57BL/6J) mice whether the appearance of RelA during liver development would coincide with the time of lethality observed in RelA knockouts. Whole cell protein samples were obtained from fetal livers of WT mice between E12 and E19. The top panel of Figure 1 ▶ demonstrates that RelA protein was present throughout liver development, from at least E12. These livers also contained the p50 subunit of NF-κB (data not shown), indicating that the components of the classic NF-κB heterodimer were present from a very early stage of liver formation.
Figure 1.

Protein expression in WT fetal liver development. Western blots using antibodies against RelA, IκBα, IκBβ, and TRAF-2 illustrate protein expression throughout liver development. Lanes 12–19: fetal liver protein from gestational days 12–19, respectively.
In many adult cell types, including hepatocytes, NF-κB is retained in an inactive complex with an inhibitory IκB protein. 17,32 Examination of IκBα and IκBβ proteins in WT fetal livers demonstrated expression from E12 through E19 (Figure 1 ▶ , middle panels). Because RelA, p50, IκBα, and IκBβ were all expressed in the fetal liver from the earliest time points analyzed, these data demonstrate that embryonic lethality in RelA−/− mice (E14–E15) does not correspond to a critical time when expression of these proteins becomes necessary in the fetal liver.
Proteins have been identified in the TNF-signaling pathway that are important for the activation of NF-κB. TRAF-2 is a TNFR-associated factor downstream of TNFR-1 that is important for NF-κB activation after TNF stimulation. 15 TRAF-2 is recruited to the TNFR-1 signaling pathway by the adapter molecule TRADD. 14 Both TRAF-2 (Figure 1 ▶ , bottom panel) and TRADD (data not shown) were expressed in the E12–E19 embryonic liver.
IκBα and IκBβ Protein Levels Are Altered during Fetal Liver Development of RelA Knockouts but Not TNFR-1 Knockouts
We next examined whether the expression of RelA, IκBα, IκBβ, TRAF-2, and TRADD was altered in fetal livers of RelA or TNFR-1 knockouts. We compared the expression of these proteins to WT mice at E12–E14 (Figure 2) ▶ . As expected, RelA was absent from RelA knockouts. In addition, the IκBα expression in the RelA−/− fetal liver was decreased, and IκBβ was undetectable. In contrast, levels of RelA, IκBα, and IκBβ in TNFR-1 knockouts were similar to those observed in WT mice. It should be noted that, in these samples, the level of the IκBβ expression is lower at E12 when compared with E13 and E14. These results suggest that, within the developing liver, NF-κB participates in the regulation of IκBα. On the other hand, there appears to be a strict dependence of the IκBβ protein on NF-κB because this protein was not detectable in RelA knockouts. All of these proteins were normally expressed in TNFR-1−/− fetal livers, indicating that TNFR-1 does not mediate the signal for the activation of these genes. The expression of TRAF-2 (Figure 2 ▶ , bottom panel) and TRADD (data not shown) was similar in WT mice and in RelA and TNFR-1 knockouts during these days of gestation. The evidence of TRADD and TRAF-2 proteins in the RelA−/− fetal liver also illustrates that not all proteins are aberrantly expressed in the developing liver of RelA knockouts.
Figure 2.

Protein expression in the fetal livers of WT mice, RelA knockouts, and TNFR-1 knockouts. Western blots were performed with RelA, IκBα, IκBβ, and TRAF-2 antibodies. Lanes indicate fetal liver protein from E12, E13 and E14 for WT fetal livers, RelA−/− fetal livers and TNFR-1−/− fetal livers. RelA−/− fetal livers illustrate absent RelA and IκBβ proteins, diminished IκBα protein, and unaltered TRAF-2 protein levels. WT and TNFR-1−/− fetal livers express all proteins.
WT and TNFR-1−/− Fetal Livers Have Active NF-κB and STAT3 during Development, whereas RelA−/− Fetal Livers Have Only Active STAT3
To determine whether the NF-κB proteins detected by Western blot were capable of binding DNA, indicative of an active protein complex, we performed EMSA using nuclear protein isolated from WT mice, TNFR-1 knockout, and RelA knockout fetal livers between 12 and 14 days gestation. The NF-κB complex formation occurred in WT (Figure 3A) ▶ and, surprisingly, also in TNFR-1−/− (Figure 3B) ▶ fetal livers. In these samples, as in adult liver, NF-κB was composed of RelA/p50 heterodimers (top band) and p50 homodimers (bottom band). Nuclear protein isolated from E12–E14 RelA−/− fetal livers only demonstrated p50 homodimer formation (Figure 3C) ▶ . This suggests that the lack of RelA is not compensated for by the formation of other NF-κB heterodimers. RelA and p50 specificity was determined by supershift analyses (shown in Figure 3, B and C ▶ ). These figures seem to indicate that, in TNFR-1−/− fetal livers, the amount of NF-κB binding decreases with age, whereas in WT samples the inverse is seen. However, this was not a consistent finding when multiple fetal liver samples were analyzed at these gestational ages. It is interesting that, during E12–E14 of fetal liver development NF-κB complexes are forming, because in the adult liver there is little to no basal NF-κB binding. Antibodies against other Rel proteins, such as c-Rel and p52 (Figure 3B) ▶ , did not cause band shifts in any samples, indicating that RelA/p50 heterodimers and p50 homodimers were the active forms of NF-κB during fetal liver development. In the RelA−/− samples, the entire band was shifted with anti-p50 antibody, indicating that other Rel proteins were not being used in the absence of RelA. We conclude from these data that the embryonic lethality in RelA−/− mice does not coincide with the timing of NF-κB activation in liver development. The RelA/p50 double knockouts 33 have a similar phenotype to that of RelA knockouts, but die earlier in development (E12). Thus, the presence of p50 homodimers might provide some protection against early lethality. The finding that TNFR-1 knockouts have active NF-κB during liver development is unexpected, as these mice fail to activate NF-κB in the regenerating liver. 3
Figure 3.

NF-κB complex formation in the fetal liver. A: Nuclear extract from WT fetal liver at E12, E13, and E14, demonstrating both RelA/p50 heterodimers (top band) and p50 homodimers (bottom band). B: Nuclear extract from TNFR-1−/− fetal livers at E12, E13, and E14, demonstrating both RelA/p50 heterodimers (top band) and p50 homodimers (bottom band). Supershift analysis was performed with E13 nuclear protein and anti-RelA antibody, anti-p50 antibody, anti-c-Rel antibody, anti-p52 antibody, or an irrelevant anti-STAT3 antibody, as indicated. Specificity of binding was determined by competition with 30-fold excess of unlabeled oligonucleotide binding sequence probe. C: Nuclear extract from RelA−/− fetal livers at E12, E13, and E14, demonstrating only p50 homodimers. Supershift analysis was performed with E13 nuclear extracts and anti-RelA antibody, which did not cause a supershift, and with anti-p50 antibody, which shifted the entire band, as indicated.
The STAT3−/− mice are embryonic lethal early in gestation (E6.5–E7.5), therefore, the role it may have in the liver or other organ development is not yet defined. 34 However, STAT3 activation is an important component in murine hepatocyte proliferation during liver regeneration as demonstrated in TNFR-1 and IL-6 knockouts. 3,25 These and other experiments suggest that STAT3 activation in the liver may depend on NF-κB through induction of IL-6. STAT3 EMSA, and supershift analyses were performed on WT, RelA knockout, and TNFR-1 knockout fetal livers from E12 through E14. Figure 4, A–C ▶ , indicates that WT, TNFR-1−/−, and RelA−/− fetal livers were capable of specifically binding STAT3. Band specificity was determined by a complete supershift with an anti-STAT3 antibody; no shift was obtained with antibodies against STAT1 or STAT5 (data not shown). Because STAT3 was active in RelA−/− mice during liver development, it appears that a functional NF-κB complex is not required for STAT3 activation in the embryonic liver. The TNFR-1 knockouts do not show a STAT3 deficit during liver development, in contrast to the findings in liver regeneration. 3
Figure 4.
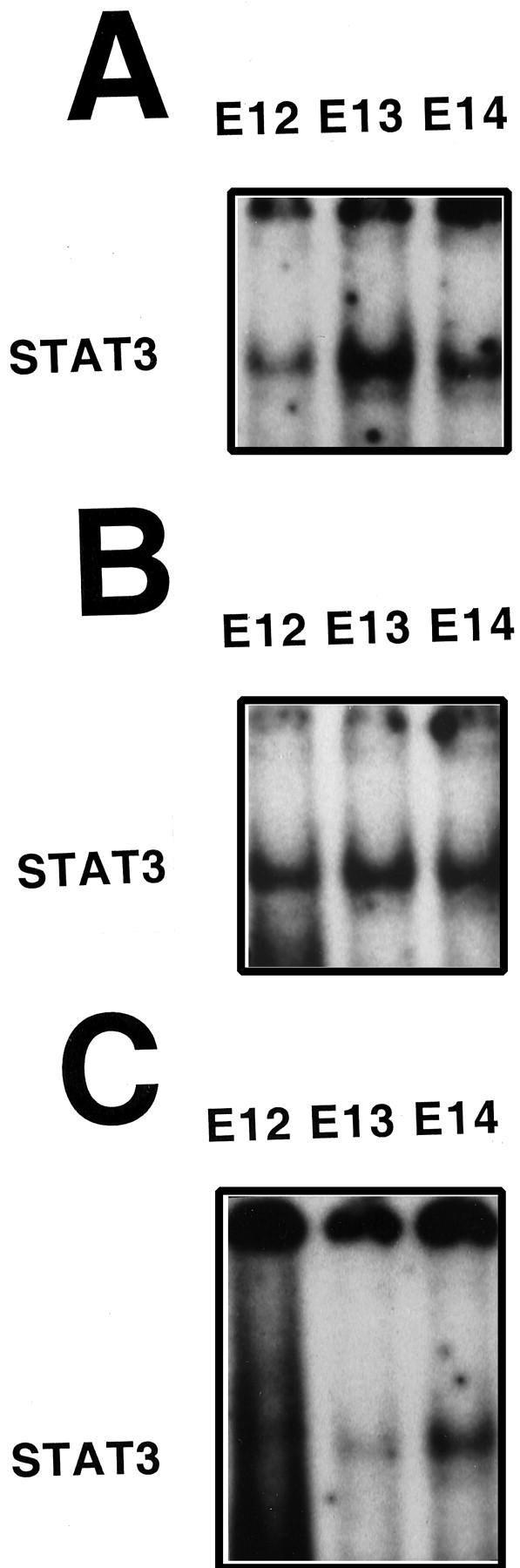
STAT3 binding in the fetal liver. Nuclear extracts were isolated from WT fetal livers (A), TNFR-1−/− fetal livers (B), and RelA−/− fetal livers (C) at E12, E13, and E14, demonstrating STAT3 binding at all time points in all animals examined.
Cell Type Expression of TNF and NF-κB Proteins Is Not Altered during Fetal Liver Development
The fetal liver contains diverse cell types, with hematopoietic cells, hepatoblasts, and endothelial cells composing the predominant types in mid-gestation. Whereas the expression of proteins and transcription factor complex assembly can be analyzed by Western blot and EMSA, respectively, they do not provide information regarding the localization of these proteins. To investigate this issue, we performed immunostaining for RelA, IκBα, TNFR-1, TRAF-2, and TNF-α in the E13 fetal liver of WT mice and RelA knockouts and the E14 TNFR-1 knockout fetal liver. At E13, RelA−/− mice have normal liver morphology similar to that of WT embryos. We found that the staining patterns observed in the fetal livers of WT mice, RelA knockouts, and TNFR-1 knockouts were remarkably similar for all the proteins tested. The results are summarized in Table 1 ▶ . In the WT mice and TNFR-1 knockouts, RelA and IκBα proteins were detected mostly in the cytoplasm of endothelial cells, hepatoblasts, and hematopoietic cells. Cytoplasmic expression of IκBα was also detected in RelA knockout fetal livers. In all instances, we saw occasional cells exhibiting nuclear staining of IκBα. The RelA−/− samples did have a decreased intensity of IκBα staining, likely due to diminished protein levels observed by Western blot (Figure 2) ▶ . Small hematopoietic cells were the primary sites of the TNFR-1 expression in the fetal livers of both the WT mice and RelA knockouts. Surprisingly, a minimal amount of nuclear staining with the TNFR-1 antibody was observed in the TNFR-1−/− fetal livers, whereas the WT and RelA−/− samples only demonstrated cytoplasmic and membrane-localized staining. However, the TNFR-1−/− mouse is the result of a functionally mutant TNFR-1 and not a completely absent gene; thus, we may be detecting a small amount of nonfunctional protein sequestered within the nucleus. In addition, the WT, RelA−/−, and TNFR-1−/− mice all had similar TNF and TRAF-2 staining patterns. These data show that the liver development of the RelA−/− and TNFR-1−/− mice is similar to that of the WT mice, at least through 13 days of gestation. After this time point, the RelA−/− mice undergo rapid liver degeneration and die, whereas the TNFR-1−/− mice proceed through gestation in a manner similar to the WT mice. Furthermore, these findings indicate that TNF and TNFR-1 are expressed during gestation in the fetal liver before the time of embryonic lethality seen in the RelA−/− mice.
Table 1.
Cell Type Expression of TNF and NF-κB Proteins during Fetal Liver Development
| Cell types | Antibodies | ||||
|---|---|---|---|---|---|
| RelA* | IκBα† | TNF | TNFR-1‡ | TRAF-2 | |
| Hepatoblasts | M | M | L | M | H |
| Hematopoietic cells | M | H | H | H | H |
| Megakaryocytes | M | H | M | M | H |
| Endothelial cells | M | H | L | M | H |
E13 WT and RelA−/− embryos and E14 TNFR-1−/− embryos were fixed in formalin, processed for parafin embedding, and used for immunostaining as described in Materials and Methods. The levels of expression indicate those seen for all samples, except where indicated. H, high levels of expression (almost all cells express); M, moderate levels of expression (many cells express but many do not); L, low levels of expression (few cells express).
*No staining in RelA knockouts.
†Decreased staining in RelA knockouts.
‡Some nuclear staining in TNFR-1 knockouts.
Signaling through TNFR-1 Is Responsible for Embryonic Lethality in the RelA−/− Mice
To determine whether signaling through TNFR-1 contributes to the embryonic lethal phenotype of RelA−/− fetuses, mice deficient in both RelA and TNFR-1 were generated as described in the Materials and Methods section. A total of 115 pups were examined by PCR and monitored for survival. Only seven of nine theoretically possible genetic outcomes were obtained in live-born animals. Table 2 ▶ summarizes the results of these crosses. From 115 newborn pups analyzed, 7 (6.1%) had homozygous mutations in RelA and TNFR-1. Nine newborns (7.8%) had homozygous WT alleles for both genes, and 33 pups (28.7%) were double heterozygous. However, only four of the remaining six genetic possibilities were observed, because newborn mice carrying a null RelA genotype were never observed unless TNFR-1 was also absent. Taking these two lethal outcomes (RelA−/−/TNFR-1+/+ and RelA−/−/TNFR+/−) into consideration, all live-born mice, including RelA−/−/TNFR-1−/−, were obtained with the expected Mendelian ratios. By monitoring the pups daily, we observed that at birth all animals appeared healthy and nursed well. However, within the first postnatal week, double-knockout mice became runted, failed to thrive despite continuing to nurse, and subsequently died. Our survival analysis on the first seven double-knockout pups revealed that the average survival time was 10 days with a range of 6 to 17 days.
Table 2.
Genetic Analysis of Offspring Obtained from RelA/TNFR-1 Double Heterozygous Matings
| Genotype | Expected number | Actual number | Expected percentage | Actual percentage |
|---|---|---|---|---|
| RelA+/+ | 6.25 | 9 | 7.2 | 7.82 |
| TNFR-1+/+ | ||||
| RelA+/− | 12.5 | 14 | 14.4 | 12.17 |
| TNFR-1+/+ | ||||
| RelA+/+ | 12.5 | 19 | 14.4 | 16.52 |
| TNFR-1+/− | ||||
| RelA+/− | 25.0 | 33 | 28.8 | 28.70 |
| TNFR-1+/− | ||||
| RelA+/+ | 6.25 | 11 | 7.2 | 9.57 |
| TNFR-1−/− | ||||
| RelA+/− | 12.5 | 22 | 14.4 | 19.13 |
| TNFR-1−/− | ||||
| RelA−/− | 6.25 | 0 | 7.2 | 0 |
| TNFR-1+/+ | ||||
| RelA−/− | 12.5 | 0 | 14.4 | 0 |
| TNFR-1+/− | ||||
| RelA−/− | 6.25 | 7 | 7.2 | 6.09 |
| TNFR-1−/− |
Genotyping was performed by PCR analysis of tail DNA using primers diagnostic for both WT and mutant alleles.
For subsequent studies, RelA+/−/TNFR-1−/− mice were used as breeders to obtain litters with a 25% chance of generating RelA−/−/TNFR-1−/− pups. RelA+/+/TNFR-1+/+ mice were used to generate age-matched WT controls. At various days after birth, pups were killed and analyzed for body weight, necropsies were performed for histology sections, and pieces of liver were harvested for whole-cell extract and RNA. Consistent with our earlier observations, all newborn pups were similar in weight at 2 and 4 days of age (Table 3) ▶ . The WT pups and TNFR-1−/− pups continued to gain weight, and by 7 days of age, they had doubled their 4-day body weight. In contrast, 7-day-old RelA−/−/TNFR-1−/− pups had gained only a minimal amount of weight and now appeared significantly smaller, resembling 4-day-old mice. This difference in size is statistically different from both the WT and TNFR-1 knockout mice at the same age. This size difference becomes even more pronounced if the RelA−/−/TNFR-1−/− pups survive to 16 days of age. However, we found that, by 16 days of age, the TNFR-1 knockout pups were also statistically different in weight from both the WT pups and the double-knockout pups at that age. Therefore, the lack of TNFR-1 alone may influence body weight to some degree. Thus, the lack of TNFR-1 rescued RelA−/− mice from embryonic lethality, demonstrating that signaling through TNFR-1 is responsible for the embryonic death observed in these animals. However, whereas RelA may not be necessary for fetal development, its absence, in combination with the lack of TNFR-1 signaling, causes death within a very short time after birth.
Table 3.
Average Weight Gain during Early Postnatal Life
| Age (days) | Average weight (grams) | ||
|---|---|---|---|
| RelA+/+/TNFR-1+/+ | RelA+/+/TNFR-1−/− | RelA−/−/TNFR-1−/− | |
| 2 | 1.4 ± 0.04 | 1.4 ± 0.1 | 1.2 ± 0.1 |
| (n = 3) | (n = 12) | (n = 2) | |
| 4 | 2.3 ± 0.1 | 2.0 ± 0.2 | 2.0 ± 0.1 |
| (n = 3) | (n = 14) | (n = 3) | |
| 7 | 4.6 ± 0.3 | 3.8 ± 0.6 | 2.5 ± 0.2*† |
| (n = 3) | (n = 8) | (n = 2) | |
| 16 | 9.6 ± 0.6 | 8.4 ± 0.2* | 5.3 ± 1.7*† |
| (n = 3) | (n = 5) | (n = 4) |
Average weight of WT, TNFR-1−/−, and RelA−/−/TNFR-1−/− pups during the first 16 postnatal days. Mice were weighed at the indicated ages. Tail samples were taken for DNA isolation and PCR genotyping. Numbers represent average ± SD.
*Statistically different from WT mice; P ≤ 0.001 to 0.01.
†Statistically different from TNFR-1 knockout mice; P ≤ 0.001 to 0.01.
All RelA−/− mice died during E14–E15 with massive hepatocyte apoptosis as previously described. 4,5 The RelA−/−/TNFR-1−/− mice survived gestation and were born with normal liver morphology. Liver histology was similar between 2-day-old WT mice and double knockouts (Figure 5, A and B) ▶ . The livers of the RelA−/−/TNFR-1−/− mice had normal architecture with a persistence of small foci of hematopoietic cells and little, if any, hepatocyte apoptosis. Between 2 and 7 days of age, foci of hematopoietic cells greatly decreased in the WT mice, but at 7 days the livers of the RelA/TNFR-1 double knockouts had more abundant and larger hematopoietic foci than the WT mice. Furthermore, within the first 2–3 postnatal weeks, the condition of the RelA/TNFR-1 double knockouts deteriorated, and, by the time of death, acute hepatitis with massive neutrophilic infiltration and necrotic foci was present. The accumulation of immature neutrophils (band cells) started in the periportal areas and progressively infiltrated the liver parenchyma (Figure 5, C–E) ▶ . Although scattered apoptotic hepatocytes were present, liver damage and, presumably, the lethality of the RelA−/−/TNFR-1−/− mice were consequences of neutrophilic infiltration and hepatocyte necrosis. The livers of the TNFR-1 knockouts were morphologically normal and were not distinguishable from those of the WT mice. In addition to severe liver abnormalities, neutrophilic infiltration, although not as extensive as that in the liver, was present in the heart (data not shown) and lungs of some RelA−/−/TNFR-1−/− mice. An example of an unusual lung lesion resembling lobar pneumonia that developed in one animal is shown in Figure 5F ▶ , in which normal alveoli are seen adjacent to the walled lesion. Spleens of the RelA−/−/TNFR-1−/− mice showed a decrease in white pulp areas and less compact germinal centers (compare Figure 5, G and H ▶ ), whereas in the thymus there were foci of cell degeneration (data not shown). It has been reported that the germinal-center formation is abnormal in TNFR-1 knockout mice. These changes appear to be accentuated in the RelA−/−/TNFR-1−/− mice.
Figure 5.
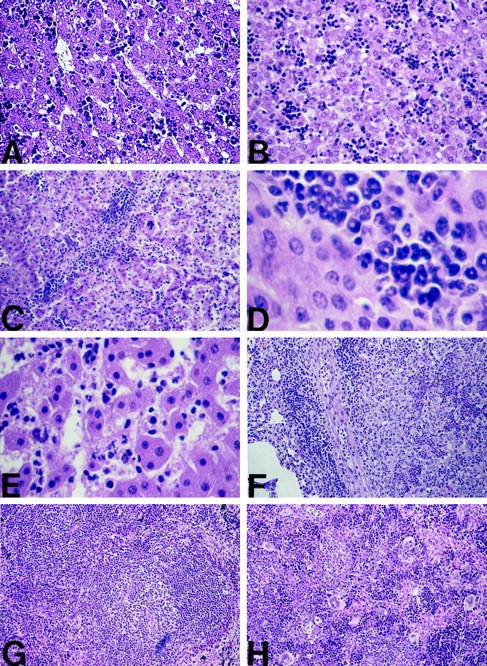
Histological analysis of WT and RelA−/−/TNFR-1−/− mice. All sections were stained with H&E. A: Liver section from a 2-day-old WT mouse showing normal hepatocytes and presence of focal areas of hematopoiesis. Original magnification, ×200. B: Liver section from a 2-day-old RelA−/−/TNFR-1−/− mouse showing normal hepatocytes and presence of focal areas of hematopoiesis. Original magnification, ×200. C: Liver section from an 8-day-old RelA−/−/TNFR-1−/− mouse illustrating the presence of massive neutrophilic infiltration. Original magnification, ×100. D: Higher magnification of the 8-day RelA−/−/TNFR-1−/− liver section, illustrating the immature neutrophils (band cells) responsible for the inflammation seen. Original magnification, ×640. E: Liver section from a 17-day-old RelA−/−/TNFR-1−/− mouse, demonstrating focal areas of necrosis. Original magnification, ×400. F: Lung lesion in a RelA−/−/TNFR-1−/− mouse killed at 16 days of age. The lesion was a solid mass that histologically resembled lobular pneumonia. The area was separated from the rest of the lung parenchyma (left side, showing alveoli) by connective tissue surrounded by inflammatory cells. The lesion itself (right side) contained closed and thickened alveoli, inflammatory cells, and cell necrosis. Original magnification, ×200. G: Spleen section from a 16-day-old WT mouse, showing normal germinal center formation. Original magnification, ×100. H: Spleen section from a 16-day-old RelA−/−/TNFR-1−/− mouse, demonstrating less defined white pulp and germinal follicle areas as well an increased numbers of megakaryocytes. Original magnification, ×200.
The embryonic survival and subsequent postnatal mechanism of death observed in the RelA/TNFR-1 knockouts suggest that the absence of RelA by itself is not responsible for fetal liver death, but that RelA−/− cells become sensitive to TNFR-1-mediated apoptotic signals. Removal of these signals in the double-knockout mice prevented embryonic lethality and did not interfere with liver development.
IκBα and IκBβ Proteins Are Altered in the Livers of RelA−/−/TNFR-1−/− Mice
To determine whether rescuing the RelA−/− lethal phenotype in the RelA/TNFR-1 knockouts would correct the defects in the IκBα and IκBβ proteins, Western blot analysis was performed. Whole-cell proteins were isolated from the livers of the WT, TNFR-1−/−, and RelA−/−/TNFR-1−/− mice at 2, 4, and 7 days of age, respectively (Figure 6) ▶ . Similar to the RelA knockouts at E12–E14, RelA/TNFR-1 double knockouts had decreased IκBα and no detectable IκBβ in comparison with the WT mice and the TNFR-1 knockouts. This suggests that a deficiency of these proteins is not causing the embryonic lethal phenotype of the RelA knockouts. Nevertheless, it could still be contributing to postnatal mortality. As expected, RelA expression was absent in the RelA/TNFR-1 knockouts, whereas TRAF-2 was unaffected in all of the mice.
Figure 6.

Protein expression in livers of WT, TNFR-1−/−, and RelA−/−/TNFR-1−/− newborn pups. Western blots for RelA, IκBα, IκBβ, and TRAF-2 were performed on liver extracts from 2-, 4-, and 7-day-old WT mice, TNFR-1−/− mice, and RelA−/−/TNFR-1−/− mice as indicated. WT and TNFR-1−/− mice express all proteins. RelA−/−/TNFR-1−/− mice have absent RelA and IκBβ proteins, diminished IκBα protein expression, and unaltered TRAF-2 protein levels.
RelA−/−/TNFR-1−/− Mice Do Not Have an Increase in TNFR-2 Messenger RNA or Incidence of Apoptosis during the First Postnatal Week
Up-regulation of TNFR-2 has been shown to result in an inflammatory response. 35 Therefore, we examined whether a TNFR-2 message was being up-regulated in the liver of the TNFR-1−/− or RelA−/−/TNFR-1−/− pups during the first week of life. We found that neither animal had up-regulated TNFR-2 messenger RNA compared with the WT mice (data not shown). We also examined whether the double knockouts had increased liver apoptosis that was contributing to their demise. Multiorgan slides of 2-, 4-, 7-, and 16-day-old WT, TNFR-1−/−, and RelA−/−/TNFR-1−/− pups were examined by terminal deoxynucleotidyltransferase-mediated uridine 5′-triphosphate end labeling assay (data not shown). We did not find an increase in incidence of apoptosis at any age, in any organ, including the liver. Therefore, TNF signaling through TNFR-2 does not result in TNFR-2 messenger RNA up-regulation or in an apoptotic phenotype in the newborn RelA−/−/TNFR-1−/− pup.
Discussion
The absence of RelA results in massive hepatocyte apoptosis and embryonic death between E14 and E15. The timing of this lethal effect may reflect the time at which NF-κB proteins are synthesized or assembled during liver development. To examine this possibility, we analyzed the expression of RelA, p50, IκBα, and IκBβ in WT mice from E12–E19. We also examined the formation of active NF-κB complexes in the livers of WT mice from E12–E14. These analyses revealed that all of the proteins and NF-κB complexes, consisting of RelA/p50 heterodimers and p50 homodimers, were present in the liver from at least E12. Because NF-κB complexes are formed in the developing liver of the WT mice at least 2 days before the onset of embryonic death in the RelA−/− mice, it is unlikely that lethality corresponds to the timing of NF-κB activation during hepatic development. An investigation of fetal livers from RelA knockouts revealed that they do not form NF-κB heterodimers, but contain only p50 homodimers. Thus, it appears that, during liver development, a RelA deficiency is not compensated for by other Rel family members. However, RelA−/−/p50−/− embryos die with a similar phenotype to the RelA−/− mice, but 2 days earlier in development. 33 It is, therefore, possible that, in the RelA knockouts, p50 partially compensates for the lack of RelA before E14.
In comparison with the WT mice, RelA−/− fetal livers from E12–E14 had lower levels of IκBα protein and no detectable IκBβ protein. This finding is similar to data reported in RelA−/− embryonic fibroblasts. 4 Here we measured IκBα and IκBβ directly in the liver and illustrated that IκBβ regulation is dependent on RelA, whereas IκBα is partially, but not completely, dependent on RelA. Similar to the WT mice, the TNFR-1−/− mice had no alterations in RelA, p50, IκBα, or IκBβ throughout liver development. TRADD and TRAF-2 proteins were not altered in any of the fetal livers examined.
TNF is a potent NF-κB activator in the adult liver, and signaling through TNFR-1 is required for the initiation of liver regeneration. Although TNF by itself is not generally apoptotic for many cell types, 36 it can cause apoptosis when given in conjunction with drugs that block transcription and translation. 37 Moreover, TNF can cause hepatocyte apoptosis in vivo and in vitro when NF-κB activation is blocked by an IκBα super-repressor. 38,39 Because the TNFR-1 knockouts develop normally, we hypothesized that the elimination of TNFR-1-mediated apoptotic signals could rescue the RelA knockouts from embryonic lethality. Therefore, we generated mice that were deficient in both RelA and TNFR-1 and showed that these animals survive embryonic development. At birth RelA−/−/TNFR-1−/− pups have normal liver morphology, but, after 4–5 days of age, most animals become runted and weigh significantly less than the WT mice of the same age. The double knockouts develop acute hepatitis and die with an average survival of 10 days. These data suggest that RelA is not critical for liver development but may serve a vital protective function against TNFR-1 apoptotic signaling. Additionally, the RelA−/−/TNFR-1−/− mice may have limited survival because RelA provides a critical postnatal function. However, the RelA−/−/TNFR-1−/− pups do not die with massive liver apoptosis as seen in the RelA knockouts. Instead, these animals develop acute hepatitis and neutrophilic infiltration leading to focal necrosis of the liver. Given the intense inflammatory infiltrate found in the livers of the dying mice, it is likely that the lack of TNF signaling and RelA makes these animals extremely susceptible to endogenous infectious agents. However, we cannot rule out the possibility that this phenotype is entirely caused by the absence of RelA in the newborn. A conditional RelA knockout will be necessary to examine which affects are due to the RelA deficiency and which are due to the combined lack of TNFR-1 and RelA. All animals, including the double knockouts, were maintained in a specific pathogen-free facility, reducing the possibility of contamination by external organisms, and were free of murine hepatitis virus. The phenotype of RelA/TNFR-1 knockouts has similarities to phenotypic abnormalities described in the RelB knockouts. 8 These animals also had neutrophilic inflammatory infiltrates and extramedullary hemopoiesis in the liver. It is puzzling that the RelA knockouts die only with liver abnormalities because RelA is ubiquitously expressed during normal development. Perhaps RelB or other Rel proteins are compensating for the loss of RelA in hematopoietic organs, although we found no evidence of this within the liver.
While this manuscript was being prepared, Doi et al reported that RelA knockouts can be rescued from embryonic lethality by crossing them with TNF−/− mice. 40 Similar to our findings, the RelA/TNF knockouts have normal liver morphology at birth; however, death does not occur until approximately 40 days of age. In RelA/TNFR-1 knockouts, we have not observed survival beyond 3 weeks of age. Histological analysis of 40-day-old RelA−/−/TNF−/− mice demonstrated lung and liver inflammation (similar to our findings in RelA/TNFR-1 knockouts), as well as kidney abnormalities that we did not observe. A lack of TNFR-1 signaling caused a more profound defect than TNF deficiency, and this might be due to TNF signaling via TNFR-2. However, signaling through TNFR-2 did not cause increased apoptosis in the double-knockout newborns, but it could be contributing to the intense inflammatory reaction. Preliminary evidence suggests that the TNFR-2−/− mice cannot rescue the RelA−/− lethal phenotype, indicating that, whereas a functional TNFR-2 may be contributing to the demise of RelA−/−/TNFR-1−/− mice, it is not playing a role in RelA−/− lethality. It has also been recently reported that TNFR-1−/− mice can rescue the embryonic lethal effects seen in IKK-2−/− mice that die from a similar phenotype to that of RelA−/− mice. 22 However, details of these TNFR-1−/−/IKK-2−/− mice were not described.
Although the main goal of this work was to examine embryonic lethality of RelA−/− mice, we also obtained interesting information about TNFR-1 knockouts. These animals develop normally, but show diminished resistance to infection by intracellular bacteria. 41 The TNFR-1 knockouts fail to activate NF-κB and STAT3 during liver regeneration, leading to high mortality and deficient DNA replication. 3 A major puzzle is to explain how these animals survive embryonic development, if we assume that, as in liver regeneration, the TNFR-1 knockouts have deficient NF-κB and STAT3 during liver development. Surprisingly, we found that both NF-κB and STAT3 were active in the liver of the TNFR-1 knockouts. This suggests that either TNF does not activate NF-κB during normal liver development or that, in the TNFR-1 knockouts, an alternate mechanism of NF-κB activation (perhaps through IL-1) is used. In any event, these results point out that hepatocyte proliferation in liver development and liver regeneration has different regulatory pathways. Thus, the absence of RelA is not directly responsible for fetal liver death, but its absence allows cells to become sensitive to TNFR-1-mediated apoptotic signals. The removal of these signals in RelA−/−/TNFR-1−/− mice prevents embryonic lethality and does not interfere with liver development.
Acknowledgments
We thank Dr. Amer Beg for RelA heterozygous breeding mice and Dr. Raj Kapur for histology consultation. We also thank Drs. Jean Campbell and Chris Franklin for helpful discussions and critical reading of the manuscript.
Footnotes
Address reprint requests to Dr. Nelson Fausto, M.D., University of Washington, 1959 NE Pacific Street, Box 357470, Seattle, WA 98195. E-mail: nfausto@u.washington.edu.
Supported by CA-23226 and CA-74131 (to N. F.); M. E. R. is the recipient of a postdoctoral fellowship from the National Institutes of Health (F32 DK09920–01).
References
- 1.Wulczyn FG, Krappmann D, Scheidereit C: The NF-κB/Rel, and IκB gene families: mediators of immune response and inflammation. J Mol Med 1996, 74:749-769 [DOI] [PubMed] [Google Scholar]
- 2.Sonenshein GE: Rel/NF-κB transcription factors, and the control of apoptosis. Sem Cancer Biol 1997, 8:113-119 [DOI] [PubMed] [Google Scholar]
- 3.Yamada Y, Kirillova I, Peschon JJ, Fausto N: Initiation of liver growth by tumor necrosis factor: deficient liver regeneration in mice lacking type 1 tumor necrosis factor receptor. Proc Natl Acad Sci USA 1997, 94:1441-1446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beg AA, Sha WC, Bronson RT, Ghosh S, Baltimore D: Embryonic lethality and liver degeneration in mice lacking the RelA component of NF-κB. Nature 1995, 376:167-170 [DOI] [PubMed] [Google Scholar]
- 5.Doi TS, Takahashi T, Taguchi O, Azuma T, Obata Y: NF-κB relA-deficient lymphocytes: normal development of T cells and B cells, impaired production of IgA and IgG1 and reduced proliferative responses. J Exp Med 1997, 185:953-961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ghosh S, May MJ, Kopp EB: NF-κB, and Rel proteins: evolutionarily conserved mediators of immune responses. Annu Rev Immunol 1998, 16:225-260 [DOI] [PubMed] [Google Scholar]
- 7.Kontgen F, Grumont RJ, Strasser A, Metcalf D, Li R, Tarlinton D, Gerondakis S: Mice lacking the c-rel proto-oncogene exhibit defects in lymphocyte proliferation, humoral immunity, and interleukin-2 expression. Genes Dev 1995, 9:1965-1977 [DOI] [PubMed] [Google Scholar]
- 8.Weih F, Carrasco D, Durham SK, Barton DS, Rizzo CA, Ryseck R-P, Lira SA, Bravo R: Multiorgan inflammation and hematopoietic abnormalities in mice with a targeted disruption of RelB, a member of the NF-κB/Rel family. Cell 1995, 80:331-340 [DOI] [PubMed] [Google Scholar]
- 9.Beg AA, Sha WC, Bronson RT, Baltimore D: Constitutive NF-κB activation, enhanced granulopoiesis, and neonatal lethality in IκBα-deficient mice. Genes Dev 1995, 9:2736-2746 [DOI] [PubMed] [Google Scholar]
- 10.Schmidt-Ullrich R, Memet S, Lilienbaum A, Feuillard J, Raphael M, Israel A: NF-κB activity in transgenic mice: developmental regulation and tissue specificity. Development 1996, 122:2117-2128 [DOI] [PubMed] [Google Scholar]
- 11.Liu Z-G, Hsu H, Goeddel DV, Karin M: Dissection of TNF receptor 1 effector functions: JNK activation is not linked to apoptosis while NF-κB activation prevents cell death. Cell 1996, 87:565-576 [DOI] [PubMed] [Google Scholar]
- 12.Baker SJ, Reddy EP: Transducers of life and death: TNF receptor superfamily and associated proteins. Oncogene 1996, 12:1-9 [PubMed] [Google Scholar]
- 13.Hsu H, Xiong J, Goeddel DV: The TNF receptor 1-associated protein TRADD signals cell death, and NF-κB activation. Cell 1995, 81:495-504 [DOI] [PubMed] [Google Scholar]
- 14.Hsu H, Shu H-B, Pan M-G, Goeddel DV: TRADD-TRAF2, and TRADD-FADD interactions define two distinct TNF receptor 1 signal transduction pathways. Cell 1996, 84:299-308 [DOI] [PubMed] [Google Scholar]
- 15.Rothe M, Sarma V, Dixit VM, Goeddel DV: TRAF2-mediated activation of NF-kappa B by TNF receptor 2, and CD40. Science 1995, 269:1424-1427 [DOI] [PubMed] [Google Scholar]
- 16.Hsu H, Huang J, Shu H-B, Baichwal V, Goeddel DV: TNF-dependent recruitment of the protein kinase RIP to the TNF receptor-1 signaling complex. Immunity 1996, 4:387-396 [DOI] [PubMed] [Google Scholar]
- 17.Henkel T, Machleidt T, Alkalay I, Kronke M, Ben-Neriah Y, Baeuerle PA: Rapid proteolysis of IκB-α is necessary for activation of transcription factor NF-κB. Nature 1993, 365:182-185 [DOI] [PubMed] [Google Scholar]
- 18.Zandi E, Rothwarf DM, Delhase M, Hayakawa M, Karin M: The IkappaB kinase complex (IKK) contains two kinase subunits, IKKalpha and IKKbeta, necessary for IkappaB phosphorylation and NF-kappaB activation. Cell 1997, 91:243-252 [DOI] [PubMed] [Google Scholar]
- 19.DiDonato JA, Hayakawa M, Rothwarf DM, Zandi E, Karin M: A cytokine-responsive IkappaB kinase that activates the transcription factor NF-kappaB. Nature 1997, 388:548-554 [DOI] [PubMed] [Google Scholar]
- 20.Traenckner EB-M, Wilk S, Baeuerle PA: A proteasome inhibitor prevents activation of NF-κB and stabilizes a newly phosphorylated form of IκB-α that is still bound to NF-κB. EMBO J 1994, 13:5433-5441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brown K, Gerstberger S, Carlson L, Franzoso G, Siebenlist U: Control of IκB-α proteolysis by site-specific, signal-induced phosphorylation. Science 1995, 267:1485-1488 [DOI] [PubMed] [Google Scholar]
- 22.Li Q, Van Antwerp D, Mercurio F, Lee K-F, Verma IM: Severe liver degeneration in mice lacking the IκB kinase 2 gene. Science 1999, 284:321-325 [DOI] [PubMed] [Google Scholar]
- 23.Tanaka M, Fuentes ME, Yamaguchi K, Durnin MH, Dalrymple SA, Hardy KL, Goeddel DV: Embryonic lethality, liver degeneration, and impaired NF-κB activation in IKK-β-deficient mice. Immunity 1999, 10:421-429 [DOI] [PubMed] [Google Scholar]
- 24.Yamada Y, Webber EM, Kirillova I, Peschon JJ, Fausto N: Analysis of liver regeneration in mice lacking type 1 or type 2 tumor necrosis factor receptor: requirement for type 1 but not type 2 receptor. Hepatology 1998, 28:959-970 [DOI] [PubMed] [Google Scholar]
- 25.Cressman DE, Greenbaum LE, DeAngelis RA, Ciliberto G, Furth EE, Poli V, Taub R: Liver failure and defective hepatocyte regeneration in interleukin-6-deficient mice. Science 1996, 274:1379-1383 [DOI] [PubMed] [Google Scholar]
- 26.Akerman P, Cote P, Yang SQ, McClain C, Nelson S, Bagby GJ, Diehl AM: Antibodies to tumor necrosis factor-α inhibit liver regeneration after partial hepatectomy. Am J Physiol 1992, 263:G579-G585 [DOI] [PubMed] [Google Scholar]
- 27.Van Antwerp DJ, Martin SJ, Kafri T, Green DR, Verma IM: Suppression of TNF-α-induced apoptosis by NF-κB. Science 1996, 274:787-789 [DOI] [PubMed] [Google Scholar]
- 28.Beg AA, Baltimore D: An essential role for NF-κB in preventing TNF-α-induced cell death. Science 1996, 274:782-784 [DOI] [PubMed] [Google Scholar]
- 29.Wang C-Y, Mayo MW, Baldwin AS, Jr: TNF-and cancer therapy-induced apoptosis: potentiation by inhibition of NF-κB. Science 1996, 274:784-787 [DOI] [PubMed] [Google Scholar]
- 30.Arsura M, FitzGerald MJ, Fausto N, Sonenshein GE: Nuclear factor-κB/Rel blocks transforming growth factor β1-induced apoptosis of murine hepatocyte cell lines. Cell Growth Differ 1997, 8:1049-1059 [PubMed] [Google Scholar]
- 31.Han Y, Brasier AR: Mechanism for biphasic Rel A NF-κB1 nuclear translocation in tumor necrosis factor α-stimulated hepatocytes. J Biol Chem 1997, 272:9825-9832 [DOI] [PubMed] [Google Scholar]
- 32.Lin Y-C, Brown K, Siebenlist U: Activation of NF-κB requires proteolysis of the inhibitor IκB-α: signal-induced phosphorylation of IκB-α alone does not release active NF-κB. Proc Natl Acad Sci USA 1995, 92:552-556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Horwitz BH, Scott ML, Cherry SR, Bronson RT, Baltimore D: Failure of lymphopoiesis after adoptive transfer of NF-κB-deficient fetal liver cells. Immunity 1997, 6:765-772 [DOI] [PubMed] [Google Scholar]
- 34.Takeda K, Noguchi K, Shi W, Tanaka T, Matsumoto M, Yoshida N, Kishimoto T, Akira S: Targeted disruption of the mouse Stat3 gene leads to early embryonic lethality. Proc Natl Acad Sci USA 1997, 94:3801-3804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Douni E, Kollias G: A critical role of the p75 tumor necrosis factor receptor (p75TNF-R) in organ inflammation independent of TNF, lymphotoxin α, or the p55TNF-R. J Exp Med 1998, 188:1343-1352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sugarman BJ, Aggarwal BB, Hass PE, Figari IS, Palladino MAJ, Shepard HM: Recombinant human tumor necrosis factor-α: effects on proliferation of normal and transformed cells in vitro. Science 1985, 230:943-945 [DOI] [PubMed] [Google Scholar]
- 37.Flick DA, Gifford GE: Comparison of in vitro cell cytotoxic assays for tumor necrosis factor. J Immunol Methods 1984, 68:167-175 [DOI] [PubMed] [Google Scholar]
- 38.Hellerbrand C, Jobin C, Iimuro Y, Licato L, Sartor RB, Brenner DA: Inhibition of NF-κB in activated rat hepatic stellate cells by proteasome inhibitors and an IκB super-repressor. Hepatology 1998, 27:1285-1295 [DOI] [PubMed] [Google Scholar]
- 39.Iimuro Y, Nishiura T, Hellerbrand C, Behrns KE, Schoonhoven R, Grisham JW, Brenner DA: NF-κB prevents apoptosis and liver dysfunction during liver regeneration. J Clin Invest 1998, 101:802-811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Doi TS, Marino MW, Takahashi T, Yoshida T, Sakakura T, Old LJ, Obata Y: Absence of tumor necrosis factor rescues RelA-deficient mice from embryonic lethality. Proc Natl Acad Sci USA 1999, 96:2994-2999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rothe J, Lesslauer W, Lotscher H, Lang Y, Koebel P, Kontgen F, Althage A, Zinkernagel R, Steinmetz M, Bluethmann H: Mice lacking the tumor necrosis factor receptor 1 are resistant to TNF-mediated toxicity but highly susceptible to infection by Listeria monocytogenes. Nature 1993, 364:798-802 [DOI] [PubMed] [Google Scholar]