

Abstract
Galectin-3 is a member of a growing family of β-galactoside-binding animal lectins. Previous studies have demonstrated a variety of biological activities for this protein in vitro, including activation of cells, modulation of cell adhesion, induction of pre-mRNA splicing, and regulation of apoptosis. To assist in fully elucidating the physiological and pathological functions of this protein, we have generated galectin-3-deficient (gal3−/−) mice by targeted interruption of the galectin-3 gene. Gal3−/− mice consistently developed fewer inflammatory cell infiltrations in the peritoneal cavities than the wild-type (gal3+/+) mice in response to thioglycollate broth treatment, mainly due to lower numbers of macrophages. Also, when compared to cells from gal3+/+ mice, thioglycollate-elicited inflammatory cells from gal3−/− mice exhibited significantly lower levels of NF-κB response. In addition, dramatically different cell-spreading phenotypes were observed in cultured macrophages from the two genotypes. Whereas macrophages from gal3+/+ mice exhibited well spread out morphology, those from gal3−/− mice were often spindle-shaped. Finally, we found that peritoneal macrophages from gal3−/− mice were more prone to undergo apoptosis than those from gal3+/+ mice when treated with apoptotic stimuli, suggesting that expression of galectin-3 in inflammatory cells may lead to longer cell survival, thus prolonging inflammation. These results strongly support galectin-3 as a positive regulator of inflammatory responses in the peritoneal cavity.
Galectins are members of a recently identified family of β-galactoside-binding animal lectins. 1 Presently more than ten members have been identified, and additional homologues are likely to be discovered. Existing information suggests that they may be involved in a multitude of biological processes including regulation of cell growth and apoptosis, neoplastic transformation, and inflammatory responses. 2-5 One of the best characterized members, galectin-3, is composed of a carboxyl-terminal lectin domain connected to an amino-terminal nonlectin part consisting primarily of short tandem repeats. 6-8 Galectin-3 is widely distributed in tissues and is found in various epithelial cells, dendritic cells, and inflammatory cells. 9 The expression of this lectin is correlated with cell proliferation, 10,11 cell differentiation, 12,13 and transactivation by viral proteins. 14
A number of biological activities of galectin-3 have been demonstrated in vitro. This lectin has been shown to activate various cells, including mast cells, 15,16 neutrophils, 17 monocytes/macrophages, 12 and lymphocytes. 14,18 The effects of this lectin in cell adhesion have also been demonstrated. 19-21 These extracellular functions suggest a possible role for this protein in modulation of immune reactions and inflammatory responses. In addition, evidence for various intracellular activities of galectin-3 are also available. It has been identified as a component of hnRNP, 22 as well as a factor in pre-mRNA splicing, 23 and shown to have anti-apoptotic activity, possibly through a mechanism involving its interactions with Bcl-2 family members, with which this lectin shares sequence similarity. 24,25
Galectin-3 has been found to be overexpressed in certain pathological conditions, including human atherosclerotic lesions. 26 The association of this lectin with neoplastic transformation has also been extensively documented. It is overexpressed in some types of cancer for which the normal parental cells do not express the protein; the examples include specific types of lymphoma, 14,27 thyroid carcinoma, 28,29 and hepatocellular carcinoma. 30 However, down-regulation of this lectin has been observed in other kinds of neoplasms, including colon, 31,32 breast, 33 ovarian, 34 and uterine carcinoma, 35 although it has also been observed that galectin-3 expression correlates positively with the progression of colon carcinoma. 36,37 Studies of cells transfected with galectin-3 cDNA have indeed provided evidence for its role in tumor transformation and metastasis. 38
Therefore, functions of galectin-3 appear to be multifaceted, extending to both intracellular and extracellular compartments. Because of its wide tissue distribution and pleiotropic effects in many systems, galectin-3 is likely to be involved in a variety of physiological and pathological processes. In an attempt to elucidate more definitively the biological functions of galectin-3, we have generated a mouse model in which this gene is inactivated through homologous recombination. We initially focused on the study of inflammatory responses in these mice and have found that the galectin-3 deficiency results in significantly altered inflammation elicited in the peritoneal cavity by thioglycollate broth.
Materials and Methods
Generation of Galectin-3-Null Mice
A vector used for homologous recombination was constructed from the cloned galectin-3 genomic DNA. 39 As shown in Figure 1, a ▶ segment from exon 4 to exon 5 within the mouse galectin-3 gene was inserted into pMC1Neo (Stratagene, La Jolla, CA) upstream of the thymidine kinase promoter-Neo cassette. Another segment from exon 5 to exon 6 followed downstream from the Neo cassette. Thus, exon 5 is interrupted by the Neo gene in this vector construct. Murine stem cells, D3, were electroporated with this vector, using procedures previously described. 40 G418 resistant cells were screened for homologous recombination by polymerase chain reaction (PCR) and Southern blotting, using procedures described below. Screening of 894 clones resulted in two with successful homologous recombination.
Figure 1.

Vector construction and homologous recombination in the galectin-3 gene. A: The targeting vector was constructed by replacing the 0.5-kb XbaI-XbaI segment at the intron 4-exon 5 junction with the Neo cassette from the pMC1NeoPLA vector. B: The homologous recombinant galectin-3 gene shown with various DNA segments used as probes. C: Predicted DNA sizes from wild-type and homologous recombinant genes in Southern blotting using HindIII and XbaI and the specific flanking probe 1. D: Southern blot of two homologous recombinant clones after digestion with HindIII (H) and XbaI (X) and detection with probe 1. The upper band in both lanes for clones 4A2 and 9A4 were detected using probe 4, specific for the Neo gene (data not shown). The profile for the wild-type parental ES cell D3 is also shown.
One clone was propagated and injected into 3.5-day-old blastocysts from C57BL/6 mice and the injected blastocysts were implanted into pseudo-pregnant CD1 mothers. Male chimeric mice were bred to C57BL/6 females to produce mice hemizygous for the galectin-3 null mutant (gal3+/−). Interbreeding gal3+/− mice resulted in mice homozygous for the galectin-3-null condition (gal3−/−). Gal3−/− mice were viable and fertile. For experimentation, wild-type (gal3+/+) mice produced by gal3+/− interbreeding were carried in a separate lineage as controls. Both gal3+/+ and gal3−/− mice were propagated and maintained in standard specific-pathogen-free environments.
Polymerase Chain Reaction and Southern Blot Analysis
Polymerase chain reactions were performed under standard conditions using Taq DNA polymerase (Promega, Madison, WI) on a Perkin-Elmer 9600 or Ericomp EZ cycler. Southern blotting analyses were performed by capillary transfer of DNA to charged nylon membranes (BioRad, Richmond, CA or Amersham, Arlington Heights, IL). Membranes were probed with an intron 3 fragment corresponding to probe 1, or probe 4 corresponding to the Neo cassette, to determine homologous recombination, as shown in Figure 1B ▶ , radiolabeled by random priming with [32P]-dATP.
Immunoblot Analysis
Tissues were extracted with 20 mmol/L Tris-HCl, pH 7.5 containing 10 mmol/L EDTA, 0.15 mol/L NaCl, 1% Triton X-100 (v/v) and protease inhibitors 0.24 u/ml Aprotinin, 1 μg/ml pepstatin, 1 μg/ml leupeptin, 1 mmol/L phenylmethylsulfonyl fluoride, and 100 μg protein from each extract was then adsorbed with lactosyl-Sepharose 4B. 41 The bound proteins were eluted, separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and analyzed by immunoblotting using specific rabbit antibodies as described. 12
Induction of Peritoneal Inflammation
Mice were injected with 1 ml of autoclaved Brewer’s thioglycollate broth as described. 42 At various intervals, peritoneal cells were harvested by lavage with minimum essential medium containing Earle’s salts (MEM). Cells were washed once with culture medium before subsequent manipulations. For quantitation of leukocyte subpopulations, the cells in the lavage fluid were allowed to attach to glass slides by cytospin, treated with soluble Wright’s stain (Leukostat, Fisher Scientific, Pittsburgh, PA) and identified as macrophages, eosinophils, neutrophils, and lymphocytes by standard morphology. Cell counts were obtained in triplicate for each sample from 100–200 cells using a 100× oil immersion objective.
Electrophoretic Mobility Shift Assay (EMSA)
Nuclear extracts were prepared by a previously described method 43 with slight modifications 44 and EMSA was performed as described. 45 Briefly, the nuclear extracts (2.5 μg protein) in 12 μl of binding buffer (5 mmol/L HEPES, pH 7.8, 5 mmol/L MgCl2, 50 mmol/L KCl, 0.5 mmol/L dithiothreitol, 0.4 mg/ml poly(dI-dC) (Pharmacia, Piscataway, NJ), 0.1 mg/ml sonicated double-stranded salmon sperm DNA, and 10% glycerol) were incubated for 10 minutes at room temperature. Subsequently, approximately 20 to 50 fmoles of 32P-labeled NF-κB-specific oligonucleotide probe (30,000–50,000 cpm) were added and the reaction mixture was incubated for 10 minutes at room temperature. The samples were analyzed on 6% polyacrylamide gels in 50 mmol/L Tris-borate buffer containing 1 mmol/L EDTA or 50 mmol/L Tris/380 mmol/L glycine/2 mmol/L EDTA, at 12 V/cm for 2 to 2.5 hours. Radioactivities were detected by exposure to X-ray film or phosphorimager plates.
Culture and Measurement of Adhesion Areas of Peritoneal Macrophages
Peritoneal macrophages were enriched by adherence onto tissue culture-treated plates in Dulbecco’s modified Eagle’s medium (DMEM), supplemented with 2 mmol/L glutamine and 10% (v/v) fetal bovine serum, at 37°C in an atmosphere of 7.5% CO2. After two hours of incubation, wells were gently pipetted to remove nonadherent cells. More than 90% of the adherent cells were macrophages as evaluated morphologically after processing with Wright’s stain. Initially nonadherent cells were obtained after two serial adherence procedures in tissue culture flasks. Culturing of these cells resulted in additional adherent macrophages. The morphology of adherent macrophages cultured for various periods were imaged from three fields of each well using a Hamamatsu XC-77 CCD camera attached to a Nikon microscope with an inverted stage. Image-1 from Universal Imaging Corporation was used to acquire and enhance image contrast, and NIH Image software was used to measure cell attachment areas from three random fields.
Induction of Apoptosis of Peritoneal Macrophages in Vitro
Inflammatory peritoneal macrophages were obtained from mice treated with 1 ml thioglycollate broth for 3 days by lavage, and cultured in RPMI/10% fetal bovine serum (RP10F) for 1 hour. Adherent cells were cultured in the presence of 10 μg/ml lipopolysaccharide (LPS, Escherichia coli 0111:B4 List Biologicals, Campbell, CA) and 10 U/ml interferon-γ (IFN-γ, Boehringer/Roche, Indianapolis, IN), and cell viability was measured by the MTT (3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide) assay 46 at regular intervals. Alternatively, inflammatory macrophages obtained from peritoneal cavities of mice 4 days after 2 ml thioglycollate treatment, were cultured for 30 minutes in RPMI 1640. The adherent cells were exposed to 0 to 60 U/ml IFN-γ in RP10F for 6 hours washed with RP10F, and then 1 μg/ml LPS in RP10F for 24 hours. 47 The cells were washed with phosphate-buffered saline, pH 7.2, fixed in 5% formaldehyde/phosphate-buffered saline, and stained with 0.05% crystal violet in 20% EtOH. Absorbance at 550 nm in methanol due to the stained adherent cells was measured after the wells were extensively washed with deionized water.
Detection of Apoptotic Macrophages by Annexin-V Binding
Inflammatory peritoneal exudate cells (5 × 106/ml) were cultured in Teflon beakers in the presence of IFN-γ for 6 hours, washed, and then incubated with 1 μg/ml LPS as described above. At various time points, cells were removed and stained with annexin-V-fluorescein isothiocyanate (PharMingen, La Jolla, CA) according to manufacturer’s directions. Processed cells were analyzed by flow cytometry on a Becton Dickinson (San Jose, CA) Facscan.
Statistical Analysis
Comparison of data from gal3+/+ and gal3−/− mice were performed with the statistical software Statview. Data were subjected to the Mann-Whitney U test or analysis of variance with Bonferroni-Dunn post hoc analysis.
Results
Homologous Recombination and Production of gal3−/− Mice
The genomic structure of galectin-3 contains six exons 39 (Figure 1A) ▶ , with exons 2 and 3 coding for the amino-terminal region and exons 4–6 coding for the carboxyl-terminal carbohydrate-binding domain. Our strategy for inactivating galectin-3 in mice was to interrupt the region coding for the carbohydrate-binding domain with a neomycin resistance gene. Specifically, a short intron 4-exon 5 segment (0.5 kb) was substituted with the antibiotic-resistance gene (Neo). Figure 1B ▶ depicts the structure of the homologous recombinant galectin-3 gene and Figure 1C ▶ predicts the restriction fragment profiles of both wild type and homologous recombinants using the specific probes shown in Figure 1B ▶ . The genomic Southern blot of two homologous recombinant gal3+/− mouse embryonic stem cell clones is shown in Figure 1D ▶ , visualized with probe 1. The upper band in each lane for clones 4A2 and 9A4 were detected with probe 4, but no bands were observed in parent D3 (data not shown).
Figure 2A ▶ shows the Southern blot analysis of galectin-3 gene from gal3−/−, gal3+/+, and gal3+/− mice. The 2.9-kb intron 3-exon 5 fragment of the galectin-3+/+ gene and the corresponding 4-kb homologous recombinant of the galectin-3−/− are evident. Hemizygous mice are characterized by the presence of both DNA fragments. To demonstrate that the galectin-3 gene was indeed inactivated in gal3−/− mice, several organ extracts were prepared and adsorbed with lactosyl-Sepharose 4B, and the adsorbed proteins were analyzed by immunoblotting. As shown in Figure 2B ▶ , although gal3+/+ mice expressed large amounts of galectin-3 in lung, spleen, and thymus tissues, gal3−/− mice were deficient in this protein in these various organs, as expected. Gal3−/− mice are viable and fertile and do not exhibit any overt defects. Various organs from gal3−/− mice were further examined histologically and no apparent alterations were detected. Organs examined included adrenal gland, brain, colon, duodenum, esophagus, gall bladder, heart, hypophysis, kidney, knee joint, liver, lung, lymph nodes, mesentery, ovary, pancreas, salivary glands, skeletal muscle, skin, small intestine, stomach, spleen, testis, thymus, thyroid, and urinary bladder. No significant differences were observed in body weights and weights of major organs, and no differences were found in blood cell counts and chemistry profiles between gal3−/− and gal3+/+ mice. Lymphocyte subpopulations of thymus, spleen, and lymph node were also examined and total numbers of lymphocytes, ratios of CD4+/CD8+ cells, and numbers of CD3+ cells, were comparable between gal3−/− and gal3+/+ mice.
Figure 2.

Confirmation of the inactivated galectin-3 gene in homologous recombinant mice. A: Southern blot analysis of DNA fragments from homozygous (gal3−/− and gal3+/+) and hemizygous (gal3+/−) mice digested with HindIII and XbaI, and analyzed using probe 1, as described in Figure 1D ▶ . The positions of DNA markers (kb) are shown at the left margin. B: Immunoblot of organ extracts from gal3+/+ and gal3−/− mice adsorbed with lactosyl-Sepharose and probed with a specific rabbit anti-galectin-3 antiserum. Each lane represents galectin-3 from 100 μg total protein. Lu, lung; Li, liver; S, spleen; T, thymus. The positions of protein Mr standards (× 10−3) are shown at the left margin.
Gal3−/− Mice Exhibit Decreased Levels of Peritoneal Inflammation
As an initial approach to assess the effect of galectin-3 deficiency on inflammatory responses, we examined the peritoneal inflammation induced by thioglycollate broth. Untreated (Day 0) gal3−/− mice consistently contained fewer leukocytes in the peritoneal cavities, although the differences were not statistically significant (P = 0.1879). Although both types of mice mounted inflammatory responses to thioglycollate broth treatment, gal3−/− mice clearly exhibited reduced inflammation (Figure 3 ▶ , Table 1 ▶ ). One day after thioglycollate broth stimulation, the yields of inflammatory leukocytes in the peritoneal cavities of gal3−/− mice were significantly lower (P < 0.05) than that of gal3+/+ mice (Figure 3) ▶ . Similar trends were observed 3 and 6 days after thioglycollate stimulation (Table 1) ▶ .
Figure 3.

Reduced peritoneal inflammatory responses are observed in gal3−/− mice when induced with thioglycollate broth. A: Mice matched by sex and age were injected intraperitoneally with 1 ml of thioglycollate broth, and inflammatory cells were harvested at various intervals. Viable leukocytes determined by trypan blue exclusion were enumerated in duplicate from individual mice. Means for each time point are shown with SE bars from 9 and 5 separate experiments, with a total of 22 and 14 mice for each genotype for days 0 (untreated) and 1, respectively. The results from gal3+/+ mice are shown in closed circles and those from gal3−/− mice are shown in open circles. B: Inflammatory peritoneal cells obtained 1 day after thioglycollate broth treatment were cytospun onto glass slides and stained with soluble Wright’s stain for differential identification of leukocytes by standard morphologies. Between 100 and 200 total cells were enumerated from random fields in triplicate using a 100× oil immersion objective. Comparisons between genotypes were tested for significance using the Mann-Whitney U test; * indicates significant differences where P < 0.05. Data are means with SE bars from 4 experiments of a total of 11 mice for each genotype.
Table 1.
Peritoneal Cell Recoveries from Wild Type and Gal-3−/− Mice Treated with Thioglycollate Broth
| Genotype | Day 0 | Day 1 | Day 2 | Day 3 | Day 4 | Day 6 |
|---|---|---|---|---|---|---|
| +/+ | 0.98 ± 0.13 | 2.01 ± 0.29 | 2.57 ± 0.23 | 2.12 ± 0.19 | 1.79 ± 0.13 | 1.49 ± 0.17 |
| −/− | 0.72 ± 0.04 | 1.31 ± 0.14 | 2.16 ± 0.27 | 1.54 ± 0.14 | 1.49 ± 0.12 | 1.058 ± 0.127 |
| n | 22 | 14 | 19 | 17 | 14 | 34 |
| P | 0.1879 | 0.0482 | 0.3578 | 0.0146 | 0.1979 | 0.0089 |
Inflammatory cells were obtained as described in Figure 3 ▶ . Recoveries are expressed as cells × 10−7 ± SE. n, number of mice used for each genotype at each day of inflammation; P, values obtained by Mann-Whitney U test comparison between genotypes. Untreated mice are represented by Day 0.
Differences between gal3+/+ and gal3−/− mice were also observed when the numbers of different leukocyte populations were examined (Figure 3 ▶ , Table 2 ▶ ). Although minor differences were observed at day 0 (untreated mice), the most striking difference is the substantially lower numbers of monocytes/macrophages and lymphocytes in gal3−/− mice 1 day after thioglycollate broth stimulation (Figure 3) ▶ . The numbers of monocytes/macrophages were comparable between the two groups of mice, however, on prolonged inflammation for several days (Table 2) ▶ . In contrast, gal3−/− mice continued to exhibit significantly lower numbers of lymphocytes on days 2 and 4 (Table 2) ▶ . On the other hand, the numbers of neutrophils were comparable, and there were consistently more eosinophils in gal3−/− mice, 2 days after the stimulation. The difference between the two groups of mice was also evident when the percentages of various leukocytes in the peritoneal cavity were compared (Table 2) ▶ . When compared with gal3+/+ mice, gal3−/− mice have lower percentages of monocytes/macrophages (day 1), lower percentages of lymphocytes (days 2 and 4), and higher percentages of eosinophils (days 1, 2, and 4).
Table 2.
Leukocyte Recoveries in Inflammatory Mouse Peritoneal Cells Induced with Thioglycollate Broth in Wild-Type and Gal-3−/− Mice
| Days | Macrophages | Neutrophils | Lymphocytes | Eosinophils |
|---|---|---|---|---|
| 1 | ||||
| +/+ | 1.54 ± 0.29 (50 ± 1) | 1.10 ± 0.24 (39 ± 2) | 0.047 ± 0.011 (2 ± 0.4) | 0.26 ± 0.057 (8 ± 1) |
| −/− | 0.67 ± 0.11* (33 ± 2)* | 0.89 ± 0.17 (51 ± 2)* | 0.020 ± 0.0030* (0.9 ± 0.2) | 0.24 ± 0.030 (14 ± 2)* |
| 2 | ||||
| +/+ | 1.52 ± 0.26 (72 ± 2) | 0.25 ± 0.081 (12 ± 2) | 0.18 ± 0.026 (8 ± 1) | 0.15 ± 0.040 (7 ± 1) |
| −/− | 1.36 ± 0.23 (69 ± 1) | 0.24 ± 0.031 (13 ± 0.8) | 0.091 ± 0.034* (4 ± 1)* | 0.26 ± 0.049* (14 ± 0.8)* |
| 4 | ||||
| +/+ | 1.47 ± 0.11 (78 ± 1) | 0.025 ± 0.007 (3 ± 0.7) | 0.20 ± 0.035 (11 ± 1) | 0.18 ± 0.030 (9 ± 0.8) |
| −/− | 1.16 ± 0.10 (79 ± 1) | 0.084 ± 0.041 (3 ± 0.6) | 0.084 ± 0.011* (6 ± 0.6)* | 0.19 ± 0.040 (12 ± 1)* |
Inflammatory cells were obtained as described in Figure 3 ▶ . Recoveries are expressed as cells × 10−7 ± SE (percentage of cell population ± SE).
*P < 0.05 by Mann-Whitney U test comparison between genotypes, n = 11 per genotype.
Peritoneal Inflammatory Cells from gal3−/− Mice Consistently Show Decreased NF-κB Responses
The results reported above suggest that gal3−/− mice develop a lower inflammatory response to thioglycollate stimulation compared with gal3+/+ mice. To substantiate this conclusion, we examined the NF-κB response in the peritoneal inflammatory cells in these mice. NF-κB is a transcription factor commonly activated in various inflammatory conditions and plays an important role in the transcription of genes for many inflammatory factors. 48 Nuclear extracts were prepared from peritoneal leukocytes obtained from untreated mice and mice treated with thioglycollate broth, and the κB binding activity was examined by EMSA. Resting peritoneal leukocytes from untreated mice of both genotypes showed low levels of κB binding activity. However, on stimulation with thioglycollate broth, leukocytes from gal3+/+ mice responded with robust κB binding activities. In contrast, leukocytes from gal3−/− mice responded weakly (Figure 4A) ▶ .
Figure 4.

Peritoneal inflammatory cells of gal3−/− mice exhibit a reduced NF-κB response. Nuclear extracts were obtained from peritoneal inflammatory cells and the κB binding activity was determined by EMSA. A: Peritoneal cells from gal3−/− and gal3+/+ mice were obtained 2 days after thioglycollate broth treatment. Each lane represents results from a single mouse. Similar results were obtained from three separate experiments. B: Peritoneal cells from untreated or thioglycollate-treated mice (1 or 2 days after the treatment) were obtained by lavage and cells from three mice for each group were pooled. They were incubated in RPMI (5 × 10 6 cells/ml) at 37°C in the absence or presence of recombinant TNF-α (40 ng/ml) for 40 minutes. The arrowheads mark the positions of DNA-protein complexes specific for NF-κB.
The lower levels of the NF-κB response in cells from gal3−/− mice are consistent with decreased levels of activation. However, another possibility is an intrinsic deficit in the NF-κB response pathway as a result of the galectin-3 deficiency. To differentiate these two possibilities, we obtained resting as well as thioglycollate-elicited peritoneal leukocytes from both genotypes, exposed them to tumor necrosis factor-α (TNF-α), and measured the NF-κB response. As shown in Figure 4B ▶ , resting cells from both genotypes exhibited nearly undetectable levels of κB binding activity, but showed comparable levels of activity on stimulation by TNFα. Similarly, while thioglycollate-elicited leukocytes from gal3−/− mice exhibited significantly reduced levels of κB binding activity compared with gal3+/+ mice, cells from both genotypes showed comparable levels of activity after stimulation by TNFα. Therefore, the gal3−/− cells do not appear to have an intrinsic defect in NF-κB response and the lower response in the peritoneal cells of gal3−/− mice following thioglycollate treatment probably reflects the lower degree of peritoneal inflammation in these mice.
Peritoneal Macrophages from gal3−/− Mice Show Decreased Areas of Adherence in Culture
When resident or thioglycollate-elicited peritoneal macrophages from gal3−/− mice and gal3+/+ mice were cultured under standard conditions, the adherent cells from both sources exhibited spread-out morphology characteristic of macrophages. However, we consistently observed that cultured resident macrophages from gal3−/− mice exhibited a lower degree of spreading. Indeed, when the cell surface areas were measured, statistically significant differences were noted (178 ± 2 vs. 151 ± 2 relative units for gal3+/+ and gal3−/− mice, respectively, from a total of 5 experiments, 13 mice, and over 3300 cells from each genotype measured, P < 0.0001). When thioglycollate-elicited peritoneal cells were cultured for 2 hours and initially nonadherent cells (containing monocytes, lymphocytes, eosinophils, and neutrophils) were further cultured, an additional macrophage population could be obtained. When these cell populations were compared, a dramatic difference in the degrees of spreading was observed (Figure 5A) ▶ . The average cell area of adherent macrophages from gal3−/− mice was less than 40% of that of cells from gal3+/+ mice (Figure 5B) ▶ . The experiments were performed four times with cells from a total of eight mice for each genotype. Cells from gal3−/− mice often exhibited spindle-shaped morphology in 22 out of 24 culture wells, while cells from gal3+/+ mice showed more uniform spread-out morphology in all 24 culture wells.
Figure 5.
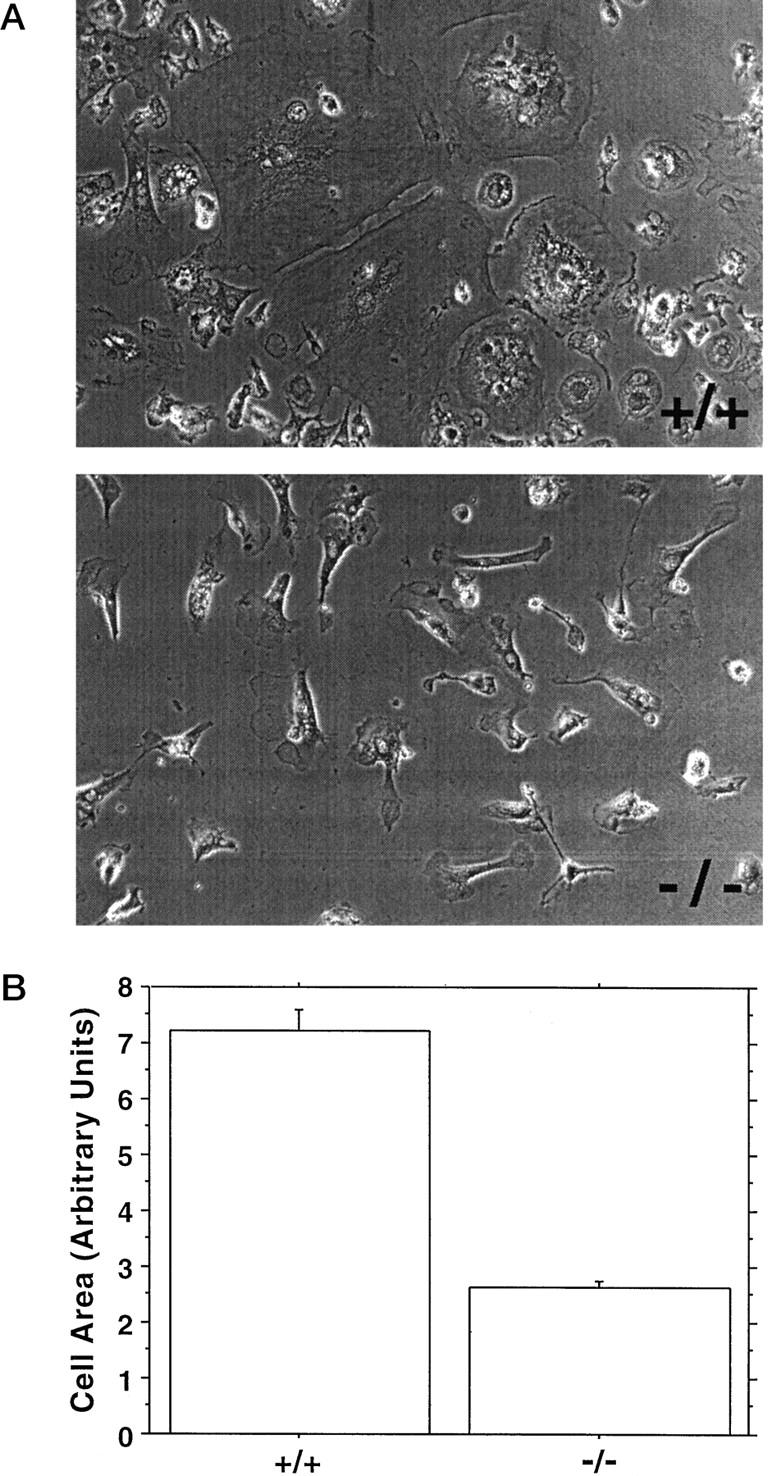
Cell areas of adherent inflammatory peritoneal macrophages from gal3−/− mice are different from macrophages of gal3+/+ mice. A: Representative phase photomicrographs of macrophages obtained from culturing initially nonadherent cells from peritoneal cavities of thioglycollate-treated gal3+/+ and gal3−/− mice. B: Mean cell areas of adherent macrophages. Cell areas of adherent cells were measured from digitized files obtained from three random fields of culture wells. Data are means from three separate experiments and are expressed in an arbitrary unit. Over 400 individual cells from each genotype were measured; a P value < 0.0001 was obtained by Mann-Whitney U comparison.
To determine whether the difference was due to the effect of galectin-3 functioning extracellularly, we cultured the cells in the presence or absence of lactose, previously shown in many studies to inhibit galectin-3 activities. 15,17,19 The difference in cell spreading were unaffected by 25 mmol/L lactose (data not shown).
Macrophages from gal3−/− Mice Are More Sensitive to Apoptotic Stimuli
Previously, we have shown that Jurkat cells transfected with galectin-3 cDNA and expressing this lectin are more resistant to apoptosis induced by anti-Fas antibodies and staurosporine compared with control transfectant not expressing the lectin, 24 and concluded that galectin-3 functions as an anti-apoptotic protein. Therefore, it is possible that the lower levels of peritoneal inflammatory cells in gal3−/− mice are at least partly attributable to the increased cell death in these mice due to the lack of this lectin. To test this possibility, we compared the rate of apoptosis of peritoneal macrophages from gal3+/+ and gal3−/− mice after treatment with IFN-γ and LPS, a procedure known to induce apoptosis in these cells. 49 We focused on macrophages because these cells are known to express galectin-3. 50 The mice were injected intraperitoneally with thioglycollate, and harvested inflammatory peritoneal macrophages were treated with 10 μg/ml LPS and 10 U/ml IFN-γ for various periods. As shown in Figure 6A ▶ , viable cells decreased with time in culture. Gal3−/− cells appeared to be more sensitive to this treatment and resulted in significantly lower numbers of surviving cells. With the alternative method of activation, where macrophages were pretreated with IFN-γ for 6 hours before LPS, significantly decreased numbers of cells were observed for doses of IFN-γ from 0.3 to 10 U/ml in a dose-dependent manner (data not shown). Because LPS/IFN-γ-induced apoptosis occurs through a nitric oxide-dependent pathway, 51-53 levels of nitrite (a stable by-product of nitric oxide) in culture supernatants were measured. No significant differences were observed between gal3+/+ and gal3−/− macrophage cultures (data not shown), suggesting that differential rate of cell death was not due to differences in the capacity to produce nitric oxide after LPS and IFN-γ activation.
Figure 6.

Peritoneal macrophages from gal3−/− mice are more sensitive to apoptotic stimuli than cells from gal3+/+ mice. A: Inflammatory macrophages obtained from peritoneal cavities of mice 3 days after treatment with 1 ml thioglycollate were cultured as adherent cells with 10 μg/ml LPS and 10 U/ml IFN-γ for the indicated periods. Surviving cells were measured by the MTT assay. 46 Data are shown from one representative experiment as means ± SE. Similar results were obtained from five separate experiments, including experiments in which macrophages elicited with 2 ml thioglycollate broth for 4 days were cultured with various amounts of IFN-γ for 6 hours followed by 1 μg/ml LPS (data not shown). A significant difference between genotypes (P < 0.0001) was obtained by analysis of variance at 5% significance with Bonferroni-Dunn post hoc analysis. B: Cells were cultured in Teflon beakers at 5 × 10 6 cells/ml and treated with 1.7 U/ml IFN-γ for 6 hours, washed, and then activated with 1 μg/ml LPS for 24 hours. The cells were stained with annexin-V-fluorescein isothiocyanate and analyzed by flow cytometry. Percentages of cells positively stained, gated within region R2, were determined, as shown in representative histograms.
We then determined whether the lower recovery of gal3−/− cells is due to increased apoptosis as a result of galectin-3 deficiency. Peritoneal macrophages from gal3+/+ and gal3−/− mice were cultured in Teflon beakers (to facilitate periodic retrieval of cells for measurements) and activated with IFN-γ and LPS as above to induce apoptosis. As shown in Figure 6B ▶ , macrophages from gal3−/− mice demonstrated significantly higher levels of apoptosis, as confirmed by higher annexin-V binding.
Discussion
We have demonstrated in this study that galectin-3 deficiency results in significantly reduced peritoneal inflammatory responses to thioglycollate stimulation. First, there are lower levels of monocyte/macrophage infiltration in the first 24 hours following thioglycollate stimulation and reduced numbers of lymphocytes throughout six days of inflammation. Second, thioglycollate-elicited leukocytes from gal3−/− mice exhibit significantly lower levels of NF-κB response. The results suggest that endogenous galectin-3 plays a role in positively regulating peritoneal inflammatory processes.
Results from a number of previous in vitro studies have suggested that galectin-3 may be an amplifier of inflammatory responses. First, galectin-3 is present in extracellular fluid and is capable of activating various cells. 8,54 Thus, it may be envisaged that in inflammatory responses in the peritoneal cavity, galectin-3 released from some of the activated cells could activate additional cells. Second, this lectin can mediate cell-cell and cell-extracellular matrix protein interactions, 19,21,55 as well as homotypic cell-cell interactions, 20 making it suited for promoting inflammatory responses. Third, galectin-3 can inhibit programmed cell death, as demonstrated by using a human T cell line transfected with galectin-3 cDNA. 24 Therefore, galectin-3 expression may lead to longer survival of inflammatory cells and thus to prolonged inflammation. The results reported here, indeed, support the previous notion that galectin-3 has a proinflammatory function. Furthermore, at least one of the above mechanisms may be contributory. Specifically, galectin-3 deficiency appears to result in the peritoneal macrophages becoming more sensitive to apoptotic stimuli, resulting in mice exhibiting decreased levels of inflammation.
The finding that galectin-3 deficiency results in increased tendency to apoptosis in macrophages in turn supports the function of galectin-3 in inhibiting apoptosis. Previously, we noted sequence similarity between galectin-3 and a well-characterized anti-apoptotic protein, Bcl-2, demonstrated binding of Bcl-2 by galectin-3, and proposed that galectin-3 inhibits apoptosis, possibly through a mechanism involving interaction with Bcl-2. Although we do not have additional information on the mechanism underlying the anti-apoptotic activity of galectin-3, we believe this molecule exerts this effect through intracellular action. One piece of supporting evidence for this notion is that recombinant galectin-3 added exogenously to cultured peritoneal macrophages did not affect the cell survival (data not shown). This finding is consistent with a previous report where galectin-3 does not induce apoptosis of thymocytes when added exogenously. 56 The mechanism of galectin-3 for regulating apoptosis is thus different from that of galectin-1, which has been shown to induce apoptosis of thymocytes through binding to cell surface glycoconjugates. 57,58
We cannot exclude the possibility that galectin-3 also affects the overall peritoneal inflammatory responses through extracellular functions, especially because abundant amounts of galectin-3 exist in the peritoneal exudate, and this protein is known to bind to cell surface glycoconjugates through lectin-carbohydrate interactions. 50 We have attempted to use anti-galectin-3 antibodies to further clarify the role of extracellular galectin-3 in the inflammatory responses. We have previously generated a number of monoclonal antibodies against galectin-3. Only one of them (B2C10) has blocking activity, whereas others (eg, A3A12) potentiate galectin-3 lectin activities. We found that, when injected into peritoneal cavity of wild-type C57BL/6 mice, A3A12 caused an increase and B2C10 caused a decrease in the numbers of inflammatory cells in the peritoneal cavity elicited by thioglycollate, compared with an isotype-matched control monoclonal antibody. Although the potentiating and suppressive effects of the two antibodies compared with the control antibody were not statistically significant, the differences between the two were (data not shown). Therefore, these preliminary data support extracellular functions of galectin-3. Further clarification would require more potent blocking antibodies or other specific inhibitors against galectin-3. Additional experiments are also required to determine the relative contribution of the intracellular functions of this lectin to its regulation of inflammatory responses. Thus, the possible mechanisms of action we propose include both intracellular and extracellular functions of galectin-3. In fact, our present view is that this lectin as well as other members of this lectin family function both inside and outside the cell. This is based on the existence of the lectin as well as galectin-3-interacting proteins in both compartments, and demonstrated extracellular (eg, activation of cells and promotion of cell adhesion) and intracellular functions (eg, engagement in pre-mRNA splicing and regulation of apoptosis) in vitro.
Because the extent of cell spreading is reflective of cell activation, the fact that macrophages from gal3+/+ mice exhibit more extended morphologies is also consistent with our proposition that cells from these mice are more activated. However, other explanations need to be considered. The most obvious is that galectin-3 released by monocytes/macrophages, a cell type known to contain high levels of the lectin, 12,59 directly mediates cell adhesion and spreading. However, this may not be in operation because the spreading of macrophages from gal3+/+ mice was unaffected when the cells were cultured in the presence of lactose (data not shown), which should inhibit extracellular galectin-3 activities. It has recently been reported that galectin-3 binds to and colocalizes with cytokeratin 60 in a fashion that is dependent on carbohydrate residues on cytokeratin. Therefore, an attractive alternative is that galectin-3-cytokeratin interactions modulate the cell shape. It is to be noted that the effect of galectin-3 deficiency on cell shape is much more pronounced in initially nonadherent cells, as compared with the initially adherent cells; the former are likely to include more immature monocytes and the latter represent mature macrophages. It is possible that during maturation of macrophages in vivo to the inflammatory state, any defects in in vitro adherence and spreading caused by galectin-3 deficiency may be partially overcome. On the other hand, conditions imposed by culturing initially nonadherent monocytes may accentuate defects in adherence and spreading properties of these cells resulting from galectin-3 deficiency. These results also suggest the possibility that cell types such as lymphocytes, neutrophils, and eosinophils included in the initially nonadherent cell population may influence the spreading of monocytes/macrophages by providing some specific signals, and these companion cells from gal3+/+ mice may be more activated than those from gal3−/− mice.
While this work was in progress, Colnot et al reported findings made with an independent line of galectin-3-deficient mice. 61 The main defect noted is that gal3−/− mice have significantly reduced numbers of neutrophils in the peritoneal cavity 4 days after thioglycollate treatment, which was not seen in our experiments. This could be due to variations in the constituents of the thioglycollate broth used as well as the amounts injected, which might have resulted in both quantitative and qualitative differences in the response. On the other hand, these investigators also observed lower numbers of macrophages in gal3−/− mice on day 1 after thioglycollate treatment, as compared with the wild-type mice (19.0 ± 2.53 vs. 31.1 ± 6.24). Although the difference is not statistically significant, it is conceivable that a statistically significant difference would be observed with a larger number of mice. Considering both studies together, the evidence is convincing that galectin-3-deficient mice mount a lower initial monocyte/macrophage response than wild-type mice.
The finding that galectin-3-deficient mice have attenuated peritoneal inflammatory responses lends strong support for the role of galectin-3 in augmentation of inflammation. The results provide new insights into mechanisms by which galectin-3 exerts its proinflammatory function, especially when considered together with information from previous in vitro studies. In particular, this lectin may contribute to inflammation by regulating apoptosis of inflammatory cells under inflammatory conditions. Additional studies with this mouse model should allow further elucidation of the function of galectin-3 in various inflammatory situations, as well as development of inhibitors of galectin-3 as therapeutic agents for suppressing inflammatory responses. In addition, the gal3−/− mice should be valuable for delineation of the role of this lectin in other pathological processes, especially the neoplasm.
Acknowledgments
We thank Dr. V. Kravchenko for providing recombinant tumor necrosis factor. This is publication number 252 from the La Jolla Institute for Allergy and Immunology.
Footnotes
Address reprint requests to Fu-Tong Liu, Ph.D., M.D., La Jolla Institute for Allergy and Immunology, 10355 Science Center Drive, San Diego, CA 92121. E-mail: ftliu@liai.org.
Supported by National Institutes of Health grants AI-20958 and AI-39620.
References
- 1.Barondes SH, Castronovo V, Cooper DNW, Cummings RD, Drickamer K, Feizi T, Gitt MA, Hirabayashi J, Hughes C, Kasai K, Leffler H, Liu F-T, Lotan R, Mercurio AM, Monsigny M, Pillai S, Poirer F, Raz A, Rigby PWJ, Rini JM, Wang JL: Galectins: A family of animal β-galactoside-binding lectins. (letter to the editor). Cell 1994, 76:597-598 [DOI] [PubMed] [Google Scholar]
- 2.Barondes SH, Cooper DNW, Gitt MA, Leffler H: Galectins: structure and function of a large family of animal lectins. J Biol Chem 1994, 269:20807-20810 [PubMed] [Google Scholar]
- 3.Kasai K, Hirabayashi J: Galectins: a family of animal lectins that decipher glycocodes. J Biochem (Tokyo) 1996, 119:1-8 [DOI] [PubMed] [Google Scholar]
- 4.Hughes RC: The galectin family of mammalian carbohydrate-binding molecules. Biochem Soc Trans 1997, 25:1194-2298 [DOI] [PubMed] [Google Scholar]
- 5.Perillo NL, Marcus ME, Baum LG: Galectins: versatile modulators of cell adhesion, cell proliferation, and cell death. J Mol Med 1998, 76:402-412 [DOI] [PubMed] [Google Scholar]
- 6.Liu F-T: Molecular biology of IgE-binding protein, IgE-binding factors and IgE receptors. CRC Crit Rev Immunol 1990, 10:289-306 [PubMed] [Google Scholar]
- 7.Hughes RC: Mac-2: a versatile galactose-binding protein of mammalian tissues. Glycobiology 1994, 4:5-12 [DOI] [PubMed] [Google Scholar]
- 8.Liu F-T: Role of galectin-3 in inflammation. Caron M Seve D eds. Lectins and Pathology. 2000, :pp 51-65 Harwood Academic Publishers, London [Google Scholar]
- 9.Flotte TJ, Springer TA, Thorbecke GJ: Dendritic cell and macrophage staining by monoclonal antibodies in tissue sections and epidermal sheets. Am J Pathol 1983, 111:112-124 [PMC free article] [PubMed] [Google Scholar]
- 10.Moutsatsos IK, Wade M, Schindler M, Wang JL: Endogenous lectins from cultured cells: nuclear localization of carbohydrate-binding protein 35 in proliferating 3T3 fibroblasts. Proc Natl Acad Sci USA 1987, 84:6452-6456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Agrwal N, Wang JL, Voss PG: Carbohydrate-binding protein 35: levels of transcription and mRNA accumulation in quiescent and proliferating cells. J Biol Chem 1989, 264:17236-17242 [PubMed] [Google Scholar]
- 12.Liu F-T, Hsu DK, Zuberi RI, Kuwabara I, Chi EY, Henderson WR, Jr: Expression and function of galectin-3, a β-galactoside-binding lectin, in human monocytes and macrophages. Am J Pathol 1995, 147:1016-1029 [PMC free article] [PubMed] [Google Scholar]
- 13.Nangia-Makker P, Ochieng J, Christman JK, Raz A: Regulation of the expression of galactoside-binding lectin during human monocytic differentiation. Cancer Res 1993, 53:1-5 [PubMed] [Google Scholar]
- 14.Hsu DK, Hammes SR, Kuwabara I, Greene WC, Liu F-T: Human T lymphotropic virus-1 infection of human T lymphocytes induces expression of the β-galactose-binding lectin, galectin-3. Am J Pathol 1996, 148:1661-1670 [PMC free article] [PubMed] [Google Scholar]
- 15.Frigeri LG, Zuberi RI, Liu F-T: εBP, a β-galactoside-binding animal lectin, recognizes IgE receptor (FcεRI), and activates mast cells. Biochemistry 1993, 32:7644-7649 [DOI] [PubMed] [Google Scholar]
- 16.Zuberi RI, Frigeri LG, Liu F-T: Activation of rat basophilic leukemia cells by εBP, an IgE-binding endogenous lectin. Cell Immunol 1994, 156:1-12 [DOI] [PubMed] [Google Scholar]
- 17.Yamaoka A, Kuwabara I, Frigeri LG, Liu F-T: A human lectin, galectin-3 (εBP/Mac-2), stimulates superoxide production by neutrophils. J Immunol 1995, 154:3479-3487 [PubMed] [Google Scholar]
- 18.Dong S, Hughes RC: Galectin-3 stimulates uptake of extracellular Ca2+ in human Jurkat T-cells. FEBS Lett 1996, 395:165-169 [DOI] [PubMed] [Google Scholar]
- 19.Kuwabara I, Liu F-T: Galectin-3 promotes adhesion of human neutrophils to laminin. J Immunol 1996, 156:3939-3944 [PubMed] [Google Scholar]
- 20.Inohara H, Akahani S, Koths K, Raz A: Interactions between galectin-3 and Mac-2-binding protein mediate cell-cell adhesion. Cancer Res 1996, 56:4530-4534 [PubMed] [Google Scholar]
- 21.Sato S, Hughes RC: Binding specificity of a baby hamster kidney lectin for H type I and II chains, polylactosamine glycans, and appropriately glycosylated forms of laminin and fibronectin. J Biol Chem 1992, 267:6983-6990 [PubMed] [Google Scholar]
- 22.Laing JG, Wang JL: Identification of carbohydrate binding protein 35 in heterogeneous nuclear ribonucleoprotein complex. Biochemistry 1988, 27:5329-5334 [DOI] [PubMed] [Google Scholar]
- 23.Dagher SF, Wang JL, Patterson RJ: Identification of galectin-3 as a factor in pre-mRNA splicing. Proc Natl Acad Sci USA 1995, 92:1213-1217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang R-Y, Hsu DK, Liu F-T: Expression of galectin-3 modulates T cell growth and apoptosis. Proc Natl Acad Sci USA 1996, 93:6737-6742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Akahani S, Nangia-Makker P, Inohara H, Kim HRC, Raz A: Galectin-3: a novel antiapoptotic molecule with a functional BH1 (NWGR) domain of Bcl-2 family. Cancer Res 1997, 57:5272-5276 [PubMed] [Google Scholar]
- 26.Nachtigal M, Al-Assaad Z, Mayer EP, Kim K, Monsigny M: Galectin-3 expression in human atherosclerotic lesions. Am J Pathol 1998, 152:1199-1208 [PMC free article] [PubMed] [Google Scholar]
- 27.Konstantinov KN, Robbins BA, Liu F-T: Galectin-3, a β-galactoside-binding animal lectin, is a marker of anaplastic large-cell lymphoma. Am J Pathol 1996, 148:25-30 [PMC free article] [PubMed] [Google Scholar]
- 28.Fernádez PL, Merino MJ, Gómez M, Campo E, Medina T, Castronovo V, Cardesa A, Liu F-T, Sobel ME: Galectin-3 and laminin expression in neoplastic and non-neoplastic thyroid tissue. J Pathol 1997, 181:80-86 [DOI] [PubMed] [Google Scholar]
- 29.Xu XC, El-Naggar AK, Lotan R: Differential expression of galectin-1 and galectin-3 in thyroid tumors: potential diagnostic implications. Am J Pathol 1995, 147:815-822 [PMC free article] [PubMed] [Google Scholar]
- 30.Hsu DK, Dowling CA, Jeng KCG, Chen JT, Yang RY, Liu FT: Galectin-3 expression is induced in cirrhotic liver and hepatocellular carcinoma. Int J Cancer 1999, 81:519-526 [DOI] [PubMed] [Google Scholar]
- 31.Lotz MM, Andrews CW, Jr, Korzelius CA, Lee EC, Steele GD, Jr, Clarke A, Mercurio AM: Decreased expression of Mac-2 (carbohydrate binding protein 35) and loss of its nuclear localization are associated with the neoplastic progression of colon carcinoma. Proc Natl Acad Sci USA 1993, 90:3466-3472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Castronovo V, Campo E, van den Brûle FA, Claysmith AP, Cioce V, Liu F-T, Fernandez PL, Sobel ME: Inverse modulation of steady state mRNA levels of two non-integrin laminin binding proteins in human colon carcinoma. J Natl Cancer Inst 1992, 84:1161-1167 [DOI] [PubMed] [Google Scholar]
- 33.Castronovo V, Van den Brule FA, Jackers P, Clausse N, Liu FT, Gillet C, Sobel ME: Decreased expression of galectin-3 is associated with progression of human breast cancer. J Pathol 1996, 179:43-48 [DOI] [PubMed] [Google Scholar]
- 34.van den Brûle FA, Berchuck A, Bast RC, Liu F-T, Pieters C, Sobel ME, Castronovo V: Differential expression of the 67-kD laminin receptor and 31-kD human laminin-binding protein in human ovarian carcinoma. Eur J Cancer 1994, 30A:1096-1099 [DOI] [PubMed] [Google Scholar]
- 35.van den Brûle FA, Buicu C, Berchuck A, Bast RC, Deprez M, Liu FT, Cooper DNW, Pieters C, Sobel ME, Castronovo V: Expression of the 67-kD laminin receptor, galectin-1, and galectin-3 in advanced human uterine adenocarcinoma. Hum Pathol 1996, 27:1185-1191 [DOI] [PubMed] [Google Scholar]
- 36.Irimura T, Matsushita Y, Sutton RC, Carralero D, Ohannesian DW, Cleary KR, Ota DM, Nicolson GL, Lotan R: Increased content of an endogenous lactose-binding lectin in human colorectal carcinoma progressed to metastatic stages. Cancer Res 1991, 51:387-393 [PubMed] [Google Scholar]
- 37.Schoeppner HL, Raz A, Ho SB, Bresalier RS: Expression of an endogenous galactose-binding lectin correlates with neoplastic progression in the colon. Cancer 1995, 75:2818-2826 [DOI] [PubMed] [Google Scholar]
- 38.Raz A, Zhu D, Hogan V, Shah N, Raz T, Karkash R, Pazerini G, Carmi P: Evidence for the role of 34-kDa galactoside-binding lectin in transformation and metastasis. Int J Cancer 1990, 46:871-877 [DOI] [PubMed] [Google Scholar]
- 39.Gritzmacher CA, Mehl VS, Liu F-T: Genomic cloning of the gene for an IgE-binding lectin reveals unusual utilization of 5′ untranslated regions. Biochemistry 1992, 31:9533-9538 [DOI] [PubMed] [Google Scholar]
- 40.Shier P, Otulakowski G, Ngo K, Panakos J, Chourmouzis E, Christjansen L, Lau CY, Fung-Leung W-P: Impaired immune responses toward alloantigens and tumor cells but normal thymic selection in mice deficient in the beta2 integrin leukocyte function-associated antigen-1. J Immunol 1996, 157:5375-5386 [PubMed] [Google Scholar]
- 41.Hsu DK, Zuberi R, Liu F-T: Biochemical and biophysical characterization of human recombinant IgE-binding protein, an S-type animal lectin. J Biol Chem 1992, 267:14167-14174 [PubMed] [Google Scholar]
- 42.Mishell BB: Selected Methods in Cellular Immunology. 1980:p 3 Freeman, San Francisco
- 43.Dignam JD, Lebovitz RM, Roeder RG: Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res 1983, 11:1475-1489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kravchenko VV, Pan Z, Han J, Herbert JM, Ulevitch RJ, Ye RD: Platelet-activating factor induces NF-kappa B activation through a G protein-coupled pathway. J Biol Chem 1995, 270:14928-14934 [DOI] [PubMed] [Google Scholar]
- 45.Browning DD, Pan ZK, Prossnitz ER, Ye RD: Cell type- and developmental stage-specific activation of NF-kappaB by fMet-Leu-Phe in myeloid cells. J Biol Chem 1997, 272:7995-8001 [DOI] [PubMed] [Google Scholar]
- 46.Hansen MB, Nielsen SE, Berg K: Re-examination and further development of a precise and rapid dye method for measuring cell growth/cell kill. J Immunol Methods 1989, 119:203-210 [DOI] [PubMed] [Google Scholar]
- 47.Shnyra A, Brewington R, Alipio A, Amura C, Morrison DC: Reprogramming of lipopolysaccharide-primed macrophages is controlled by a counterbalanced production of IL-10 and IL-12. J Immunol 1998, 160:3729-3736 [PubMed] [Google Scholar]
- 48.Baeuerle PA, Henkel T: Function and activation of NF-kappaB in the immune system. Annu Rev Immunol 1994, 12:141-179 [DOI] [PubMed] [Google Scholar]
- 49.Lakics V, Vogel SN: Lipopolysaccharide and ceramide use divergent signaling pathways to induce cell death in murine macrophages. J Immunol 1998, 161:2490-2500 [PubMed] [Google Scholar]
- 50.Frigeri LG, Liu F-T: Surface expresson of functional IgE binding protein, an endogenous lectin, on mast cells and macrophages. J Immunol 1992, 148:861-869 [PubMed] [Google Scholar]
- 51.Messmer UK, Brune B: Nitric oxide-induced apoptosis: p53-dependent and p53-independent signalling pathways. Biochem J 1996, 319:299-305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Messmer UK, Ankarcrona M, Nicotera P, Brune B: p53 expression in nitric oxide-induced apoptosis. FEBS Lett 1994, 355:23-26 [DOI] [PubMed] [Google Scholar]
- 53.Albina JE, Cui S, Mateo RB, Reichner JS: Nitric oxide-mediated apoptosis in murine peritoneal macrophages. J Immunol 1993, 150:5080-5085 [PubMed] [Google Scholar]
- 54.Liu F-T: S-type mammalian lectins in allergic inflammation. Immunol Today 1993, 14:486-490 [DOI] [PubMed] [Google Scholar]
- 55.Bao Q, Hughes RC: Galectin-3 expression and effects on cyst enlargement and tubulogenesis in kidney epithelial MDCK cells cultured in three-dimensional matrices in vitro. J Cell Sci 1995, 108:2791-2800 [DOI] [PubMed] [Google Scholar]
- 56.Wada J, Ota K, Kumar A, Wallner EI, Kanwar YS: Developmental regulation, expression, and apoptotic potential of galectin-9, a β-galactoside binding lectin. J Clin Invest 1997, 99:2452-2461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Perillo NL, Pace KE, Seilhamer JJ, Baum LG: Apoptosis of T cells mediated by galectin-1. Nature 1995, 378:736-739 [DOI] [PubMed] [Google Scholar]
- 58.Perillo NL, Uittenbogaart CH, Nguyen JT, Baum LG: Galectin-1, an endogenous lectin produced by thymic epithelial cells, induces apoptosis of human thymocytes. J Exp Med 1997, 185:1851-1858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cherayil BJ, Weiner SJ, Pillai S: The Mac-2 antigen is a galactose-specific lectin that binds IgE. J Exp Med 1989, 170:1959-1972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Goletz S, Hanisch F-G, Karsten U: Novel αGalNAc containing glycans on cytokeratins are recognized in vitro by galectins with type II carbohydrate recognition domains. J Cell Sci 1997, 110:1585-1596 [DOI] [PubMed] [Google Scholar]
- 61.Colnot C, Ripoche MA, Milon G, Montagutelli X, Crocker PR, Poirier F: Maintenance of granulocyte numbers during acute peritonitis is defective in galectin-3-null mutant mice. Immunology 1998, 94:290-296 [DOI] [PMC free article] [PubMed] [Google Scholar]