

Abstract
Most colorectal cancers have loss-of-function mutations in the adenomatosis polyposis coli (APC) tumor suppressor gene. This leads to the accumulation of nuclear β-catenin, which, together with the DNA-binding protein TCF-4, functions as a transcriptional activator. The recently defined target genes c-myc, cyclin D1, and matrilysin are responsible for tumor proliferation or malignant progression and explain the oncogenic potential of nuclear β-catenin. To investigate its role in early colon carcinogenesis, we analyzed the expression of β-catenin, its target gene c-myc, and the proliferative activity in 88 colorectal adenomas of varying size and grade of dysplasia. The results revealed i) the most significant correlation of nuclear β-catenin and c-myc expression was not with the grade of dysplasia but with the size of the colon adenoma; ii) perfect correlation of nuclear β-catenin and c-myc expression; iii) no significant correlation of adenoma size with the proliferative activity; and iv) no significant correlation of proliferative activity and the nuclear expression of β-catenin and c-myc. These results imply that APC mutations have additional β-catenin-independent functions; APC mutations alone are not sufficient for nuclear overexpression of β-catenin; and nuclear β-catenin has additional important functions for exceeding a threshold tumor size.
The loss-of-function mutations in the adenomatosis polyposis coli (APC) tumor suppressor gene are the first event in colorectal carcinogenesis and have been found in more than 80% of early colon adenomas. 1 This leads to decreased APC-dependent degradation of the oncoprotein β-catenin, thereby increasing its cytoplasmatic free pool. 2,3 In a cell membrane-associated fraction bound to E-cadherin, β-catenin participates in cell-cell adhesion. However, the oncogenic potential of β-catenin is derived from a nuclear pool associated with the T cell factor (TCF) family of transcription factors. The resulting transcription complex can activate target genes associated with proliferation and invasion, linking APC gene defects to these processes. Recently defined target genes of β-catenin/TCF are c-myc, 4 cyclin D1, 5 and the matrix metalloproteinase matrilysin. 6,7
However, there is increasing evidence that APC gene mutations alone are not responsible for the increased nuclear expression of β-catenin. For instance, most colorectal carcinomas show a heterogenous distribution pattern of nuclear β-catenin, suggesting additional genetic or epigenic events regulating β-catenin distribution. 8,9 On the other hand, β-catenin-independent functions of APC gene mutations are suggested for early colorectal carcinogenesis. 10
Because some of the identified β-catenin target genes are potentially associated with proliferation, we wanted to correlate the proliferative activity, nuclear β-catenin expression, and the expression of the target gene c-myc in colon adenoma. We used a collection of 88 different cases, from small, early to late colon adenoma with beginning invasive growth, for immunohistochemical analysis. The results revealed i) a highly heterogenous expression pattern of nuclear β-catenin, with the most significant correlation of nuclear β-catenin and c-myc expression not with the grade of dysplasia but with the size of the colon adenoma; ii) a perfect correlation of nuclear β-catenin and c-myc expression; iii) no significant correlation of adenoma size with the proliferative activity; and iv) no significant correlation of proliferative activity and the nuclear expression of β-catenin and c-myc. These results have impact on the functions of APC gene mutations and nuclear β-catenin in early colon carcinogenesis.
Materials and Methods
Tissue Specimens
Formalin-fixed, paraffin-embedded tissues from patients who underwent polypectomy were retrieved from the archive of the Institute for Pathology, University of Erlangen-Nürnberg. All adenomas were totally removed, and the size was determined after formalin fixation. The 88 investigated colorectal adenomas had a size distribution from 2 to 25 mm in diameter, with 33 showing mild, 37 showing medium, and 18 showing severe grades of dysplasia. To exclude statistical results based simply on the strong correlation between adenoma size and grades of dysplasia, normally found in colorectal adenomas, high-grade dysplastic small adenomas and low-grade dysplastic large adenomas were also included in the collective.
Immunohistochemistry
Serial sections (3 μmol/L) were deparaffinized, rehydrated, and incubated with the different mouse monoclonal antibodies for 2 hours. Antigen retrieval was used for α-mib-1 and α-c-myc (microwave treatment, 10 minutes at 800 W and 10 minutes at 600 W). Dilutions were 1:30 for α-mib-1 (Dako, Hamburg, Germany), 1:200 for anti-c-myc (clone 9E11 from Novocastra, Newcastle, UK), and 1:100 for α-β-catenin (Transduction Laboratories, Lexington, KY). Biotinylated rabbit α-mouse Ig antiserum diluted 1:50 was used as the secondary antibody. After washing, the slides were incubated for 30 minutes. at room temperature with streptavidin-coupled alkaline phosphatase (Dako) and developed for 12 minutes at 37°C, using Fast Red (Sigma, Deisenhofen, Germany) as substrate. After rinsing in water, the sections were counterstained with hemalaun (Merck, Darmstadt, Germany), dehydrated, and coverslipped.
Evaluation of Immunostaining
The sections were examined by two independent researchers. Normal colonic epithelia adjacent to the adenomas were used as internal controls of staining efficiency and evaluation. Because the nuclear expression of β-catenin and c-myc was never homogeneous but varied in both intensity and distribution in the adenomas, we used a scoring system that included an evaluation of both the staining intensity and the percentage of stained cells. Staining intensity was graded as no staining (value 0, as in normal colonic epithelia), weak staining (value 1), moderate staining (value 2), or strong staining (value 3). The percentages of tumor cells with nuclear staining of β-catenin and c-myc were assigned scores as follows: 1, <5%; 2, 5–20%; 3, 21–50%; 4, >50%. Multiplication of the intensity value and the percentage value gave a multiplication value from 0 to 12. The multiplication values were grouped in four immunoreactive scores defined as 0 (negative; multiplication values 0, 1), 1 (low; multiplication values 2, 3), 2 (medium; multiplication values 4, 6), or 3 (high; multiplication values 8, 9, 12). For the proliferative activity, we determined the percentage of mib-1-positive tumor cells and scored 0, <5%; 1, 5–20%; 2, 21–50%; and 3, >50% mib-1-positive cells. Because the proliferative activity of normal colon epithelium was scored as 1, no adenoma was scored as 0.
Statistics
Statistical analysis was performed with the SPSS software (SPSS Standard version 8.0.0, SPSS Inc., Chicago, IL). The correlations between absolute size (in millimeters) and immunoreactivity were evaluated by the Mann-Whitney U test. The correlations among sizes, groups, and grades of dysplasia with immunoreactivity were evaluated by χ 2 test (linear-by-linear association).
Results
To correlate the nuclear expression of β-catenin in colorectal adenoma with the expression of its target gene c-myc, the proliferative activity, the adenoma size, and the grade of dysplasia, we performed an immunohistochemical analysis of β-catenin, c-myc, and mib-1 expression. Of 88 cases, 40 adenomas (45.4%) showed nuclear overexpression of β-catenin, 39 adenomas (44.3%) showed overexpression of c-myc, and 76 adenomas (86.3%) had a higher proliferative activity than normal colon epithelium.
Statistical analysis confirmed a very strong correlation (P < 0.001) of nuclear β-catenin-immunoreactive scores 0, 1, and 2 with tumor size. Median adenoma diameters were 3, 7, 13, and 17 mm for the scores 0, 1, 2, and 3, respectively (Figure 1A) ▶ . According to the observation that c-myc is a target gene of β-catenin/TCF4, we found an almost identical correlation of nuclear c-myc expression with adenoma size; median adenoma diameters were 3.5, 7.2, 14, and 18 mm for the immunoreactive scores 0, 1, 2, and 3, respectively (Figure 1B) ▶ . It is interesting that we found no significant correlation of the proliferative activity with the size of the adenomas (Figure 1C) ▶ and, therefore, not with nuclear β-catenin and c-myc expression.
Figure 1.
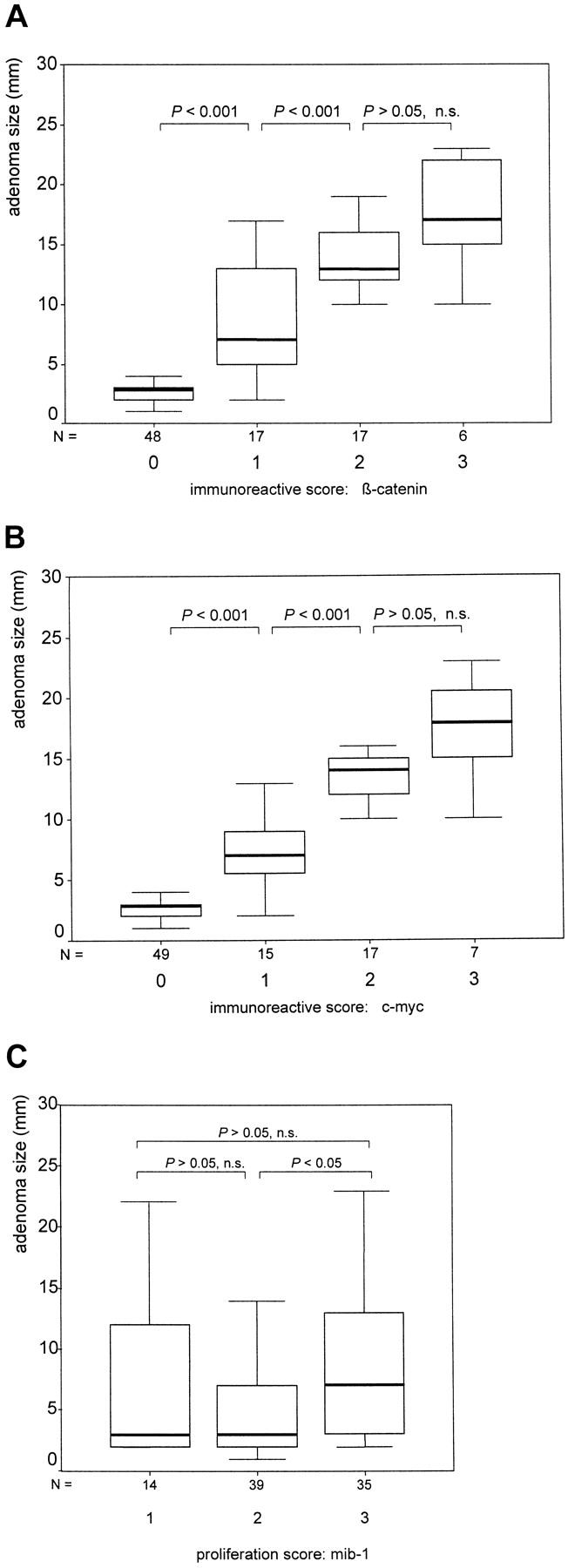
Correlation between adenoma size and nuclear β-catenin (A), c-myc (B), and mib-1 (C) expression. Boxplot charts (50% of values are within the box; horizontal bar, median; vertical bar, range of values). Included are the P values of the correlations between the indicated staining scores. A P value <0.05 was considered significant. n.s., not significant.
Based on these results, we could define two size groups of adenomas with a minimum (<5-mm) and a maximum (>8-mm) β-catenin expression (Table 1) ▶ . Of the adenomas <5 mm in size, 92% showed no nuclear β-catenin expression, and no case in this size group had medium (2) or high (3) immunoreactive scores. In contrast, all examined adenomas >8 mm in size showed nuclear overexpression of β-catenin, with scores of 2 or 3. We also defined an intermediate size group between 5 and 8 mm with beginning and increasing expression of nuclear β-catenin. The statistical significance of these results was again very high (P < 0.001). As expected, we again found almost identical correlations of the defined three size groups with c-myc expression (P < 0.001; Table 2 ▶ ) and no significant correlation with the proliferative activity (Table 3) ▶ .
Table 1.
Correlation between Adenoma Size and Nuclear Expression of β-Catenin
| Size (mm) | Immunoreactive score | Total | |||
|---|---|---|---|---|---|
| 0 | 1 | 2 | 3 | ||
| <5 | 46 (92) | 4 (8) | 0 | 0 | 50 |
| 5–8 | 2 (20) | 8 (80) | 0 | 0 | 10 |
| >8 | 0 | 5 (17.8) | 17 (60.7) | 6 (21.3) | 28 |
Table shows absolute numbers (percentages); P < 0.001.
Table 2.
Correlation between Adenoma Size and Nuclear Expression of c-myc
| Size (mm) | Immunoreactive score | Total | |||
|---|---|---|---|---|---|
| 0 | 1 | 2 | 3 | ||
| <5 | 47 (94) | 3 (6) | 0 | 0 | 50 |
| 5–8 | 2 (20) | 8 (80) | 0 | 0 | 10 |
| >8 | 0 | 4 (14.3) | 17 (60.7) | 7 (25) | 28 |
Table shows absolute numbers (percentages); P < 0.001.
Table 3.
Correlation between Adenoma Size and Expression of mib-1
| Size (mm) | Proliferative activity score | Total | |||
|---|---|---|---|---|---|
| 0 | 1 | 2 | 3 | ||
| <5 | 0 | 9 (18) | 26 (52) | 15 (30) | 50 |
| 5–8 | 0 | 1 (10) | 4 (40) | 5 (50) | 10 |
| >8 | 0 | 4 (14.3) | 9 (32.1) | 15 (53.6) | 28 |
Table shows absolute numbers (percentages); P > 0.05, not significant.
Previous investigations demonstrated a dependence of nuclear β-catenin expression on the grade of dysplasia, 11 which we could confirm by grouping the adenomas as mildly, moderately, and severely dysplastic (Table 4) ▶ . Again, we also found a correlation with c-myc expression (Table 5) ▶ and no significant correlation with proliferative activity (Table 6) ▶ . However, compared with the statistical significance of the correlations with adenoma size (P < 0.001), the correlation of nuclear β-catenin and c-myc with the grade of dysplasia was weaker (P < 0.005). The adenomas that were <5 mm in size with severe dysplasia did not express nuclear β-catenin and c-myc.
Table 4.
Correlation between Grade of Adenoma Dysplasia and Nuclear Expression of β-Catenin
| Dysplasia | Immunoreactive score | Total | |||
|---|---|---|---|---|---|
| 0 | 1 | 2 | 3 | ||
| Mild | 28 (84.8) | 3 (9.1) | 2 (6.1) | 0 | 33 |
| Moderate | 15 (40.5) | 9 (24.3) | 9 (24.3) | 4 (11.8) | 37 |
| Severe | 5 (27.8) | 5 (27.8) | 6 (33.3) | 2 (11.1) | 18 |
Table shows absolute numbers (percentages); P < 0.005.
Table 5.
Correlation between Grade of Adenoma Dysplasia and Expression of c-myc
| Dysplasia | Immunoreactive score | Total | |||
|---|---|---|---|---|---|
| 0 | 1 | 2 | 3 | ||
| Mild | 28 (84.8) | 3 (9.1) | 2 (6.1) | 0 | 33 |
| Moderate | 16 (43.2) | 9 (24.3) | 9 (24.3) | 3 (8.1) | 37 |
| Severe | 5 (27.8) | 3 (16.6) | 6 (33.3) | 4 (22.2) | 18 |
Table shows absolute numbers (percentages); P < 0.005.
Table 6.
Correlation between Grade of Adenoma Dysplasia and Expression of mib-1
| Dysplasia | Proliferative activity score | Total | |||
|---|---|---|---|---|---|
| 0 | 1 | 2 | 3 | ||
| Mild | 0 | 7 (21.2) | 15 (45.5) | 11 (33.3) | 33 |
| Moderate | 0 | 4 (10.8) | 19 (51.4) | 14 (37.8) | 37 |
| Severe | 0 | 3 (16.7) | 5 (27.8) | 10 (55.5) | 18 |
Table shows absolute numbers (percentages); P > 0.05, not significant.
As already demonstrated, the nuclear expression of β-catenin in most colorectal carcinomas is heterogeneous with focal overexpression. 8,9 We found similar heterogeneous expression patterns for nuclear β-catenin, c-myc, and mib-1 in the investigated colorectal adenomas. As expected, the expression of c-myc was always found in the same tumor area, which expressed nuclear β-catenin (example in Figure 2, B and C ▶ ). In contrast, many cases of tumor cells in the mib-1-positive proliferative area did not express nuclear β-catenin or c-myc (Figure 2, A ▶ -C) and vice versa (Figure 2, D ▶ -F). Thus, as has been shown statistically, for the whole collection of adenomas there was no perfect, spatial correlation between immunohistochemically detectable nuclear β-catenin or c-myc and the proliferative active zones in the individual adenomas.
Figure 2.
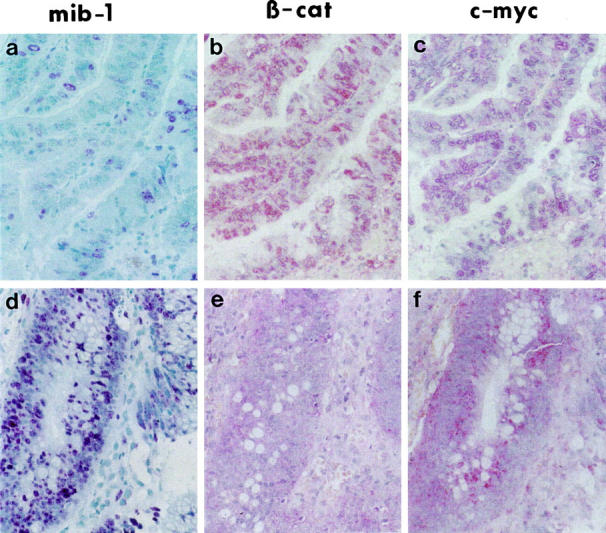
Correlation of β-catenin expression with the expression of c-myc but not with proliferative activity. Immunohistochemical staining for mib-1, β-catenin, and c-myc (red staining). Serial section of the same area of two colorectal adenomas with low (a−c) and high (d−f) proliferative activity. Strongly correlated expression of nuclear β-catenin (b) and c-myc (c) in the low-proliferating adenoma and no expression of both nuclear β-catenin (e) and c-myc (f) in the tumor cells of the high-proliferating adenoma. Original magnification, ×400.
Discussion
A collection of 88 different cases from small, early to late colorectal adenomas with beginning invasive growth was used for immunohistochemical analysis. The results revealed i) the most significant correlation of nuclear β-catenin and c-myc expression not with histological grading, but with the size of the colon adenomas; ii) a perfect correlation of nuclear β-catenin and c-myc expression; iii) no significant correlation of adenoma size with the proliferative activity; and iv) no significant correlation of proliferative activity with the nuclear expression of β-catenin and c-myc.
The functional consequence of the strong correlation between nuclear β-catenin amounts and the adenoma size must be discussed critically, because already low amounts of immunohistochemically undetectable transcriptional activators could be functionally active. However, we also found a perfect correlation between the expression level of nuclear β-catenin and its target gene c-myc in individual adenomas, which implies a direct dependence of the amounts of expressed c-myc on the concentration of nuclear β-catenin. This is also confirmed by functional analyses in cell culture, in which the transcriptional activity of β-catenin target gene promoters depends on the amounts of β-catenin (T. Brabletz, A. Jung, and T. Kirchner, unpublished data). Recent results could explain this dose-dependent function of β-catenin. Without nuclear β-catenin, TCF proteins are bound by a corepressor, as demonstrated for the repressor groucho in Drosophila species, and this complex acts as transcriptional repressor of the target genes. 12,13 Nuclear β-catenin competes with the repressor for TCF binding in a dose-dependent manner. Higher amounts of β-catenin could build up more activator complexes, which would lead to a more efficient de-repression of the target genes. Furthermore, it was demonstrated that high amounts of β-catenin are necessary to overcome a repression of TCF activity by the histone acetylase C-terminal-binding protein. 14 Thus, the concentration of nuclear β-catenin is an important parameter.
We detected no significant correlation between the nuclear expression of β-catenin or c-myc and proliferative activity. This was demonstrated both for large adenomas, in which highly proliferative areas often did not show nuclear β-catenin, and for strongly proliferating small adenomas, which did not express detectable amounts of nuclear β-catenin and c-myc at all. The question is how hyperproliferation is induced in colon adenomas. One possibility is that low, nondetectable nuclear amounts of β-catenin are sufficient to induce proliferation genes like cyclin D1, and high amounts are necessary to derepress genes associated with tumor progression. Alternatively, both β-catenin and c-myc may not be directly associated with the induction of proliferation but with later steps in tumor progression. For instance, c-myc overexpression has been described as promoting genomic instability, thereby increasing the rate of genetic alterations. 15 More than 80% of colorectal adenomas have APC gene defects as an initial event, leading to an increase of the cytoplasmatic free pool of β-catenin. However, our results demonstrate that early adenomas do not express detectable amounts of nuclear β-catenin. It is expected that there are β-catenin-independent effects of mutated APC. One could speculate that induction of proliferation is one of these β-catenin independent functions of mutated APC. It is interesting that, in a recent publication, Samowitz et al reported that dominant, active mutations in the β-catenin gene, which also lead to a reduced degradation of β-catenin and are associated with normal APC, are more often found in small, early adenomas than in late adenomas and colorectal carcinomas. 10 Particularly, there seemed to be a threshold adenoma size of about 5 mm, above which the rate of β-catenin mutations rapidly decreases. This is exactly the size above which we defined an increase of nuclear β-catenin (see discussion below). This could mean that low amounts of nuclear β-catenin, perhaps enough to induce proliferation, can be found on the basis of both β-catenin and APC mutations. However, a further increase of nuclear β-catenin, potentially necessary for tumor progression (see discussion below), could not be achieved simply by mutated β-catenin, but would need mutant APC as basic genetic alteration. This implies further functions of mutant APC in addition to simple reduction of β-catenin degradation.
The most stringent correlation we found was between adenoma size and the nuclear expression of β-catenin and c-myc (P < 0.001), which was higher than the already described correlation between the grade of dysplasia and nuclear β-catenin (P < 0.005). 11 Thus the already high correlation of nuclear β-catenin with dysplasia can be based mainly on the strong dependence of the dysplasia grade on adenoma size. This observation could be simply explained by a bystander effect, in which genetic alterations leading to an increase of tumor size and progression also lead to an increase of nuclear β-catenin without a functional consequence. However, it is more likely that nuclear β-catenin is necessary for exceeding a certain tumor size. The observed sharp size boundaries for low or absent (<5 mm) and medium to high (>8 mm) nuclear expression of β-catenin, with a transition size between 5 and 8 mm, support this hypothesis. What could be the role of β-catenin for the transition above this certain adenoma size level? It is known that this is also a critical threshold for the supply of tumors by pre-existing blood vessels. Beyond this size, colon adenomas increase in blood vessel density. 16 It is tempting to speculate that high amounts of nuclear β-catenin derepress genes necessary for tumor angiogenesis and subsequent progression. There are already other recently defined target genes of β-catenin involved in later steps of colorectal carcinogenesis. We and others have described the activation of the matrix metalloproteinase matrilysin, which is important for tumor growth and invasion by β-catenin in colorectal cancers. 6,7 Also, CD44, 17 fibronectin, 18 and the urokinase-type plasminogen activator receptor, 19 important factors for invasion or metastasis, are potential target genes of β-catenin/TCF. Thus, we would suggest that nuclear β-catenin is responsible for the progression of colorectal carcinogenesis, not only by affecting the grade of dysplasia but also exceeding a certain threshold tumor size.
One question that remains is how nuclear β-catenin increases during adenoma progression. One possibility is that, based on the APC gene defect, additional genetic alterations lead to a redistribution of the membranous or cytoplasmatic pool to the nucleus. Both dominant ras mutations 20 and E-cadherin gene defects 7 are described to have an influence on the β-catenin localization and activity. Dominant K-ras gene mutations are found in about 50% of colon adenomas >10 mm in size and are correlated with the adenoma size like the expression of nuclear β-catenin. 21 However, the nuclear overexpression of β-catenin, both in late adenomas and colon carcinomas, is heterogenous 9 and often found in contact zones to the extracellular matrix. 8 This also implies that extracellular matrix-induced epigenetic modulations may be responsible for the nuclear translocation of β-catenin in larger adenomas. For instance, the integrin-linked kinase, which is associated with the α5/β1 integrin fibronectin receptor, was shown to induce nuclear translocation of β-catenin. 22 Further work will show whether K-ras mutations or the expression of the α5/β1-integrin is also strongly correlated with the size of colorectal adenomas.
Our results, demonstrating a strong correlation of nuclear β-catenin and c-myc with adenoma size, but not with proliferative activity, imply that i) APC mutations have additional β-catenin-independent functions; ii) APC mutations alone are not sufficient for nuclear overexpression of β-catenin; and iii) nuclear β-catenin has additional important functions for exceeding a threshold tumor size.
Acknowledgments
We thank L. McKenna for critical reading of the manuscript.
Footnotes
Address reprint requests to Thomas Brabletz, Department of Pathology, University of Erlangen-Nürnberg, Krankenhausstr. 8–10, D-91054 Erlangen, Germany. E-mail: Thomas.Brabletz@Patho.Med.Uni-Erlangen.de.
T. B. and K. H. contributed equally to this publication.
References
- 1.Kinzler KW, Vogelstein B: Lessons from hereditary colorectal cancer. Cell 1996, 87:159-170 [DOI] [PubMed] [Google Scholar]
- 2.Korinek V, Barker N, Morin PJ, van Wichen D, de Weger R, Kinzler KW, Vogelstein B, Clevers H: Constitutive transcriptional activation by a β-catenin-Tcf complex in APC−/− colon carcinoma. Science 1997, 275:1784-1787 [DOI] [PubMed] [Google Scholar]
- 3.Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, Kinzler KW: Activation of β-catenin-Tcf signaling in colon cancer by mutations in β-catenin or APC. Science 1997, 275:1787-1790 [DOI] [PubMed] [Google Scholar]
- 4.He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, Morin PJ, Vogelstein B, Kinzler KW: Identification of c-myc as a target of the APC pathway. Science 1998, 281:1509-1512 [DOI] [PubMed] [Google Scholar]
- 5.Tetsu O, McCormick F: Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature 1999, 398:422-426 [DOI] [PubMed] [Google Scholar]
- 6.Brabletz T, Jung A, Dag S, Hlubek F, Kirchner T: β-Catenin regulates the expression of the matrix metalloproteinase-7 in human colorectal cancer. Am J Pathol 1999, 155:1033-1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Crawford HC, Fingleton BM, Rudolph-Owen LA, Heppner Goss KJ, Rubinfeld B, Polakis P, Matrisian LM: The metalloproteinase matrilysin is a target of β-catenin transactivation in intestinal tumors. Oncogene 1999, 18:2883–2891 [DOI] [PubMed]
- 8.Brabletz T, Jung A, Hermann K, Gunther K, Hohenberger W, Kirchner T: Nuclear overexpression of the oncoprotein β-catenin in colorectal cancer is localized predominantly at the invasion front. Pathol Res Pract 1998, 194:701-704 [DOI] [PubMed] [Google Scholar]
- 9.Hugh TJ, Dillon SA, Taylor BA, Pignatelli M, Poston GJ, Kinsella AR: Cadherin-catenin expression in primary colorectal cancer: a survival analysis. Br J Cancer 1999, 80:1046-1051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Samowitz WS, Powers MD, Spirio LN, Nollet F, van Roy F, Slattery ML: Beta-catenin mutations are more frequent in small colorectal adenomas than in larger adenomas and invasive carcinomas. Cancer Res 1999, 59:1442-1444 [PubMed] [Google Scholar]
- 11.Hao X, Tomlinson I, Ilyas M, Palazzo JP, Talbot IC: Reciprocity between membranous and nuclear expression of β-catenin in colorectal tumours. Virchows Arch 1997, 431:167-172 [DOI] [PubMed] [Google Scholar]
- 12.Cavallo RA, Cox RT, Moline MM, Roose J, Polevoy GA, Clevers H, Peifer M, Bejsovec A: Drosophila Tcf and Groucho interact to repress Wingless signaling activity. Nature 1998, 395:604-608 [DOI] [PubMed] [Google Scholar]
- 13.Roose J, Molenaar M, Peterson J, Hurenkamp J, Brantjes H, Moerer P, van de Wetering M, Destree O, Clevers H: The Xenopus Wnt effector XTcf-3 interacts with Groucho-related transcriptional repressors. Nature 1998, 395:608-612 [DOI] [PubMed] [Google Scholar]
- 14.Waltzer L, Bienz M: Drosophila CBP represses the transcription factor TCF to antagonize Wingless signalling. Nature 1998, 395:521-525 [DOI] [PubMed] [Google Scholar]
- 15.Yin XY, Grove L, Datta NS, Long MW, Prochownik EV: C-myc overexpression and p53 loss cooperate to promote genomic instability. Oncogene 1999, 18:1177-1184 [DOI] [PubMed] [Google Scholar]
- 16.Skinner SA, Frydman GM, O’Brien PE: Microvascular structure of benign and malignant tumors of the colon in humans. Dig Dis Sci 1995, 40:373-384 [DOI] [PubMed] [Google Scholar]
- 17.Wielenga VJ, Smits R, Korinek V, Smit L, Kielman M, Fodde R, Clevers H, Pals ST: Expression of CD44 in Apc and Tcf mutant mice implies regulation by the WNT pathway. Am J Pathol 1999, 154:515-523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gradl D, Kuhl M, Wedlich D: The Wnt/Wg signal transducer β-catenin controls fibronectin expression. Mol Cell Biol 1999, 19:5576-5587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mann B, Gelos M, Siedow A, Hanski ML, Gratchev A, Ilyas M, Bodmer WF, Moyer MP, Riecken EO, Buhr HJ, Hanski C: Target genes of β-catenin-T cell-factor/lymphoid-enhancer-factor signaling in human colorectal carcinomas. Proc Natl Acad Sci USA 1999, 96:1603-1608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Potempa S, Ridley AJ: Activation of both MAP kinase and phosphatidylinositide 3-kinase by Ras is required for hepatocyte growth factor/scatter factor-induced adherens junction disassembly. Mol Biol Cell 1998, 9:2185-2200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vogelstein B, Fearon ER, Hamilton SR, Kern SE, Preisinger AC, Leppert M, Nakamura Y, White R, Smits AM, Bos JL: Genetic alterations during colorectal tumor development. N Engl J Med 1988, 319:525-532 [DOI] [PubMed] [Google Scholar]
- 22.Novak A, Hsu SC, Leung-Hagesteijn C, Radeva G, Papkoff J, Montesano R, Roskelley C, Grosschedl R, Dedhar S: Cell adhesion and the integrin-linked kinase regulate the LEF-1 and β-catenin signaling pathways. Proc Natl Acad Sci USA 1998, 95:4374-4379 [DOI] [PMC free article] [PubMed] [Google Scholar]