

Abstract
To determine whether adult cardiac myocytes are capable of multiple divisions and whether this form of growth is restricted to a subpopulation of cells that retain this capacity with age, telomere lengths were measured in myocyte nuclei isolated from the left ventricle of fetal and neonatal Fischer 344 rats and rats at 4, 12, and 27 months after birth. Two independent methodologies were used for this analysis: laser scanning cytometer and confocal microscopy. In each case, fluorescence intensity of a peptide nucleic acid probe specific for telomeric sequence was evaluated. The two techniques yielded comparable results. Telomeric shortening increased with age in a subgroup of myocytes that constituted 16% of the entire cell population. In the remaining nondividing cells, progressive accumulation of a senescent associated nuclear protein, p16INK4, was evidenced. In conclusion, a significant fraction of myocytes divides repeatedly from birth to senescence, counteracting the continuous death of cells in the aging mammalian rat heart.
The recognition that cellular aging may occur in vitro and in vivo has been discussed in recent reviews. 1-3 A characteristic of replicating cells is shortening of telomeres, which may trigger growth arrest and senescence. 4-6 Erosion of telomeres may be interpreted as DNA damage, activating p53, p53-inducible genes, and programmed cell death. 7-9 This condition is operative only in cells that are not terminally differentiated and experience multiple divisions during their lifespan. Telomere shortening should not be considered a molecular trait of cardiac myocyte aging, because this cell population is believed to be permanent and irreplaceable. 10,11 According to the dogma, the number of muscle cells in the mammalian heart is defined at birth, and in the absence of cardiac diseases, these myocytes persist throughout the life of an individual or animal. However, recent observations suggest that the heart is characterized by ongoing cell death and cell regeneration during the entire life of a human being or an animal. 12,13 On this basis, the assessment of the length distribution of telomeres in myocytes with aging may provide information concerning the capacity of adult myocytes of undergoing multiple mitotic divisions. Additionally, whether cell regeneration affects all or a subgroup of myocytes and whether cellular aging involves in a uniform manner cardiac myocytes in vivo may be identified by this analysis. For this purpose, laser scanning cytometer and confocal microscopy were used to measure telomere lengths in cardiac myocytes from fetal, neonatal, adult, and senescent Fischer 344 rats. Specifically, animals at 4, 12, 16, and 27 months after birth were studied, because these rats are considered to be young adults at 4 to 6 months, fully mature adults at 10 to 12 months, aged at 16 to 18 months, and senescent at 22 to 27 months. Moreover, to establish whether the age of the entire parenchymal cell population of the heart corresponded to the age of the animals, the expression of p16INK4, a marker of cellular aging, 14-18 was examined. p16 is a member of the INK4 family of proteins which inhibits cdk4 and cdk6, maintaining Rb in its hypophosphorylated form. 19,20 Changes in the quantity of this protein in myocytes as a function of age were evaluated by immunoprecipitation, whereas changes in p16INK4 distribution in the cells were determined by confocal microscopy.
Materials and Methods
Myocyte Isolation
Myocytes were enzymatically dissociated from the left ventricle of Fischer 344 rats at gestational day 19 and at 1 day after birth, and at 4, 12, 16, and 27 months of age. In each isolation of fetal and neonatal myocytes, an entire litter in each case had to be used. A total of 10 litters, 5 for fetal and 5 for neonatal myocytes, were used. Rats were decapitated and their hearts were quickly removed and placed in calcium- and magnesium-free Hanks’ balanced salt solution (HBSS). Atria and right ventricle were dissected free and left ventricular tissue was cut in small pieces, which were transferred into 10 ml of dissociation medium (0.1% trypsin, 1:250, and 0.01% deoxyribonuclease I in HBSS) and stirred (approximately 60 rpm) at 37°C. The material dissociated from the tissue during first 15 minutes of trypsinization was discarded. An aliquot of dissociation medium (10 ml) was added to the remaining fragments of myocardium, and after 10 minutes of gentle stirring at 37°C the supernatant was collected. The cells contained in this fraction were centrifuged at 300 × g for 4 minutes, resuspended in Eagle’s minimal essential medium (MEM) supplemented with 10% fetal calf serum, and stored on ice. The procedure was repeated 4 to 6 times. Isolated cells were preplated in a 150-mm petri dish (Corning) for 1 hour at 37°C, and myocytes which did not attach to the dish during this time were collected. 21,22
A different approach was followed for the collection of myocytes from rats at 4 (n = 5), 12 (n = 5), 16 (n = 5), and 27 (n = 5) months. Hearts were placed on a plastic cannula for retrograde perfusion through the aorta. The solutions were supplements of modified commercial MEM Eagle Joklik. HEPES-MEM contained (in mmol/L) NaCl 117, KCl 5.7, NaHCO3 4.4, KH2PO4 1.5, MgCl2 17, HEPES 21.1, and glucose 11.7, with amino acids and vitamins, 2 mmol/L L-glutamine, and 21 mu/ml insulin; pH was adjusted to 7.2 with NaOH. Osmolarity of this solution is 292 mOsm. Washing solution was HEPES-MEM with the addition of 0.5 mmol/L EGTA. Resuspension medium was HEPES-MEM supplemented with 0.5% bovine serum albumin, 0.3 mmol/L calcium chloride, adjusted to 292 mOsm. The cell isolation procedure consisted of three main steps. Low calcium perfusion: blood washout in the presence of EGTA for about 10 minutes, and collagenase (selected Type I, Worthington Biochemical Corp., Freehold, NJ) perfusion of the myocardium was carried out at 37°C with HEPES-MEM gassed with 85% O2/15% N2. Mechanical tissue dissociation: after removing the myocardium from the cannula, the left ventricle was cut into small pieces and subsequently shaken in resuspension medium at 37°C. Supernatant cell suspensions were washed and resuspended in resuspension medium. Separation of intact cells: intact cells were enriched by centrifugation through Percoll (Sigma, St. Louis, MO). Approximately 10 6 cells were suspended in 10 ml of isotonic Percoll (final concentration, 41% in resuspension medium) and centrifuged for 10 minutes at 34 × g. Intact cells were recovered from the pellet and washed. Rectangular, trypan blue-excluding cells constituted nearly 80% of all myocytes. The extent of non-myocytes present in each preparation was determined by preparing smears of the isolated myocytes and staining them with α-sarcomeric actin and propidium iodide (PI). 23-25
Telomere Length
Myocytes were fixed in methanol/acetic acid (3/1) and resuspended in 50% acetic acid. Smears of nuclei were fixed in 4% formaldehyde and digested with pepsin. Ten microliters of hybridization solution containing 7 μl formamide, 3 ng fluorescein isothiocyanate (FITC)-labeled (C3TA2)3 peptide nucleic acid (PNA) probe (PE Biosystems, Foster City, CA), 0.5 mg blocking reagent (Boehringer Mannheim, Indianapolis, IN), and 3 μl 10 mmol/L Tris, pH 7.5, were added, DNA denatured, and hybridization performed. 26 Subsequently, slides were washed with 70% formamide and 10 mmol/L Tris, pH 7.5, and with 150 mmol/L NaCl and 50 mmol/L Tris, pH 7.5. Slides were then incubated with PI (10 μg/ml phosphate buffered saline) and RNase A (1 mg/ml). Total fluorescence of FITC-PNA probe, which corresponded to the content of telomeric sequences per nucleus, was determined by laser scanning Compucyte cytometer (CompuCyte Corp., Cambridge, MA) and confocal microscopy (BioRad 1000, Hercules, CA). In laser scanning cytometer, fluorescence excitation was provided by a 488 nm laser line. The green fluorescence of FITC-PNA probe was measured using a combination of dichroic mirrors and filters transmitting light at 530 ± 20 nm wavelength. The red fluorescence of PI was measured by long pass filters transmitting at >610 nm. Software of the cytometer was used to deconvolute histograms and estimate cells with specific telomeric lengths. 27,28 Approximately 5000 nuclei were measured by the Compucyte cytometer in each left ventricle. By confocal microscopy, FITC-PNA fluorescence was assessed by optically sectioning the entire thickness of each nucleus and recording the intensity of fluorescence in each section. Total fluorescence in each myocyte was calculated. In each animal, approximately 300 nuclei were measured.
Immunoprecipitation and Western Blot
Nuclear extracts were prepared by incubation of myocytes with hypotonic buffer. Lysates were mixed with 0.5% NP-40, centrifuged, and the nuclear pellets incubated in high-salt buffer; 300 μg protein were incubated with 3 μg of mouse monoclonal p16INK4 (F-12, Santa Cruz Biotechnology, Santa Cruz, CA) and 250 μl of HNTG buffer (20 mmol/L Hepes, pH 7.5, 150 mmol/L NaCl, 0.1% Triton X-100, 10% glycerol), containing protease inhibitors 0.2 mmol/L PMSF, 2 μg/μL aprotinin, and 0.2 mmol/L Na3VO4 overnight at 4°C. Subsequently, 50 μl of protein A-Agarose (Pierce, Rockford, IL) was added to each sample. 29 After several washings with a buffer containing 20 mmol/L Tris-HCl, pH 7.4, 300 mmol/L NaCl, 2 mmol/L EDTA, and 2 mmol/L EGTA, samples were centrifuged at 14,000 rpm and proteins were separated by 15% sodium dodecyl sulfate-polyacrylamide gel electrophoresis, transferred onto nitrocellulose filters, and exposed to mouse monoclonal p16INK4 antibody, 1 μg/ml of Tris-buffered saline/Tween 20 (TBST).
p16INK4 Distribution
Five hearts each from 1-day-old neonatal rats and rats at 4, 12, and 27 months of age were excised and the left ventricle frozen at −70°C. Cryostat sections were fixed in 2% paraformaldehyde for 10 minutes and incubated with p16INK4 antibody (F-12, Santa Cruz), diluted 1:30 in phosphate buffered saline for 1 hour at 37°C. FITC-labeled anti-mouse IgG was used as secondary antibody. Nuclei were stained with PI and the cytoplasm with α-sarcomeric actin (clone 5C5, Sigma). Myocyte nuclei labeled by p16INK4 were evaluated by confocal microscopy. A total of 500 myocyte nuclei was examined in each heart.
Data Analysis
Results are presented as mean ± SD. Statistical significance in multiple comparisons in which analysis of variance and the Fisher test indicated the presence of significant differences, was determined by the Bonferroni method. 30 P values <0.05 were considered to be significant.
Results
The average yield of left ventricular myocytes in the fetal and neonatal heart was 0.5 × 10 6 and 0.8 × 10 6 cells, respectively. Corresponding values in rats at 4, 12, 16, and 27 months were 7.6 × 10, 6 7.2 × 10, 6 6.5 × 106, and 4.0 × 10 6 myocytes. Fibroblasts accounted for 3 to 4% of cells in fetal and neonatal myocytes, and 1 to 2% of cells in young adult, adult, aged, and senescent myocyte preparations (Figure 1A) ▶ . After enzymatic dissociation of cardiac muscle cells and Percoll treatment, nuclei were isolated (Figure 1B) ▶ and stained by in situ hybridization with a fluorescent PNA probe. This probe, which is specific for telomeric sequence, was labeled with FITC (Figure 1C) ▶ . During this procedure, 25 to 30% of myocytes were lost, resulting in the preservation of nearly 70 to 75% of myocyte nuclei. Total nuclear DNA was assessed by PI (red fluorescence) and distinguished from the amount of fluorescence emitted by the PNA probe (green fluorescence). The latter corresponded to the total telomeric length present in each nucleus. 26 These parameters were used to obtain a bivariate distribution of telomeric length versus DNA in each nucleus; nearly 5000 nuclei were examined in each left ventricle by laser scanning cytometer. Length of telomeres was rather uniform in fetal and neonatal myocytes, but telomere shortening was evident in myocyte nuclei at 4 and 12 months and was even more apparent at 27 months (Figure 2) ▶ . Additionally, 300 nuclei were evaluated in each left ventricle by confocal microscopy. This consisted in the assessment of the aggregate fluorescence of the PNA probe (Figure 1C) ▶ in each nucleus. The results collected with these two independent methodologies are described below.
Figure 1.
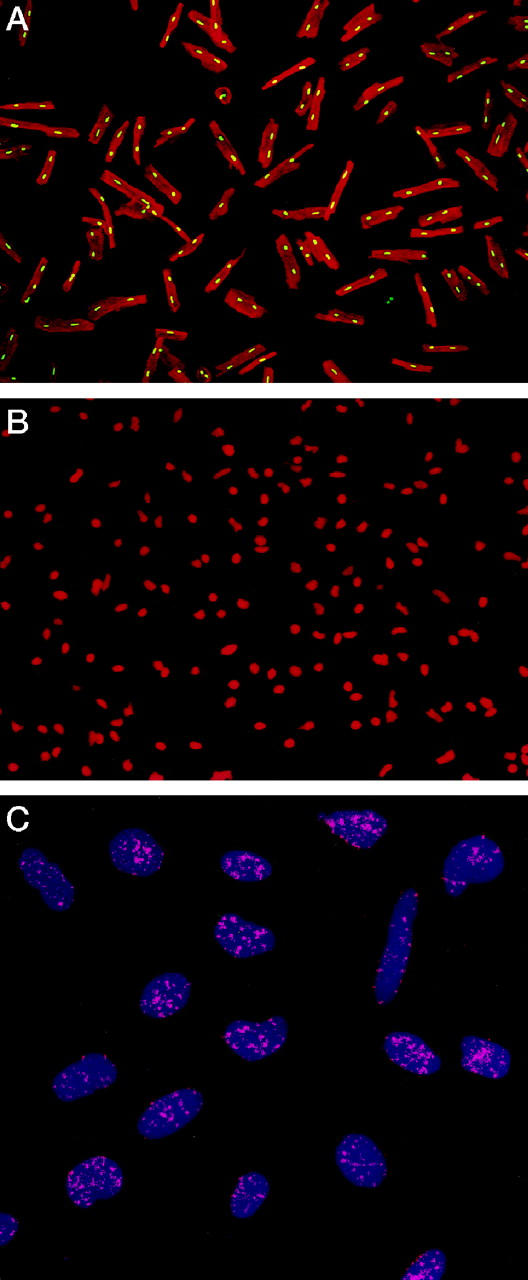
Percoll-treated myocytes (A). Nuclei are stained by PI (yellow fluorescence) and the cytoplasm by α-sarcomeric actin (red fluorescence). Isolated myocyte nuclei are depicted by red fluorescence of PI (B). Nuclei (blue fluorescence) after in situ hybridization with a PNA probe specific for telomeric sequence (C). Red fluorescent dots correspond to individual telomeres. Confocal microscopy; original magnifications, ×150 (A), ×300 (B), ×1500 (C).
Figure 2.

Bivariate distribution of DNA content and telomere length in myocyte nuclei from the left ventricle of fetal (F) and 1-day-old neonatal (N) rats, and rats at 4 (4M), 12 (12M), and 27 (27M) months of age.
The telomeric length frequency histograms obtained by laser scanning Compucyte cytometer and confocal microscopy were analyzed, and the fraction of nuclei with shorter telomeres was calculated. This was achieved by using fetal myocyte nuclei as baseline; telomeres shorter than the average telomeric length in the fetal heart, minus two standard deviations, were considered to have experienced loss of repeat sequences. By laser scanning cytometer and confocal microscopy, telomere shortening was detectable in a small group of fetal (0.5–0.7%) and neonatal (1.2–1.7%) myocytes (Figure 3) ▶ . However, nuclei with shorter telomeres increased at 4 and 12 months, involving 5 to 7% of the entire myocyte population. This value reached approximately 16% at 27 months. Additionally, telomere shortening in senescent myocytes was 2.3- to 2.5-fold greater than in cells at 12 months, and this difference was significant (Figure 3) ▶ .
Figure 3.

Measurements of the percentage of myocyte nuclei with shorter telomeres by laser scanning cytometer and confocal microscopy (see Figure 2 ▶ for symbols). Asterisks indicate statistical differences (P < 0.05) from F (*), N (**), 4M (***), and 12M (****) values; n = 5 in each group.
Telomere shortening was restricted to a subgroup of myocytes, indicating that this marker of cellular aging was applicable to a limited number of cells in the heart. This observation suggested that 16% of myocytes reentered the cell cycle and underwent mitotic division. To characterize the process of myocyte aging in nondividing cells, changes in the expression of p16INK4 in myocyte nuclear proteins were evaluated by immunoprecipitation and Western blot. p16INK4 optical density (OD) increased in myocytes as a function of age (Figure 4) ▶ . This protein was barely detectable in neonatal myocytes (OD, 0.04 ± 0.03, n = 5), becoming more apparent at 4 months (OD, 0.29 ± 0.09, n = 5). However, this difference did not reach statistical significance. Conversely, the quantity of p16INK4 increased markedly at 12 months (OD, 0.71 ± 0.21, n = 5). In comparison with neonatal myocytes a 19-fold upregulation of this protein was measured, and this change was significant (P < 0.002). Even greater were the increases in p16INK4 at 16 (OD, 1.01 ± 0.26, n = 5) and 27 (OD, 1.27 ± 0.31, n = 5) months. From 4 to 16 months, p16INK4 increased 3.5-fold (P < 0.002), and from 4 to 27 months, it increased 4.4-fold (P < 0.0001).
Figure 4.

Detection of p16INK4 in myocyte nuclear proteins by immunoprecipitation and Western blot. See Figure 2 ▶ for symbols.
Measurements of the amount of p16INK4 by immunoprecipitation did not provide information on the relative distribution of this protein at the cellular level. Thus, the biochemical determinations were complemented with the quantitative estimation of the percentage of myocyte nuclei labeled by p16INK4 antibody. This analysis was performed by confocal microscopy (Figure 5) ▶ . The fraction of p16INK4-positive myocytes was low at birth, averaging 9 ± 3% (n = 5) of the cells. This parameter increased significantly with age. Values of 25 ± 7% (n = 5), 51 ± 12% (n = 5), and 82 ± 5% (n = 5) were found at 4, 12, and 27 months, respectively. These morphometric data paralleled the changes detected by immunoprecipitation, documenting that the increases in p16 quantity reflected the progressive involvement of a larger number of cells with age.
Figure 5.
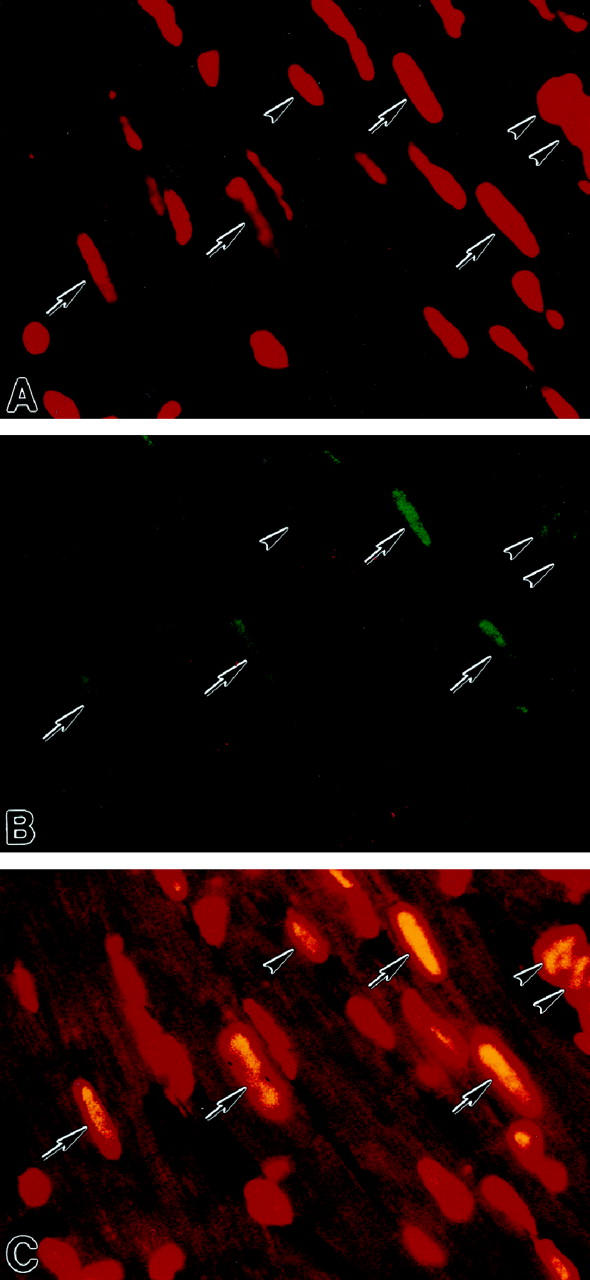
Localization of p16INK4 in myocyte nuclei by confocal microscopy in a rat heart at 27 months. A illustrates by red fluorescence nuclei stained by propidium iodide and B shows by green fluorescence p16INK4 labeling of the majority of nuclei. In C, red fluorescence corresponds to α-sarcomeric actin antibody staining of the myocyte cytoplasm and yellow fluorescence reflects the combination of PI and p16INK4 labeling of nuclei. Arrows, p16-positive myocyte nuclei; arrowheads, p16-positive nonmyocyte nuclei. Original magnifications, ×1200.
Discussion
The results of the current study indicate that, from birth to senescence, the mammalian rat heart is composed of nondividing and dividing myocytes. This distinction is suggested by the length of telomeres, which remains constant in a large fraction of myocyte nuclei from 1 day to 27 months of age. Loss of telomeric DNA is minimal in fetal and neonatal myocytes, suggesting that cell division during gestation and in the immediate postnatal period may be characterized by an almost complete reconstitution of telomeric repeat sequences by telomerase. 31 This is consistent with the high level of bromodeoxyuridine labeling of myocytes in the fetal and 1-day-old rat heart. 22 Conversely, telomeric shortening in this early phase of active cardiac growth may be below the sensitivity of the methodology used. Loss of telomeric DNA per cell division is approximately 50–100 bp 32 and a few population doublings may not produce an appreciable decrease in fluorescence intensity of the PNA probe.
Telomeric shortening occurred in 5 to 7% of adult myocytes, and a value of 16% was found in senescent hearts; 16% of myocytes experienced multiple divisions, and this percentage may reflect the maximum number of cells that can replicate in life. The formation of new myocytes by dividing cells and the development of aging-associated events in nondividing cells complicate the understanding of the time required for cellular senescence to occur. However, p16INK4, a marker of cellular senescence, was detected in 9% of 1-day-old myocytes, indicating that this condition can be reached in approximately 1 month in nonreplicating cells of the rat heart. Gestation lasts 3 weeks in rats. A postnatal rate of cellular senescence of 2.7% myocytes per month (r = 0.98; P < 0.001) could be computed from the immunocytochemistry data obtained at various age intervals. p16 quantity, measured by immunoprecipitation, increased progressively with age, at a rate of 3.6% per month (r = 0.96; P < 0.001), confirming the quantitative results. There are some apparent inconsistencies concerning p16INK4 localization in myocytes. This member of the INK4 family of proteins is a marker of replicative senescence. 16 However, p16INK4 was detected in more than 80% of myocytes in the old heart, when telomere shortening involved 16% of this cell population. The concomitant presence of intact telomeres and p16INK4 staining suggests that this nuclear protein may be involved in senescence of differentiated myocytes. p16INK4 and its mRNA are extremely stable, 19 and they may accumulate in nonreplicating cells. Understanding the high percentage of myocytes labeled by p16INK4 late in life is complex. Because they increase by 2.7% per month, senescent myocytes may have a long life span, accumulating with age. Apoptosis and necrosis affect the aging heart 33 and extensive areas of myocardial damage, involving death of large groups of myocytes, are commonly observed across the ventricular wall. 23 The age-dependent increase in massive cell loss contrasts with the orderly appearance of p16INK4-positive cardiac muscle cells. Although this phenomenon remains unclear and cellular aging is particularly evident in the old heart, animals do not have to be old to possess senescent myocytes.
Shortening of telomeres in myocyte nuclei in vivo has provided critical information against the dogma that the mammalian heart is composed of terminally differentiated myocytes only and tissue regeneration occurs exclusively by hypertrophy of existing cells. 34,35 Loss of telomeric repeat sequences strengthens documentation that cardiac myocytes synthesize DNA 36 and undergo karyokinesis 12,36,37 and cytokinesis. 13 Myocyte nuclei with constant PNA fluorescence intensity vary from 98% to 84% of the cells from birth to senescence. This indicates that the majority of myocytes exits the cell cycle permanently in the early postnatal period 34 and the regenerative reserve is restricted to a small group of muscle cells. Results favoring 12,13 and opposing 38 this possibility have been published.
Culture studies have proposed that telomere shortening may be used as a marker of cellular aging. 4-6 When cells have reached their limit in population doublings, telomeres shorten, growth arrest occurs, and senescence is reached. 1,2,39 Telomeres may continue to shorten and cells may activate their suicide program, undergoing apoptosis. 8 A decrease in length of telomeres with age has been observed in vivo in peripheral blood mononuclear cells. 40 Short telomeres have been detected in fibroblasts from patients with Hutchinson-Gilford progeria 41 and in lymphocytes from subjects with Down’s syndrome. 42 Both diseases are characterized by premature aging. Telomeric repeat sequences are lost in a small fraction of myocytes and the predominant number maintains the initial telomeric length. However, the majority of cells is characterized by p16INK4 labeling, and this larger population may be more susceptible to other age-associated events, including the accumulation of lipofuscin 43,44 and gene products promoting apoptosis, such as p53 and Bax. 45,46
The consequence of fibroblast contamination in the preparation must be acknowledged and discussed. As indicated in Results, 1 to 2% of nonmyocytes were present in the isolated cells. Five thousand nuclei were measured by laser scanning Compucyte cytometer in each case. Because nearly 90% of myocytes are binucleated and 10% are mononucleated, 23,33 5000 nuclei reflected 2250 binucleated myocytes and 500 mononucleated myocytes. A 2% fraction of fibroblasts would imply that 55 nuclei of the 5000 measured belonged to nonmyocytes. This level of contamination corresponds to 1% of the total population of nuclei. This value is well within the SD of the 16% of cells experiencing telomeric shortening. Identical results are obtained when this computation is applied to nuclei measured by confocal microscopy. An additional issue to be addressed concerns nuclear division without cytokinesis, which would lead to an increased number of nuclei per cell and telomeric shortening in the absence of formation of new myocytes. However, this possibility can be excluded because the proportion of mononucleated cells and binucleated cells is minimally affected by age in Fischer 344 rats from 4 to 29 months after birth. 23,33 The percentage of mononucleated cells increases from 5% at 4 months to 11% at 29 months, while binucleated cells decrease from 95 to 88%. 23 These data strongly suggest that karyokinesis without cytokinesis was, at most, a rare event.
Footnotes
Address reprint requests to Jan Kajstura, Ph.D., Department of Medicine, Vosburgh Pavilion, Room 302, New York Medical College, Valhalla, NY 10595. E-mail: jan_kajstura@nymc.edu.
Supported by grants HL-38132, HL-39902, NCI-28704, HL-43023, AG-15746, and AG-17042 from the National Institutes of Health.
References
- 1.Campisi J: Replicative senescence: an old lives’ tale? Cell 1996, 84:497-500 [DOI] [PubMed] [Google Scholar]
- 2.Guarente L: Do changes in chromosomes cause aging? Cell 1996, 86:9-12 [DOI] [PubMed] [Google Scholar]
- 3.Warner HR, Hodes RJ, Pocinki K: What does cell death have to do with aging? J Am Geriatr Soc 1997, 45:1140-1146 [DOI] [PubMed] [Google Scholar]
- 4.Harley CB, Futcher AB, Greider CW: Telomeres shorten during ageing of human fibroblasts. Nature 1990, 345:458-460 [DOI] [PubMed] [Google Scholar]
- 5.Hastie ND, Dempster M, Dunlop MG, Thompson AM, Green DK, Allshire RC: Telomere reduction in human colorectal carcinoma and with ageing. Nature 1990, 346:866-868 [DOI] [PubMed] [Google Scholar]
- 6.Kruk PA, Rampino NJ, Bohr VA: DNA damage and repair in telomeres: relation to aging. Proc Natl Acad Sci USA 1995, 92:258-262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Allsopp RC, Vaziri H, Patterson C, Goldstein S, Younglai EV, Futcher AB, Greider CW, Harley CB: Telomere length predicts replicative capacity of human fibroblasts. Proc Natl Acad Sci USA 1992, 89:10114-10118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huang L, Clarkin KC, Wahl GM: Sensitivity and selectivity of the DNA damage sensor responsible for activating p53-dependent G1 arrest. Proc Natl Acad Sci USA 1996, 93:4827-4832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Karlseder J, Broccoli D, Dai Y, Hardy S, de Lange T: p53- and ATM-dependent apoptosis induced by telomeres lacking TRF2. Science 1999, 283:1321-1325 [DOI] [PubMed] [Google Scholar]
- 10.Soonpaa MH, Field LJ: Assessment of cardiomyocyte DNA synthesis during hypertropy in adult mice. Am J Physiol 1994, 266:H1439−H1445 [DOI] [PubMed]
- 11.Kirshenbaum LA, Schneider MD: The cardiac cell cycle, pocket proteins and p300. Trends Cardiovasc Med 1995, 5:230-235 [DOI] [PubMed] [Google Scholar]
- 12.Anversa P, Kajstura J: Ventricular myocytes are not terminally differentiated in the adult mammalian heart. Circ Res 1998, 83:1-14 [DOI] [PubMed] [Google Scholar]
- 13.Kajstura J, Leri A, Finato N, Di Loreto C, Beltrami CA, Anversa P: Myocyte proliferation in end-stage cardiac failure in humans. Proc Natl Acad Sci USA 1998, 95:8801-8805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hara E, Smith R, Parry D, Hidetoshi T, Stone S, Peters G: Regulation of p16CDKN2 expression and implication for cell immortalization and senescence. Mol Cell Biol 1996, 16:859-867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Uhrbom L, Nistger M, Westermark B: Induction of senescence in human malignant glioma cells by p16INK4A. Oncogene 1997, 15:505-514 [DOI] [PubMed] [Google Scholar]
- 16.Brenner A, Stampfer MR, Aldaz MC: 1998: Increased p16 expression with first senescence arrest in human mammary epithelial cells and extended growth capacity with p16 inactivation. Oncogene 1998, 17:199-205 [DOI] [PubMed] [Google Scholar]
- 17.Huschtscha LI, Noble JR, Neumann AA, Moy EL, Barry P, Melki R, Clark SJ, Reddel RR: Loss of p16INK4 expression by methylation is associated with lifespan extension of human mammary epithelial cells. Cancer Res 1998, 58:139-146 [PubMed] [Google Scholar]
- 18.Vogt M, Haggblom C, Yeargin J, Christiansen-Weber T, Hass M: Independent induction of senescence by p16INK4A and p21CIP1 in spontaneously immortalized human fibroblasts. Cell Growth Differ 1998, 9:139-146 [PubMed] [Google Scholar]
- 19.Sherr CJ, Roberts JM: Inhibition of mammalian G1 cyclin-dependent kinases. Genes Dev 1995, 9:1149-1163 [DOI] [PubMed] [Google Scholar]
- 20.McConnell BB, Starborg M, Brooks S, Peters G: Inhibitors of cyclin-dependent kinases induce features of replicative senescence in early passage human diploid fibroblasts. Curr Biol 1998, 8:351-354 [DOI] [PubMed] [Google Scholar]
- 21.Kajstura J, Cheng W, Reiss K, Anversa P: The IGF-1-IGF-1 receptor system modulates myocyte proliferation but not myocyte cellular hypertrophy in vitro. Exp Cell Res 1994, 215:273-283 [DOI] [PubMed] [Google Scholar]
- 22.Cheng W, Reiss K, Kajstura J, Kowal K, Quaini F, Anversa P: Downregulation of the IGF-1 system parallels the attenuation in the proliferative capacity of rat ventricular myocytes during postnatal development. Lab Invest 1995, 72:646-655 [PubMed] [Google Scholar]
- 23.Anversa P, Palackal T, Sonnenblick EH, Olivetti G, Meggs LG, Capasso JM: Myocyte cell loss and myocyte cellular hyperplasia in the hypertrophied aging rat heart. Circ Res 1990, 67:871-885 [DOI] [PubMed] [Google Scholar]
- 24.Cheng W, Reiss K, Li P, Chun MJ, Kajstura J, Olivetti G, Anversa P: Aging does not affect the activation of the myocyte insulin-like growth factor-1 autocrine system after infarction and ventricular failure in Fischer 344 rats. Circ Res 1996, 78:536-546 [DOI] [PubMed] [Google Scholar]
- 25.Leri A, Claudio PP, Li Q, Wang X, Reiss K, Wang S, Malhotra A, Kajstura J, Anversa P: Stretch-mediated release of angiotensin II induces myocyte apoptosis by activating p53 that enhances the local renin-angiotensin system and decreases the Bcl-2-to-Bax protein ratio in the cell. J Clin Invest 1998, 101:1326-1342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.DePauw ESD, Verwoerd NP, Duinkerken N, Willemze R, Raap AK, Fibbe WE, Tanke HJ: Assessment of telomere length in hematopoietic interphase cells using in situ hybridization and digital fluorescence microscopy. Cytometry 1998, 32:163-169 [DOI] [PubMed] [Google Scholar]
- 27.Bedner E, Burfeind P, Hsieh T, Wu JM, Aguero-Rosenfeld ME, Melamed MR, Horowitz HW, Wormser GP, Darzynkiewicz Z: Cell cycle effects and induction of apoptosis caused by infection of HL-60 cells with human granulocytic ehrlichiosis pathogen measured by flow and laser scanning cytometry. Cytometry 1998, 33:47-55 [PubMed] [Google Scholar]
- 28.Deptala A, Bedner E, Gorczyca W, Darzynkiewicz Z: Activation of nuclear factor kappa B (NF-κB) assayed by laser scanning cytometry (LSC). Cytometry 1998, 33:376-382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leri A, Liu Y, Wang X, Kajstura J, Malhotra A, Meggs LG, Anversa P: Overexpression of IGF-1 attenuates the myocyte renin-angiotensin system in transgenic mice. Circ Res 1999, 84:752-762 [DOI] [PubMed] [Google Scholar]
- 30.Wallenstein S, Zucker CL, Fleiss JL: Some statistical methods useful in circulation research. Circ Res 1980, 47:1-9 [DOI] [PubMed] [Google Scholar]
- 31.Wright WE, Piatyszek MA, Rainey WE, Byrd W, Shay JW: Telomerase activity in human germline and embronic tissues and cells. Dev Genet 1996, 18:173-179 [DOI] [PubMed] [Google Scholar]
- 32.Vaziri H, Dragowska W, Allsopp RC, Thomas TE, Harley CB, Landsorp PM: Evidence for a mitotic clock in human hematopoietic stem cells: loss of telomeric DNA with age. Proc Natl Acad Sci USA 1994, 91:9857-9860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kajstura J, Cheng W, Sarangarajan R, Li P, Li B, Nitahara JA, Chapnick S, Reiss K, Olivetti G, Anversa P: Necrotic and apoptotic myocyte cell death in the aging heart of Fischer 344 rats. Am J Physiol 1996, 271:H1215−H1228 [DOI] [PubMed]
- 34.Zak R: Development and proliferative capacity of cardiac muscle cells. Circ Res 1974, 35(Suppl V):17-26 [PubMed] [Google Scholar]
- 35.Tam SKC, Gu W, Mahdavi V, Nadal-Ginard B: Cardiac myocyte terminal differentiation: potential for cardiac regeneration. Ann NY Acad Sci 1995, 752:72-79 [DOI] [PubMed] [Google Scholar]
- 36.Beltrami CA, Di Loreto C, Finato N, Rocco M, Artico D, Cigola E, Gambert SR, Olivetti G, Kajstura J, Anversa P: Proliferating cell nuclear antigen (PCNA), DNA synthesis and mitosis in myocytes following cardiac transplantation in man. J Mol Cell Cardiol 1997, 29:2789-2802 [DOI] [PubMed] [Google Scholar]
- 37.Quaini F, Cigola E, Lagrasta C, Saccani G, Quaini E, Rossi C, Olivetti G, Anversa P: End-stage cardiac failure in humans is coupled with the induction of proliferating cell nuclear antigen and nuclear mitotic division in ventricular myocytes. Circ Res 1994, 75:1050-1063 [DOI] [PubMed] [Google Scholar]
- 38.Soonpaa MH, Field LJ: Survey of studies examining mammalian cardiomyocyte DNA synthesis. Circ Res 1998, 83:15-26 [DOI] [PubMed] [Google Scholar]
- 39.Allsopp RC, Chang E, Kashefi-Aazam M, Rogaev EI, Piatyszek MA, Shay JW, Harley CB: Telomere shortening is associated with cell division in vitro and in vivo. Exp Cell Res 1995, 220:194-200 [DOI] [PubMed] [Google Scholar]
- 40.Iwama H, Ohyashiki K, Ohyashiki JH, Hayashi S, Yahata N, Ando K, Toyama A, Hoshika M, Takasaki M, Mori M, Shay J: Telomeric length and telomerase activity vary with age in peripheral blood cells obtained from normal individuals. Hum Genet 1998, 102:397-402 [DOI] [PubMed] [Google Scholar]
- 41.Johnson FB, Sinclair DA, Guarente L: Molecular biology of aging. Cell 1999, 96:291-302 [DOI] [PubMed] [Google Scholar]
- 42.Vaziri H, Schachter F, Uchida I, Wei L, Zhu X, Effros R, Cohen D, Harley CB: Loss of telomeric DNA during aging of normal and trisomy 21 human lymphocytes. Am J Hum Genet 1993, 52:661-667 [PMC free article] [PubMed] [Google Scholar]
- 43.Ivy GO, Schottler F, Wenzel J, Baudry M, Lynch G: Inhibitors of lysosomal enzymes: accumulation of lipofuscin-like dense bodies in the brain. Science 1984, 226:985-987 [DOI] [PubMed] [Google Scholar]
- 44.von Zglinicki T, Nilsson E, Docke WD, Brunk UT: Lipofuscin accumulation and ageing of fibroblasts. Gerontology 1995, 41:95-105 [DOI] [PubMed] [Google Scholar]
- 45.Sugrue MM, Shin DY, Lee SW, Aaronson SA: Wild-type p53 triggers a rapid senescence program in human tumor cells lacking functional p53. Proc Natl Acad Sci USA 1997, 94:9648-9653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Oltvai ZN, Milliman CL, Korsmeyer SJ: Bcl-2 heterodimerizes in vivo with a conserved homolog, bax, that accelerates programmed cell death. Cell 1993, 73:609-619 [DOI] [PubMed] [Google Scholar]