

Abstract
Gastrointestinal stromal tumors (GISTs) are the most common mesenchymal neoplasms arising in the gastrointestinal tract. GISTs express the KIT receptor tyrosine kinase, and many cases have activating mutations in the KIT juxtamembrane region. We now report an analysis of KIT cDNA and genomic sequences in eight GISTs that lack juxtamembrane region mutations. Six cases contained heterozygous exon 9 mutations in which six nucleotides, encoding Ala-Tyr, were duplicated. The other two cases contained homozygous exon 13 missense mutations, resulting in substitution of Glu for Lys642, that were associated with constitutive KIT tyrosine phosphorylation. Sequence analysis of DNAs from nonneoplastic companion tissues revealed that both the exon 9 and exon 13 mutations were somatic. These are the first descriptions, in any tumor, of mutations in KIT exons encoding the C-terminal end of the extracellular domain and the first part of the split kinase domain. These findings indicate that KIT may be activated by mutations in at least three domains—extracellular, juxtamembrane, and kinase—in GISTs.
The KIT protein is a type III receptor tyrosine kinase (RTK). 1 Interactions between KIT and its ligand, SCF, are important in the development and maintenance of hematopoietic cells, melanocytes, germ cells, and the interstitial cells of Cajal. 2-5 Although expression is restricted to a relatively small number of cell types, KIT nonetheless plays a major oncogenic role. Activating KIT mutations have been identified in human mast cell tumors, 6-9 myelofibrosis, 10 chronic myelogenous leukemia, 10 germ cell tumors, 11 and gastrointestinal stromal tumors (GISTs). 12,13 To date, KIT oncogenic mutations have been identified in exon 2 (myelofibrosis and chronic myelogenous leukemia), the exon 11 juxtamembrane domain (mast cell tumors and GISTs), and the exon 17 phosphotransferase domain (mast cell tumors and germ cell tumors). In the present study we evaluated eight GISTs that expressed KIT (as determined immunohistochemically) but lacked exon 11 mutations. We reasoned that some of these cases might have novel KIT mutations and that detection of such mutations might lead to a fuller understanding of KIT activating mechanisms.
Materials and Methods
Patient Samples
The study group consisted of eight primary intraabdominal neoplasms (one gastric, five small bowel, one mesenteric, one abdominal wall) biopsied at Brigham and Women’s Hospital and the Dana-Farber Cancer Institute. All cases were evaluated histologically by two of the authors (BPR and CDMF) and were diffusely and strongly immunoreactive with KIT antibody (A-4052, rabbit polyclonal antiserum, 1:100; DAKO Corporation, Carpinteria, CA), according to analysis by an avidin-biotin-peroxidase complex method after microwave antigen retrieval. All cases lacked mutations of KIT exons 10 and 11, a region containing the entire coding sequence for the KIT juxtamembrane domain, as determined by sequencing of genomic DNAs isolated from frozen tumor materials.
RT-PCR Analysis of KIT Transcripts
RNAs were isolated from frozen tissue, using TRIzol (GIBCO BRL Life Technologies, Gaithersburg, MD), and cDNAs were synthesized using AMV reverse transcriptase and random 9-mer primers (TaKaRa Shuzo Co., Seoul, Korea), according to the manufacturer’s protocols. Polymerase chain reaction (PCR) amplifications were performed using Taq DNA polymerase and oligonucleotide primer sequences as described by Furitsu et al. 6 DNAs were amplified in 20-μl PCR reactions of 0.5 minute at 94°C, 0.5 minute at 60°C, and 1 minute at 72°C for 35 cycles. Amplified products were purified with the QIAquick gel extraction kit (Qiagen, Valencia, CA) and directly sequenced in the forward and reverse directions with ABI BigDye terminators (Applied Biosystems, Foster City, CA). Sequences were analyzed on an ABI Prism 377 sequencer (Applied Biosystems), and alignments and mutation scanning were performed using Sequence Navigator (Applied Biosystems) and BLAST (National Center for Biotechnology Information) software.
Genomic DNA Sequencing
DNAs were isolated from frozen GIST specimens by the use of NaOH boiling preps. DNAs were isolated from paraffin sections of nonneoplastic companion tissues (evaluated to exclude the possibility of constitutional polymorphisms), using standard proteinase K digestion methods. Intronic primers were chosen using the Whitehead Genome Center Primer3 software, and all intronic primers were numbered according to KIT genomic sequence Genbank number U63834. The PCR cycling conditions were identical to those in the cDNA amplifications.
Exon 9: F/KIT/74056, 5′-ATTTATTTTCCTAGAGTAAGCCAGGG-3′ R/KIT/74360, 5′-ATCATGACTGATATGGTAGACAGAGC-3′
Exons 12–13: F/KIT/75961, 5′-ATTTTGAAACTGCACAAATGGTCCTT-3′ R/KIT/76499, 5′-GCAAGAGAGAACAACAGTCTGGGTAA-3′
The PCR products were purified, as described above, and cycle sequencing was performed using the following intronic primer pairs:
Exon 9: Same as for PCR
Exons 12–13: F/KIT/76032, 5′-CACCATCACCACTTACTTGTTGTCT-3′ R/KIT/76403, 5′-GACAGACAATAAAAGGCAGCTTGGAC-3′
Fluorescence in Situ Hybridization
Total yeast DNA was isolated from CEPH yeast artificial chromosome clone, 840_E_11, containing the human KIT locus at chromosome band 4q12. Two hundred nanograms of 840_E_11 DNA was biotin labeled by random-octamer priming (BioPrime Kit; Gibco, Rockville, MD), and 500 ng of the labeled YAC was cohybridized with a digoxigenin-labeled chromosome 4 pericentromeric probe (D4Z1) against cytogenetic preparations of GIST cells. YAC and centromere probe detection was carried out with strepavidin-FITC (Zymed Laboratories, South San Francisco, CA) and rhodamine anti-digoxigenin (Zymed Laboratories), respectively.
Evaluation of KIT Tyrosine Phosphorylation
GIST and malignant peripheral nerve sheath tumor (MPNST) cells were lysed in ice-cold 1% NP-40, 50 mmol/L Tris (pH 8.0), 100 mmol/L sodium fluoride, 30 mmol/L sodium pyrophosphate, 2 mmol/L sodium molybdate, 5 mmol/L EDTA, 2 mmol/L sodium vanadate, 5 μg/ml aprotinin, 5 μg/ml leupeptin, and 50 μg/ml phenylmethylsulfonyl fluoride (lysis buffer). Lysates were incubated with anti-KIT (C-19; Santa Cruz) for 2 hours, followed by 20 μl of protein A-Sepharose (Zymed Laboratories) for 1 hour at 4°C. Immunoprecipitates were washed three times in lysis buffer, then eluted at 100°C into 40 μl of sodium dodecyl sulfate-polyacrylamide gel electrophores (SDS-PAGE) loading buffer and resolved by SDS-PAGE under reducing conditions (4–12% gradient gels). Immunoblotting was accomplished by electrophoretic transfer to polyvinyl pyrrolidine fluoride membranes (Millipore), blocking in phosphate-buffered saline containing 0.2% Tween-20 and 5% dry milk for 1 hour, then sequential incubation with murine anti-phosphotyrosine (PY99; Santa Cruz) and horseradish peroxidase anti-mouse Ig (Amersham, Piscataway, NJ). Detection was made by chemoluminescence (SuperSignal West Femto Maximum Sensitivity Substrate; Pierce, Rockford, IL).
Results and Discussion
Initial mutation screening was carried out by sequence analysis of the entire KIT coding sequence, and potential mutations were then confirmed by genomic sequencing. Each of the eight GISTs contained a single mutation. Six cases had heterozygous exon 9 mutations (1530ins6) in which six nucleotides were duplicated (Figure 1A) ▶ . In one of these cases, the 1530ins6 mutation was demonstrated both in the primary tumor and in a subsequently biopsied metastasis. cDNA and genomic sequencing indicated a 50:50 ratio, for wild-type:mutant transcripts and alleles, respectively, in the 1530ins6 mutation cases (Figure 1A) ▶ . These findings suggest that transcriptional regulation of the mutant 1530ins6 allele is unperturbed, compared to that of the nonmutant allele. In contrast, the remaining two GISTs contained homozygous exon 13 missense mutations, 1945A>G. Wildtype KIT genomic and cDNA sequences were undetectable in both of these cases (Figure 1B) ▶ , and loss of KIT allelic heterozygosity was evidenced by loss of constitutional polymorphisms in exon 10, intron 16, and intron 17 (case 7) and in exon 10 and intron 17 (case 8). Dual-color fluorescence in situ hybridization, using a KIT-containing YAC clone, showed no evidence of KIT deletion, or other rearrangement, in either copy of chromosome 4 from the GISTs with exon 13 mutations (Figure 1C) ▶ . Together, these data demonstrate loss of the wild-type KIT allele and are consistent with duplication of the 1945A>G mutant allele.
Figure 1.
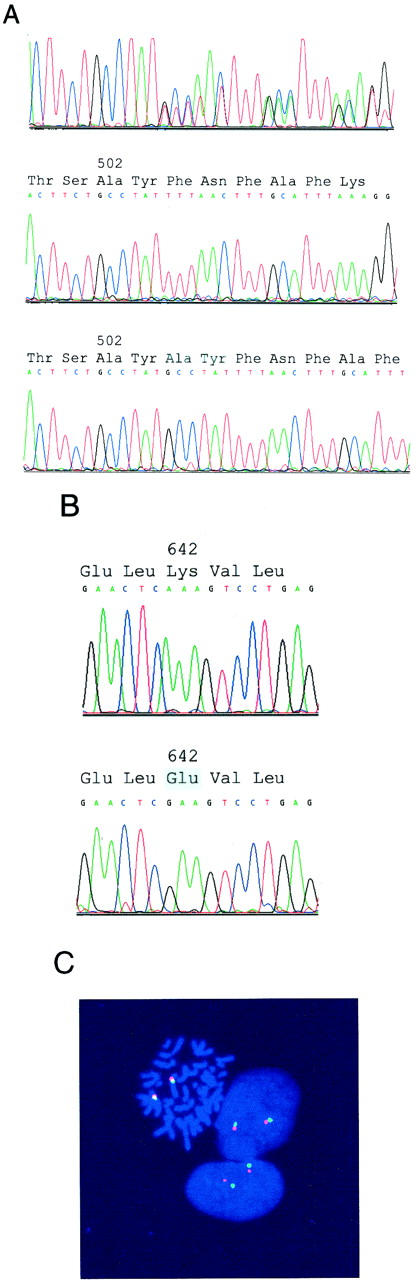
Genomic evaluations of KIT by sequencing (A and B) and fluorescence in situ hybridization (C). A: Sequence analysis of total genomic DNA demonstrates a heterozygous exon 9 6-bp insertion/duplication in a GIST (top). Sequence analysis of cloned PCR products, from the same GIST as shown at top, demonstrates the wild-type (middle) and mutant (bottom) alleles. B: Sequence analysis of total genomic DNA demonstrates a homozygous exon 13 A>G missense mutation. The GIST sequence is at bottom, and the wild-type sequence, obtained from adjacent nonneoplastic tissue, is at top. C: Fluorescence in situ hybridization demonstrates nondeleted KIT, in the same GIST as in B, consistent with loss of the chromosome 4 homolog containing the wild-type KIT allele and duplication of the chromosome 4 homolog containing the mutant KIT allele. FITC signals (green) identify the KIT loci at chromosome band 4q12, and rhodamine signals (red) identify the chromosome 4 pericentromeric region.
The exon 9 1530ins6 mutations are predicted to encode KIT proteins with duplication of amino acid residues Ala502 and Tyr503. The exon 13 1945A>G mutations result in substitution of a Glu at Lys642. To confirm that the exon 9 and 13 mutations were acquired, we sequenced nonneoplastic cell genomic DNAs from the same patients. The companion nonneoplastic specimens contained only wild-type KIT sequences, indicating that the mutations were somatic—and presumably tumor-restricted—rather than being germline mutations or polymorphisms (Figure 1B) ▶ .
Most GISTs are characterized by diffuse KIT protein expression, and many GISTs contain oncogenic KIT mutations. 12 To date, GIST KIT mutations have been reported only in the exon 11 juxtamembrane domain. 12-17 Oncogenic KIT exon 11 mutations are associated with constitutive ligand-independent receptor dimerization and activation of the kinase domain. 9,12 Although KIT exon 11 juxtamembrane domain mutations are found in most GISTs, a subset of cases lack such mutations. In our experience (B. P. Rubin and J. A. Fletcher, unpublished data) 32 of 45 GISTs expressed KIT (as determined immunohistochemically) and contained exon 11 mutations. The remaining 13 cases expressed KIT but lacked exon 11 mutations. We now report that eight of these 13 GISTs contain somatic mutations in KIT exon 9 (extracellular domain) or exon 13 (first part of the split tyrosine kinase domain). Clinicopathological correlations, for the entire series, will be reported separately.
Although KIT exon 9 mutations have not been described previously, it is likely that the 1530ins6 mutations activate the receptor via ligand-independent oligomerization. Extracellular region mutations have been characterized in several other receptor tyrosine kinase genes, including FGFR2, FGFR3, and RET, and in all cases these mutations are associated with constitutive receptor oligomerization. 18-21 Although the mechanistic implications of the novel exon 13 (1945A>G) mutations are unclear, it is worth noting that Lys642 is conserved in all members of the type III RTK family, highlighting its likely functional importance. KIT is constitutively tyrosine-phosphorylated, in a ligand-independent manner, in a cell line established from one of our exon 13 mutant (1945A>G) GISTs (Figure 2) ▶ . Therefore, the exon 13 mutation is almost certainly activating. The Lys642→Glu (K642E) substitution imparts a negative charge and would likely alter the three-dimensional structure of the mutant protein. This potential conformational change might either encourage receptor oligomerization or, alternatively, might be associated with constitutive kinase activation in the absence of receptor oligomerization. 22 It is intriguing that wild-type KIT sequences were undetectable in either of the two GISTs with exon 13 mutations. This finding is notable, because heterodimerization—i.e., interaction between mutant and nonmutant receptors—is a potential activating mechanism for receptor tyrosine kinase oncoproteins. The homozygous nature of the K642E mutations demonstrates that heterodimerization is not requisite, in vivo, for oncogenic activation. It is conceivable that heterodimerization blunts the activating impact of the K642E mutation, thereby providing selective advantage to cells in which the wild-type allele has been deleted. Alternatively, this mutation might be intrinsically less activating than the generally heterozygous mutations found in exons 9 and 11. GIST precursors might require two copies of the mutant allele (loss of the wild-type allele followed by duplication of the mutant) for neoplastic transformation.
Figure 2.

Demonstration of high-level, ligand-independent, KIT tyrosine phosphorylation in a GIST cell line, established from case 7, expressing the KIT K642E oncoprotein. Immunoprecipitations and Western blotting were with KIT and phosphotyrosine antibodies, respectively. K642E mutant KIT is tyrosine phosphorylated in GIST cells cultured in 15% fetal bovine serum (second lane), serum-free media with stem cell factor 100 ng/ml (third lane), and serum-free media without stem cell factor (fourth lane). Low-level KIT activation, in a malignant peripheral nerve sheath tumor cell line (ST88-14), 25,26 is not detected on this 10-second film exposure (first lane).
In summary, our finding of novel KIT exon 9 and 13 mutational hotspots underscores the central role of KIT oncogenic activation in GIST pathogenesis. Functional characterization of the KIT exon 9 and 13 mutations should provide new insights into the varied mechanisms of KIT-mediated oncogenesis in GISTs and other tumors. KIT signal transduction pathways regulate proliferation, differentiation, migration, and survival of various cell types, including hematopoietic stem cells, mast cells, melanocytes, germ cells, and the interstitial cells of Cajal. 2-4,6,23,24 Therefore anti-KIT oncological therapies will likely be toxic, particularly to the bone marrow compartment. Future elucidation of KIT oncogenic mechanisms might enable development of highly specific therapies that target particular KIT mutational pathways and minimize toxicity to KIT-dependent nonneoplastic cell lineages.
Footnotes
Address reprint requests to Dr. Brian P. Rubin or Dr. Jonathan A. Fletcher, Department of Pathology, Brigham and Women’s Hospital, 75 Francis Street, Boston, MA 02115. E-mail: bprubin@bics.bwh.harvard.edu
Drs. Lux and Rubin contributed equally to this work.
References
- 1.Fleischman RA: From white spots to stem cells: the role of the Kit receptor in mammalian development. Trends Genet 1993, 9:285-290 [DOI] [PubMed] [Google Scholar]
- 2.Russell ES: Hereditary anemias of the mouse: a review for geneticists. Adv Genet 1979, 20:357-459 [PubMed] [Google Scholar]
- 3.Kitamura Y, Go S: Decreased production of mast cells in S1/S1d anemic mice. Blood 1979, 53:492-497 [PubMed] [Google Scholar]
- 4.Huizinga JD, Thuneberg L, Kluppel M, Malysz J, Mikkelsen HB, Bernstein A: W/kit gene required for interstitial cells of Cajal and for intestinal pacemaker activity. Nature 1995, 373:347-349 [DOI] [PubMed] [Google Scholar]
- 5.Isozaki K, Hirota S, Nakama A, Miyagawa J, Shinomura Y, Xu Z, Nomura S, Kitamura Y: Disturbed intestinal movement, bile reflux to the stomach, and deficiency of c-kit-expressing cells in Ws/Ws mutant rats. Gastroenterology 1995, 109:456-464 [DOI] [PubMed] [Google Scholar]
- 6.Furitsu T, Tsujimura T, Tono T, Ikeda H, Kitayama H, Koshimizu U, Sugahara H, Butterfield JH, Ashman LK, Kanayama Y: Identification of mutations in the coding sequence of the proto-oncogene c-kit in a human mast cell leukemia cell line causing ligand-independent activation of c-kit product. J Clin Invest 1993, 92:1736-1744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tsujimura T, Furitsu T, Morimoto M, Isozaki K, Nomura S, Matsuzawa Y, Kitamura Y, Kanakura Y: Ligand-independent activation of c-kit receptor tyrosine kinase in a murine mastocytoma cell line P-815 generated by a point mutation. Blood 1994, 83:2619-2626 [PubMed] [Google Scholar]
- 8.Tsujimura T, Furitsu T, Morimoto M, Kanayama Y, Nomura S, Matsuzawa Y, Kitamura Y, Kanakura Y: Substitution of an aspartic acid results in constitutive activation of c-kit receptor tyrosine kinase in a rat tumor mast cell line RBL-2H3. Int Arch Allergy Immunol 1995, 106:377-385 [DOI] [PubMed] [Google Scholar]
- 9.Tsujimura T, Morimoto M, Hashimoto K, Moriyama Y, Kitayama H, Matsuzawa Y, Kitamura Y, Kanakura Y: Constitutive activation of c-kit in FMA3 murine mastocytoma cells caused by deletion of seven amino acids at the juxtamembrane domain. Blood 1996, 87:273-283 [PubMed] [Google Scholar]
- 10.Nakata Y, Kimura A, Katoh O, Kawaishi K, Hyodo H, Abe K, Kuramoto A, Satow Y: c-kit point mutation of extracellular domain in patients with myeloproliferative disorders. Br J Haematol 1995, 91:661-663 [DOI] [PubMed] [Google Scholar]
- 11.Tian Q, Frierson HF, Jr, Krystal GW, Moskaluk CA: Activating c-kit gene mutations in human germ cell tumors. Am J Pathol 1999, 154:1643-1647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hirota S, Isozaki K, Moriyama Y, Hashimoto K, Nishida T, Ishiguro S, Kawano K, Hanada M, Kurata A, Takeda M, Muhammad Tunio G, Matsuzawa Y, Kanakura Y, Shinomura Y, Kitamura Y: Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science 1998, 279:577–580 [DOI] [PubMed]
- 13.Nakahara M, Isozaki K, Hirota S, Miyagawa J, Hase-Sawada N, Taniguchi M, Nishida T, Kanayama S, Kitamura Y, Shinomura Y, Matsuzawa Y: A novel gain-of-function mutation of c-kit gene in gastrointestinal stromal tumors. Gastroenterology 1998, 115:1090-1095 [DOI] [PubMed] [Google Scholar]
- 14.Ernst SI, Hubbs AE, Przygodzki RM, Emory TS, Sobin LH, O’Leary TJ: KIT mutation portends poor prognosis in gastrointestinal stromal/smooth muscle tumors. Lab Invest 1998, 78:1633-1636 [PubMed] [Google Scholar]
- 15.Lasota J, Jasinski M, Sarlomo-Rikala M, Miettinen M: Mutations in exon 11 of c-Kit occur preferentially in malignant versus benign gastrointestinal stromal tumors and do not occur in leiomyomas or leiomyosarcomas. Am J Pathol 1999, 154:53-60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moskaluk CA, Tian Q, Marshall CR, Rumpel CA, Franquemont DW, Frierson HF, Jr: Mutations of c-kit JM domain are found in a minority of human gastrointestinal stromal tumors. Oncogene 1999, 18:1897-1902 [DOI] [PubMed] [Google Scholar]
- 17.Taniguchi M, Nishida T, Hirota S, Isozaki K, Ito T, Nomura T, Matsuda H, Kitamura Y: Effect of c-kit mutation on prognosis of gastrointestinal stromal tumors. Cancer Res 1999, 59:4297-4300 [PubMed] [Google Scholar]
- 18.d’Avis PY, Robertson SC, Meyer AN, Bardwell WM, Webster MK, Donoghue DJ: Constitutive activation of fibroblast growth factor receptor 3 by mutations responsible for the lethal skeletal dysplasia thanatophoric dysplasia type I. Cell Growth Differ 1998, 9:71-78 [PubMed] [Google Scholar]
- 19.Bongarzone I, Vigano E, Alberti L, Borrello MG, Pasini B, Greco A, Mondellini P, Smith DP, Ponder BA, Romeo G, Pierotti MA: Full activation of MEN2B mutant RET by an additional MEN2A mutation or by ligand GDNF stimulation. Oncogene 1998, 16:2295-2301 [DOI] [PubMed] [Google Scholar]
- 20.Robertson SC, Meyer AN, Hart KC, Galvin BD, Webster MK, Donoghue DJ: Activating mutations in the extracellular domain of the fibroblast growth factor receptor 2 function by disruption of the disulfide bond in the third immunoglobulin-like domain. Proc Natl Acad Sci USA 1998, 95:4567-4572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Webster MK, Donoghue DJ: FGFR activation in skeletal disorders: too much of a good thing. Trends Genet 1997, 13:178-182 [DOI] [PubMed] [Google Scholar]
- 22.Kitayama H, Kanakura Y, Furitsu T, Tsujimura T, Oritani K, Ikeda H, Sugahara H, Mitsui H, Kanayama Y, Kitamura Y: Constitutively activating mutations of c-kit receptor tyrosine kinase confer factor-independent growth and tumorigenicity of factor-dependent hematopoietic cell lines. Blood 1995, 85:790-798 [PubMed] [Google Scholar]
- 23.Kitamura Y, Go S, Hatanaka K: Decrease of mast cells in W/Wv mice and their increase by bone marrow transplantation. Blood 1978, 52:447-452 [PubMed] [Google Scholar]
- 24.Maeda H, Yamagata A, Nishikawa S, Yoshinaga K, Kobayashi S, Nishi K, Nishikawa S: Requirement of c-kit for development of intestinal pacemaker system. Development 1992, 116:369-375 [DOI] [PubMed] [Google Scholar]
- 25.Ryan JJ, Klein KA, Neuberger TJ, Leftwich JA, Westin EH, Kauma S, Fletcher JA, DeVries GH, Huff TF: Role for the stem cell factor/KIT complex in Schwann cell neoplasia and mast cell proliferation associated with neurofibromatosis. J Neurosci Res 1994, 37:415-432 [DOI] [PubMed] [Google Scholar]
- 26.Badache A, Muja N, De Vries GH: Expression of Kit in neurofibromin-deficient human Schwann cells: role in Schwann cell hyperplasia associated with type 1 neurofibromatosis. Oncogene 1998, 17:795-800 [DOI] [PubMed] [Google Scholar]