

Abstract
Activation of extracellular signal-regulated kinases (ERK) has been associated with the advent of asbestos-associated apoptosis and proliferation in mesothelial and alveolar epithelial cells and may be linked to the development of pulmonary fibrosis. The objective of studies here was to characterize the development of inflammation, cellular proliferation, and fibrosis in asbestos-exposed C57Bl/6 mice in relationship to patterns of ERK phosphorylation. Inflammation occurred after 10 and 20 days of asbestos exposure as evidenced by increases in total protein and neutrophils in bronchoalveolar lavage fluid. Increases in cell proliferation were observed at 30 days in bronchiolar epithelia and at 4, 14, and 30 days in the alveolar compartment of the lung. Trichrome-positive focal lesions of pulmonary fibrosis developed at 30 days in the absence of elevations in lung hydroxyproline or procollagen mRNA levels. Striking increases in ERK phosphorylation were observed within pulmonary epithelial cells at sites of developing fibrotic lesions after 14 and 30 days of inhalation. In addition to characterizing a murine inhalation model of asbestosis, we provide the first evidence showing activation of ERK signaling within lung epithelium in vivo, following inhalation of asbestos fibers.
Patterns of pulmonary inflammation, cell proliferation and fibrosis following chronic inhalation of asbestos fibers have been characterized by our laboratory in a rat model of asbestosis. Low-dose (≤0.2 mg/m 3 air) inhalation exposures to crocidolite or chrysotile asbestos fibers induce transient inflammation in bronchoalveolar lavage (BAL) samples and reversible inflammatory lesions in lung typified by focal aggregations of fiber-laden alveolar macrophages and maintenance of a normal lung architecture. 1,2 In contrast, high-dose (≥7.0 mg/m 3 air) exposures result in a more intense and protracted inflammation as demonstrated by an increased proportion of neutrophils in BAL and cellular foci along terminal bronchiolar and alveolar ductal regions. 2,3 Rapid and reversible proliferation of bronchiolar and alveolar type II epithelial cells, as indicated by enhanced incorporation of 5-bromo-2′-deoxyuridine (BrdU), is observed within days of high-dose inhalation exposures to asbestos. 3 Proliferation within pulmonary interstitial cells, increases in lung hydroxyproline content, a biomarker of collagen synthesis, and morphological evidence of pulmonary fibrosis become apparent with prolonged high-dose exposure (≥14 days) to crocidolite asbestos. 1
Recent evidence indicates that asbestos can stimulate gene expression in a variety of cell types in vitro via downstream intracellular signaling cascades. 4 These signaling cascades include the mitogen-activated protein kinases (MAPK) which can lead to increased transcriptional activation of genes intrinsic to cell proliferation, apoptosis and inflammation. The MAPK cascade is characterized by a sequential series of phosphorylation events catalyzed by the extracellular signal-regulated kinases (ERK), c-jun NH2-terminal kinases (JNK) or stress-activated protein kinases (SAPK), and p38. 5 In vitro exposure to asbestos fibers selectively induces ERK phosphorylation and activity in rat mesothelial cells, leading to apoptosis and increases in BrdU incorporation. 6,7 Furthermore, preferential activation of ERK by asbestos in rat mesothelial cells can be mediated through the phosphorylation of the Epidermal Growth Factor receptor. 6 Ultimately, asbestos-induced MAPK signaling events may be critical factors in gene transcription governing phenotypic outcomes of lung injury.
In the present study, we used a murine inhalation model of chrysotile asbestos exposure to enhance our understanding of the ERK signaling cascade in relationship to the development of asbestosis. Our primary objective was to demonstrate the spatial patterns of ERK phosphorylation during the development of epithelial cell proliferation, inflammation and asbestosis. Our findings support the hypothesis that MAPK signaling cascades are activated during the development of fibroproliferative lung diseases.
Materials and Methods
Animals
Two- to three-month-old male C57Bl/6 mice were obtained from the National Institutes of Health (Bethesda, MD). Animals were housed and allowed to acclimate for 1 week in suspended cages under controlled conditions of temperature, humidity, and light and provided food and water ad libitum before the initiation of inhalation exposures. Animal and exposure facilities are approved by the American Association for Accreditation of Laboratory Animal Care and operated under the supervision of the Institutional Animal Care and Use Committee of the University of Vermont.
Experimental Protocol and Inhalation Exposures
The experimental protocol consisted of two experiments as depicted in Figure 1 ▶ . In the first experiment 4 animals per group were exposed to either ambient air or NIEHS reference samples of chrysotile asbestos for 6 hours per day, 5 days a week for a total of 10 or 20 days. These animals were assigned for bronchoalveolar lavage and lung procollagen mRNA analyses. In the second experiment, 5 animals per group were exposed as above for a total of 4, 14, or 30 days and an additional group was exposed for 30 days and allowed to recover with exposure to ambient air for an additional 28 days. These animals were analyzed for lung hydroxyproline content, histopathology and by immunohistochemistry.
Figure 1.

Experimental protocols of National Institute of Environmental Health Sciences chrysotile asbestos inhalation experiments (∼10 mg/m3, 6 hours/day, 5 days/week) in C57Bl/6 mice (n = 5/group/time period).
The chemical and physical characteristics of the NIEHS chrysotile asbestos have been previously described. 8 Asbestos fibers were aerosolized using a modified Timbrell generator to generate a target concentration of 10 mg/m 3 air, as previously described. 9 Aerosol characteristics and concentrations were measured daily using a Sierra cascade impactor. The average chrysotile fiber concentrations (mean ± SD) were 8.43 ± 1.95 mg/m 3 and 9.35 ± 1.83 mg/m 3 air, respectively, for the two experiments with an aerosol diameter of 0.55 μm.
Bronchoalveolar Lavage (BAL)
Following asbestos exposure for 10 and 20 days, 4 mice from each exposure group received a lethal dose of pentobarbital. Chest cavities were opened and then lungs were cannulated via the trachea with polyethylene tubing. Lungs were then lavaged in situ 6 times with sterile Ca2+- and Mg2+-free phosphate-buffered saline (PBS) at a volume of 1 ml for each lavage. One lung lobe was excised following lavages and flash-frozen in liquid nitrogen and stored at −70°C for Northern blot analysis. BAL fluids were centrifuged to obtain a cell pellet for total and differential cell counts and a cell-free supernatant for total protein determination. Cell numbers and differentials were determined from hemacytometer counting and cytocentrifuge preparation of resuspended cells, respectively. Cytocentrifuge preparations were stained with Giemsa and May-günwald stains and 500 cells were counted in each preparation. Total protein content was determined in cell-free BAL fluid supernatants stored at −20°C with the Bio-Rad protein assay (Bio-Rad Laboratories, Hercules, CA).
Northern Blot Analysis
Total RNA was extracted from lung lobes taken from 20 day sham- and chrysotile-exposed animals and prepared for Northern blotting as previously described. 10 Briefly, 15 μg of RNA was denatured and fractionated by electrophoresis on a 1.0% agarose-formaldehyde gel and transferred onto nitrocellulose filters. A murine procollagen type I cDNA probe (obtained from Dr. David Rowe, University of Connecticut Health Center, Farmington, CT) was labeled with α[32P]-dCTP (New England Nuclear, Cambridge, MA) by using a Prime-a-Gene labeling system (Promega, Madison, WI). Hybridizations were performed on duplicate RNA samples and signals were quantitated by PhosphorImage analysis (Bio-Rad Laboratories).
Histopathology
Following asbestos exposures of 4, 14, 30, and 30 + 28 days, 5 mice from each exposure group also received a lethal dose of pentobarbital and chest cavities were opened. The lungs were then perfused with PBS via the right ventricle and the right lung lobes were clamped off at the main bronchi and removed for hydroxyproline analysis as previously described. 2,11 The left lung lobes were then fixed by intratracheal instillation of 4% paraformaldehyde at a constant pressure of 14 cmH2O. The lungs were then immersed in fixative overnight at 4°C before embedding tissue blocks in paraffin. Lung sections were cut 3 μm thick for immunohistochemistry, described below, or stained using the Masson’s trichrome technique for detection of collagen.
Immunohistochemistry for BrdU and Phosphorylated ERK
To measure cell proliferation, all animals (n = 40; 5/group/time period) in the second experiment were intraperitoneally administered BrdU (100 mg/kg; Boehringer Mannheim, Indianapolis, IN) 48 and 24 hours before sacrifice. 12 Lung sections from sham- and chrysotile-exposed animals were deparaffinized in xylene for 5 minutes three times, rehydrated through graded ethanols and equilibrated in PBS. BrdU immunoreactivity was detected with the Zymed streptavidin-biotin based BrdU staining kit (Zymed Laboratories, San Francisco, CA) and 3,3′-diaminobenzidine (DAB) as a chromogen (Vector Laboratories, Burlingame, CA) according to the manufacturer’s protocol. Following color development, sections were rinsed in distilled water, counterstained with hematoxylin, dehydrated through graded ethanols, cleared with xylene, and mounted with Histomount (Zymed Laboratories) before microscopy. BrdU incorporation by cells undergoing DNA synthesis was quantitated with image analysis (BioQuant, Nashville, TN). The number of BrdU immunoreactive epithelial cells were determined in 10 bronchial and/or bronchiolar airways for duplicate lung sections per animal and normalized to airway epithelial length. The number of BrdU immunoreactive alveolar cells were also determined in 20 random 400× field areas (15,700 μm2) for duplicate lung sections per animal.
Phosphorylation (activation) of ERK was also determined by immunohistochemistry in lung sections from all animals in the second experiment. Lung sections were deparaffinized, rehydrated, equilibrated, as described above, permeabilized with 100% methanol at −20°C for 10 minutes and washed with 0.1% Triton X-100 in PBS (Tr-PBS). Endogenous peroxidase activity was dampened by treatment with 3% hydrogen peroxide in methanol for 10 minutes, followed by a 10-minute wash in PBS. Sections were encircled with a hydrophobic film (PAP PEN, Electron Microscopy Sciences, Ft. Washington, PA) and nonspecific protein binding was obstructed with a 200-μl overlay of 2% normal goat serum (Jackson ImmunoResearch Labs, West Grove, PA) in Tr-PBS for 20 minutes 3 times. Excess buffer was absorbed before overlaying with 200 μl of polyclonal rabbit anti-phosphorylated-p44/42 MAPK (phosphorylated ERK; New England Biolabs, Beverly, MA) 1:250 in Tr-PBS containing 2% normal goat serum overnight at 4°C in a humidor. Specificity of the phosphorylated ERK primary antibody was confirmed by Western blot analysis of proteins isolated from mouse lung homogenates and from murine alveolar type II epithelial cells stimulated with epidermal growth factor or hydrogen peroxide (data not shown). Mouse intestinal sections were treated identically as positive controls and negative controls consisted of lung and intestinal sections incubated with a rabbit IgG type-matched monoclonal antibody (Zymed Laboratories) in place of the phosphorylated-ERK primary antibody. Immunoreactivity was then detected using the anti-rabbit-IgG Vectastain ABC Elite kit (Vector Laboratories) and DAB as a chromogen according to the manufacturer’s protocols. Following color development, sections were rinsed in distilled water, counterstained with hematoxylin, dehydrated, cleared, and mounted with VectaMount (Vector Laboratories).
Light Microscopic Image Capture and Processing
Masson’s trichrome-stained and immunostained lung sections were imaged using Kodak T160 color slide film and an Olympus PM-30 photomicrograph system attached to an Olympus BX-50 microscope. Color slide images (35 mm) were scanned at a resolution of 320 pixels per inch into .tif format using a Nikon LS 2000 slide scanner and imported into Adobe Photoshop version 5.0 for cropping, assembly, and labeling. Finally, image montages were printed with a Fujix Pictrography 3000 video printer.
Statistical Analysis
All data are presented as means ± SE. Differences between means for group data were tested for significance by analysis of variance. A Student-Newman-Keuls test was used to identify significant differences between chrysotile asbestos-exposed and sham-exposed groups at each time point. All statistical analyses were performed blindly by Dr. Pamela M. Vacek in the Department of Biostatistics at the University of Vermont, and P values ≤ 0.05 were considered significant.
Results
Lung Injury, Inflammation, and Pulmonary Fibrosis
In comparison to sham controls, chrysotile inhalation resulted in pulmonary inflammation after 10 and 20 days of exposure, as indicated by elevations in the percentages of neutrophils in BAL and a concomitant decrease in percentages of alveolar macrophages (Figure 2) ▶ . A slight humoral inflammatory response was suggested by a lymphocytic infiltration after 20 days of exposure, as compared to control animals (Figure 2B) ▶ . However, the increased presence of inflammatory cells were not accompanied by a change in the total number of leukocytes present in the BAL fluid (data not shown). Evidence of pulmonary inflammation after 10 and 20 days of asbestos exposure was supported by increases in total protein levels in BAL (Figure 2D) ▶ , indicative of plasma extravasation into luminal spaces.
Figure 2.

Differential cell counts and total protein levels in BAL samples from sham- and chrysotile asbestos-exposed C57Bl/6 mice. No significant differences in total cell numbers were observed between exposure groups (data not shown). Values are means ± SE; n = 4 mice/group. *Significant difference from time-matched control, P ≤ 0.05.
Histological analysis of lung sections from chrysotile-exposed animals revealed no remarkable changes in lung architecture following 4 days of high-dose inhalation, with respect to controls. After 14 days of chrysotile exposure, moderate areas of cellular inflammation were noted in the distal alveolar regions accompanied by sporadic increases in alveolar-ductal cellularity. Bronchial airways of sham and chrysotile-exposed mice appeared similar. In comparison to sham controls (Figure 3A) ▶ , the development of focal pulmonary fibrotic lesions became apparent after 30 days of exposure (Figure 3B) ▶ , but were not associated with increases in lung hydroxyproline content (Figure 4) ▶ or elevations in steady-state procollagen type I mRNA levels (data not shown). Masson’s trichrome staining revealed increases in peribronchiolar cellularity and collagen deposition, whereas larger airways appeared unaffected. Developing fibrotic lesions were consistently confined to bronchiolar and to bronchiolar-alveolar ductal bifurcations. The severity and extent of lung injury and pulmonary fibrosis at 30 days did not appear to progress during a 4-week recovery period (Figure 3C) ▶ .
Figure 3.
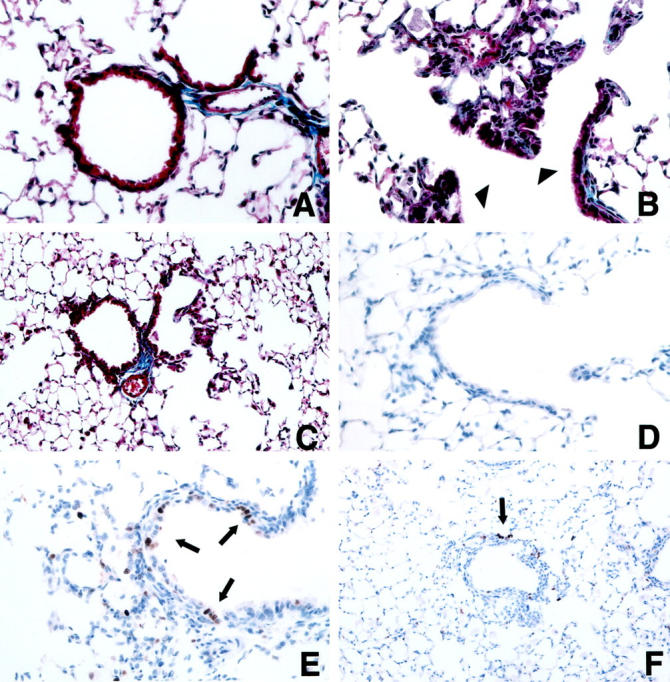
Representative images of lung tissue sections using Masson’s trichrome staining (A−C) or an antibody to BrdU (D−F). A and D: 30-day sham exposure; B, E, and F: 30-day chrysotile exposure; C: 30-day chrysotile exposure + 28-day recovery period. Focal increases in fibrosis were noted in chrysotile-exposed lungs at 30 days (B) at bronchiolar-alveolar ductal bifurcations (arrowheads). Lesions did not appear to increase in severity or extent in the 30-day chrysotile exposure + 28-day recovery group (C). Note increased BrdU immunostaining (arrows) within a developing alveolar ductal lesion (E) and peribronchiolar lesion (F) after 30 days of chrysotile exposure. Original magnifications,×400 (A, B, D, E) and ×200 (C and F).
Figure 4.

Hydroxyproline content of C57Bl/6 mouse lungs after inhalation of chrysotile asbestos. Values are means ± SE; n = 5 mice/group.
Immunohistochemical Detection of BrdU Incorporation
In comparison to lung tissue sections from control mice (Figure 3D) ▶ , increased numbers of cells undergoing DNA synthesis, as indicated by BrdU incorporation, were seen in asbestos-exposed lungs. BrdU immunoreactivity (DAB-positive) was primarily localized to bronchiolar epithelial (Clara cells) and alveolar ductal cells. Focal alveolar staining was observed within and adjacent to developing fibrotic lesions after 30 days of chrysotile inhalation (Figure 3E, 3F) ▶ . Quantitation of BrdU immunoreactivity revealed increased cellular proliferation in the alveolar regions of animals after 4, 14, and 30 days of chrysotile exposure (Figure 5A) ▶ . In contrast, elevations in proliferating bronchiolar epithelial cells were not statistically significant until 30 days of exposure (Figure 5B) ▶ . The numbers of proliferating cells decreased substantially but remained elevated, in comparison to control animals, in bronchiolar epithelium and alveoli during the 4-week recovery period after 30 days of chrysotile inhalation.
Figure 5.

Quantitative image analysis of BrdU incorporation in proliferating lung cells in C57Bl/6 mice after inhalation of chrysotile asbestos. Values are means ± SE; n = 5 mice/group. *Statistically significant difference from time-matched control, P ≤ 0.05.
Immunohistochemical Localization of ERK Phosphorylation
MAPK activation following asbestos exposure was assessed by immunostaining for phosphorylated ERK in lung sections from sham- and chrysotile-exposed animals. Negative control tissues, which were incubated with a rabbit IgG type-matched monoclonal antibody in place of the phosphorylated-ERK specific primary antibody, did not reveal any immunoreactivity (Figure 6A) ▶ . In contrast, phosphorylated ERK was detected in clusters of epithelial cells in the crypts of intestinal mucosa (positive control; Figure 6B ▶ ). Increases in ERK phosphorylation were not noted in the lungs of sham-exposed mice, with the exception of an occasional interstitial cell (Figure 6C) ▶ . However, sporadic clustering of cells immunoreactive for phosphorylated ERK were noted within bronchiolar bifurcations and epithelium after 14 and 30 days of chrysotile exposure (Figure 6D, 6E) ▶ . Focal ERK phosphorylation also was apparent within perivascular and peribronchiolar fibrotic lesions after 30 days of chrysotile inhalation (Figure 6F) ▶ . As with BrdU incorporation, ERK phosphorylation after 30 days of exposure increased within bronchiolar epithelium and developing fibrotic lesions, primarily confined to alveolar ductal regions (Figure 6G, 6H) ▶ . ERK phosphorylation was comparable to levels of immunoreactivity in sham-exposed animals after a 4-week recovery period (observations not shown).
Figure 6.
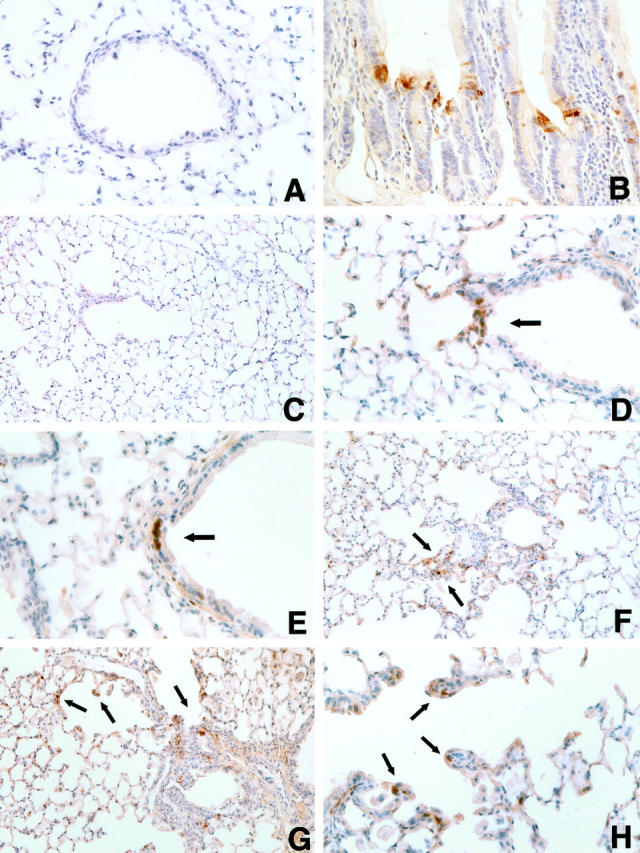
Representative images illustrating phosphorylated-ERK immunoreactivity (arrows) in lung tissue sections: A, 30-day chrysotile exposure (negative staining control); B, mouse intestine (positive staining control); C, 30-day sham exposure; D, 14-day chrysotile exposure; E−H, 30-day chrysotile exposure. Note increased clustering of ERK phosphorylation in bronchiolar epithelial and bifurcation cells after 14 and 30 days of chrysotile exposure (D and E). Increased focal ERK phosphorylation was also noted in developing peribronchiolar fibrotic lesions and alveolar ductal lesions after 30 days of exposure (F−H). Hematoxylin counterstain; original magnifications, ×400 (A, B, D, E, and H) and ×200 (C, F, and G).
Discussion
We show here increases in phosphorylated ERK proteins at sites of fibrogenesis. The majority of published asbestos-related research in animals has been performed in rats. Because those results suggest that the rat may be more susceptible than other rodent species and humans to particle-induced lung injury, 13-15 we first characterized cell proliferation, inflammation, and fibrosis in a subchronic inhalation model using C57Bl/6 mice. This strain of mice exhibits transient increases in BrdU labeling at alveolar duct bifurcations and terminal bronchioles, as well as focal bronchiolar thickening after brief 5-hour exposures to chrysotile asbestos. 16 The responses of C57Bl/6 mice to chrysotile asbestos reported here were generally consistent with our previously described model using Fischer 344 rats. Rats receiving a similar high-dose inhalation exposure to chrysotile asbestos also demonstrated an early and protracted inflammation with proportions of neutrophils in BAL elevated after 3, 9, and 20 days of exposure. 2 In the present study, pulmonary inflammation was supported by the morphological appearance of increased lung cellularity at 14 days and followed by Masson’s trichrome-positive fibrotic lesions after 30 days of exposure. Rats also developed increases in lung cellularity and focal fibrotic lesions after 20 days of exposure. 2,3 In contrast to our murine model, the severity and extent of fibrotic lesions progressed in rats during a 20-day recovery period. Statistical increases in hydroxyproline content, a biochemical indicator of increased collagen synthesis, were not observed in the lungs of chrysotile-exposed animals in either species. However, inhalation of similar concentrations of crocidolite asbestos causes increases in hydroxyproline levels in rats. 3 In addition to evaluating inflammation and fibrosis, we also characterized cell proliferation (a phenotypic endpoint associated with cytokines inducing ERK activation in a number of cell types). Significant increases in proliferation were protracted in mice after subchronic inhalation of chrysotile, first occurring at 4 days in the alveolar compartment and persisting during the 4-week recovery period. These findings are comparable with results of asbestos inhalation studies using A/J mice 17 and Sprague-Dawley rats. 18 In contrast, transient and reversible elevations in the number of cells incorporating BrdU in the alveolar duct region and bronchiolar epithelium are seen in mice exposed briefly to chrysotile 16,19 or in Fischer 344 rats inhaling chrysotile over a 30-day time period. 20
The characterization of inflammation, cell proliferation, and fibrotic endpoints now allows the utilization of molecular strategies in transgenic mice to elucidate the critical signaling cascades that are activated following asbestos exposure. For example, epidermal growth factor (EGF), a known inducer of ERK, causes proliferation of epithelial cells in a number of models. 21,22 Increased EGF receptor (EGF-R) expression is observed in asbestos patients 23 and in human mesothelial cells exposed to asbestos. 20 Following inhalation of asbestos by rats, transforming growth factor-α (TGF-α), which can bind EGF-R, is also up-regulated with corresponding increases in cellular proliferation within developing asbestotic lesions. 24 Furthermore, in vitro studies have demonstrated that asbestos and TGF-α exposure can preferentially induce activation of ERK, as opposed to JNK, in pleural mesothelial cells. 6 Activation of ERK signaling was subsequently observed to be mediated through phosphorylation of EGF-R and linked to apoptosis and/or cell proliferation. 6,7,25,26 Our observation that ERK phosphorylation is focally induced in developing fibrotic lesions after 14 and 30 days of asbestos inhalation provides further evidence that ERK signaling cascades may be key mediators in the development of asbestos-associated lung disease through regulation of cell proliferation or apoptosis.
The induction of ERK phosphorylation at sites of developing fibrotic lesions at peribronchiolar and at bronchiolar-alveolar junctions are consistent with the deposition patterns of inhaled asbestos in acute inhalation models. After brief inhalation of chrysotile asbestos, fibers are primarily deposited on alveolar duct bifurcations in both rats and mice. 27,28 It is believed that the deposition patterns of inhaled asbestos dictate the sites of pulmonary inflammatory and early cell proliferation in asbestosis.
Proliferation of type II and bronchiolar epithelial cells is a prominent feature of lung injury, including pulmonary fibrosis. 29 Our work and that of others show bronchiolar and alveolar epithelial cell proliferation, as measured by increased numbers of cells incorporating BrdU, 3H-thymidine, and proliferating cell nuclear antigen (PCNA) immunoreactivity at sites of developing fibrotic lesions following inhalation of chrysotile asbestos. 2,3,16,19,24 The onset of increased cell proliferation could possibly be mediated by a number of proinflammatory or growth factor molecules. Many members of the cytokine, chemokine, and growth factor families are candidates for the critical mediators of asbestos-associated lung diseases including pulmonary fibrosis. 19,23,24,30,31 This mediators include platelet-derived growth factors (PDGF), 32 TGF-α 24 and -β1, 16,33 and tumor necrosis factor-α (TNF-α) 16,21 which are up-regulated or activated at the bronchiolar-alveolar sites of chrysotile-induced fibrotic lesions in rats and mice. However, a number of overlapping intracellular signaling cascades shared by these and other extracellular stimuli 6,34-36 may be inducible agents of ERK activation in lung cells. Thus, ERK phosphorylation and subsequent downstream signaling events may represent a common pathway for protection and/or repair after lung injury.
There is increasing evidence that intracellular MAPK signaling pathways may be critical determinants of the phenotypic outcome of lung diseases. 37-40 Patterns of focal ERK phosphorylation at sites of chrysotile-induced cell proliferation and fibrosis may reflect paracrine signaling by the aforementioned cytokines and growth factors at sites of fiber deposition and subsequent lung injury. A similar pattern of focal ERK activation by autocrine/paracrine signaling growth factors has been postulated to occur in low grade gliomas. 41 In addition, ERK activation appears to be an important signaling pathway in a murine model of urethane-induced lung injury and carcinogenesis. 38 It might be that ERK-mediated signaling is a shared pathway by which inflammatory mediators and growth factors form an autocrine/paracrine system to repair or compensate for injured lung tissue. However, persistent activation of this pathway may override cell growth checkpoints, eventually leading to the development of pulmonary fibrosis and carcinogenesis.
Generation of transgenic mouse models is expanding the knowledge of intracellular signaling events from cellular to physiological phenotypes of disease. Our murine model of asbestosis with quantifiable outcomes of inflammation and cell proliferation will be invaluable in determining whether MAPK signaling is directly related to the development of these endpoints and their relationship to fibrogenesis. Ongoing experiments are using this inhalation model with transgenic mice expressing a mutant form of EGF-R 42 or dominant-negative ERK kinase (MEK-1) in bronchiolar epithelial cells to further delineate the signaling cascades involved in the development of asbestosis.
Footnotes
Address reprint requests to Brooke T. Mossman, Department of Pathology, College of Medicine, University of Vermont, Burlington, VT 05405-0068. E-mail: bmossman@zoo.uvm.edu.
Supported by National Institutes of Health grants T32ES07122, RO1ES/HL09213, RO1HL39469, and RO1ES06499.
References
- 1.Quinlan TR, Marsh JP, Janssen YMW, Leslie KO, Hemenway D, Vacek P, Mossman BT: Dose-responsive increases in pulmonary fibrosis after inhalation of asbestos. Am J Crit Care Med 1994, 150:200-206 [DOI] [PubMed] [Google Scholar]
- 2.Quinlan TR, BéruBé KA, Marsh JP, Janssen YMW, Taishi P, Leslie KO, Hemenway D, O’Shaughnessy PT, Vacek P, Mossman BT: Patterns of inflammation, cell proliferation, and related gene expression in lung after inhalation of chrysotile asbestos. Am J Pathol 1995, 147:728-739 [PMC free article] [PubMed] [Google Scholar]
- 3.BéruBé KA, Quinlan TR, Moulton G, Hemenway D, O’Shaughnessy P, Vacek P, Mossman BT: Comparative proliferative and histopathologic changes in rat lungs after inhalation of chrysotile or crocidolite asbestos. Toxicol Appl Pharm 1996, 137:67-74 [DOI] [PubMed] [Google Scholar]
- 4.Mossman BT, Faux S, Janssen Y, Jiménez LA, Timblin C, Zanella C, Goldberg J, Walsh E, Barchowsky A, Driscoll K: Cell signaling pathways elicited by asbestos. Environ Health Perspect 1997, 105(suppl 5):1121-1125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Karin M: The regulation of AP-1 activity by mitogen-activated protein kinases. J Biol Chem 1995, 270:16483-16486 [DOI] [PubMed] [Google Scholar]
- 6.Zanella CL, Posada J, Tritton TR, Mossman BT: Asbestos cause stimulation of the extracellular signal-related kinase 1 mitogen-activated protein kinase cascade after phosphorylation of the epidermal growth factor receptor. Cancer Res 1996, 56:5334-5338 [PubMed] [Google Scholar]
- 7.Jiménez LA, Zanella C, Fung H, Janssen YMW, Vacek P, Charland C, Goldberg J, Mossman BT: Role of extracellular signal-related protein kinases in apoptosis by asbestos and H2O2. Am J Physiol 1997, 273:L1029-L1035 [DOI] [PubMed] [Google Scholar]
- 8.Campbell WJ, Huggins CW, Wylie AG: Chemical and physical characterization of amosite, chrysotile, crocidolite, and nonfibrous tremolite for oral ingestion studies by the National Institute of Environmental Health Sciences. Washington, DC, US Bureau of Mines Report of Investigations no. 8452, 1980
- 9.Hemenway DR, MacAskill SM: Design, development, and test results of a horizontal flow facility. Am Ind Hyg Assoc J 1982, 43:874-879 [DOI] [PubMed] [Google Scholar]
- 10.Janssen YMW, Marsh JP, Absher MP, Hemenway D, Vacek PM, Leslie KO, Borm PJA, Mossman BT: Expression of antioxidant enzymes in rat lungs after inhalation of asbestos or silica. J Biol Chem 1992, 267:10625-10630 [PubMed] [Google Scholar]
- 11.Woessner JF: Determinations of hydroxyproline in tissue and protein samples containing small proportions of the amino acid. Arch Biochem Biophys 1961, 93:440-447 [DOI] [PubMed] [Google Scholar]
- 12.Eldridge SR, Tilbury LF, Goldsworthy TL, Butterworth BE: Measurement of chemically induced cell proliferation in rodent liver and kidney: A comparison of 5-bromo-2′-deoxyuridine and [3H]-thymidine administered by injectionor osmotic pump. Carcinogenesis 1990, 11:2245-2251 [DOI] [PubMed] [Google Scholar]
- 13.Heinrich U, Fuhst R, Rittinghausen S, Creutzenberg O, Bellman B, Koch W, Levsen K: Chronic inhalation exposure of Wistar rats and two different strains of mice to diesel engine exhaust, carbon black, and titanium dioxide. Inhal Toxicol 1995, 7:533-556 [Google Scholar]
- 14.Heinrich U, Muhle H, Takenada S, Ernst H, Fuhst R, Mohr U, Pott F, Stöber W: Chronic effects on the respiratory tract of hamsters, mice and rats after long-term inhalation of high concentrations of filtered and unfiltered diesel engine emissions. J Appl Toxicol 1986, 6:383-395 [DOI] [PubMed] [Google Scholar]
- 15.Mauderly JL, Banas DA, Griffith WC, Hahn FH, Henderson RF, McClellan RO: Diesel exhaust is not a pulmonary carcinogen in CD-1 mice exposed under conditions carcinogenic to F344 rats. Fundam Appl Toxicol 1996, 30:233-242 [DOI] [PubMed] [Google Scholar]
- 16.Brass DM, Hoyle GW, Poovey HG, Liu J-Y, Brody AR: Reduced tumor necrosis factor-α and transforming growth factor-β1 expression in the lungs of inbred mice that fail to develop fibroproliferative lesions consequent to asbestos exposure. Am J Pathol 1999, 154:853-862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dixon D, Bowser AD, Badgett A, Hasseman JK, Brody AR: Incorporation of bromodeoxyuridine (BrdU) in the bronchiolar-alveolar regions of the lungs following two inhalation exposures to chrysotile asbestos in strain A/J mice. J Environ Pathol Toxicol Oncol 1995, 14:205-213 [PubMed] [Google Scholar]
- 18.Donaldson K, Brown DM, Miller BG, Brody AR: Bromodeoxyuridine (BrdU) uptake in the lungs of rats inhaling amosite asbestos or vitreous fibers at equal airborne fibre concentrations. Exp Toxicol Pathol 1995, 47:207-211 [DOI] [PubMed] [Google Scholar]
- 19.Liu J-Y, Brass DM, Hoyle GW, Brody AR: TNF-α receptor knockout mice are protected from the fibroproliferative effects of inhaled asbestos fibers. Am J Pathol 1998, 153:1839-1847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pache J-C, Janssen YMW, Walsh ES, Quinlan TR, Zanella CL, Low RB, Taatjes DJ, Mossman BT: Increased epidermal growth factor-receptor protein in a human mesothelial cell line in response to long asbestos fibers. Am J Pathol 1998, 152:333-340 [PMC free article] [PubMed] [Google Scholar]
- 21.Schmidt-Ullrich RK, Mikkelsen RB, Dent P, Todd DG, Valerie K, Kavanagh BD, Contessa JN, Rorrer WK, Chen PB: Radiation-induced proliferation of the human A341 squamous carcinoma cells is dependent on EGFR tyrosine phosphorylation. Oncogene 1997, 15:1191-1197 [DOI] [PubMed] [Google Scholar]
- 22.Carpenter G: Employment of the epidermal growth factor receptor in growth factor-independent signaling pathways. J Cell Biol 1999, 146:697-702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Partanen R, Hemminki K, Koskinen H, Luo J-C, Carney WP, Brandt-Rauf PW: The detection of increased amounts of the extracellular domain of the epidermal growth factor receptor in serum during carcinogenesis in asbestosis patients. J Med 1994, 36:1324-1328 [DOI] [PubMed] [Google Scholar]
- 24.Liu J-Y, Morris GF, Lei W-H, Corti M, Brody AR: Up-regulated expression of transforming growth factor-α in the bronchiolar-alveolar duct regions of asbestos-exposed rats. Am J Pathol 1996, 149:205-217 [PMC free article] [PubMed] [Google Scholar]
- 25.Goldberg JL, Zanella CL, Janssen YMW, Timblin CR, Jimenez LA, Vacek P, Taatjes DJ, Mossman BT: Novel cell imaging techniques show induction of apoptosis and proliferation in mesothelial cells by asbestos. Am J Respir Cell Mol Biol 1997, 17:265-271 [DOI] [PubMed] [Google Scholar]
- 26.Zanella CL, Timblin CR, Cummins A, Jung M, Goldberg J, Raabe R, Tritton TR, Mossman BT: Asbestos-induced phosphorylation of the epidermal growth factor receptor (EGF-R) is linked causally to c-fos expression and apoptosis in mesothelial cells. Am J Physiol 1999, 277:L684-693 [DOI] [PubMed] [Google Scholar]
- 27.Brody AR, Roe MW: Deposition pattern of inorganic particles at the alveolar level in the lungs of rats and mice. Am Rev Respir Dis 1983, 128:724-729 [DOI] [PubMed] [Google Scholar]
- 28.Brody AR, Hill LH, Adkins B, O’Connor RW: Chrysotile asbestos inhalation in rats: deposition pattern and reaction of alveolar epithelium and pulmonary macrophages. Am Rev Respir Dis 1981, 123:670-679 [DOI] [PubMed] [Google Scholar]
- 29.Crouch E: Pathobiology of pulmonary fibrosis. Am J Physiol 1990, L159–L184 [DOI] [PubMed]
- 30.Robledo R, Mossman B: Cellular and molecular mechanisms of asbestos-induced fibrosis. J Cell Physiol 1999, 180:158-159 [DOI] [PubMed] [Google Scholar]
- 31.Lasky JA, Tonthat B, Liu J-Y, Friedman M, Brody AR: Upregulation of the PDGF-alpha receptor precedes asbestos-induced lung fibrosis in rats. Am J Respir Cri Care Med 1998, 157:1652-1657 [DOI] [PubMed] [Google Scholar]
- 32.Liu J-Y, Morris GF, Lei W-H, Hart CE, Lasky JA, Brody AR: Rapid activation of PDGF-A and PDGF-B expression at sites of lung injury in asbestos-exposed rats. Am J Respir Cell Mol Biol 1997, 17:129-140 [DOI] [PubMed] [Google Scholar]
- 33.Perdue TD, Brody AR: Distribution of transforming growth factor-β1, fibronectin, and smooth muscle actin in asbestos-induced pulmonary fibrosis in rats. J Histochem Cytochem 1994, 42:1061-1070 [DOI] [PubMed] [Google Scholar]
- 34.Hu Y, Schett G, Zou Y, Dietrich H, Xu Q: Abundance of platelet-derived growth factors (PDGFs), PDGF receptors and activation of mitogen-activated protein kinases in brain decline with age. Brain Res Mol Brain Res 1998, 53:252-259 [DOI] [PubMed] [Google Scholar]
- 35.Chin Y, Petrache I, Choi AM, Choi ME: Transforming growth factor beta 1 rescues serum deprivation-induced apoptosis via the mitogen-activated protein kinase (MAPK) pathway in macrophages. J Biol Chem 1999, 274:11362-11368 [DOI] [PubMed] [Google Scholar]
- 36.Janssen-Heininger YM, Macara I, Mossman BT: Cooperativity between oxidants and tumor necrosis factor in the activation of nuclear factor (NF)-kappaB: requirement of ras/mitogen-activated protein kinases in the activation of nf-kappa-b by oxidants. Am J Respir Cell Mol Biol 1999, 20:942-952 [DOI] [PubMed] [Google Scholar]
- 37.Carter AB, Monick MM, Hunninghake GW: Both Erk and p38 kinases are necessary for cytokine gene transcription. Am J Respir Cell Mol Biol 1999, 20:751-758 [DOI] [PubMed] [Google Scholar]
- 38.Yano T, Yano Y, Nagashima Y, Yuasa M, Yajima S, Horikawa S, Hagiwara K, Kishimoto M, Ichikawa T, Otani S: Activation of extracellular signal-regulated kinase in lung tissues of mice treated with carcinogen. Life Sci 1999, 64:229-236 [DOI] [PubMed] [Google Scholar]
- 39.Weintraub SJ: Dormant tumor-suppressor pathways in tumors. Am J Respir Cell Mol Biol 1999, 20:541-542 [DOI] [PubMed] [Google Scholar]
- 40.Ravi RK, Thiagalingam A, Weber E, McMahon M, Nelkin BD, Mabry M: Raf-1 causes growth suppression and alteration of neuroendocrine markers in DMS53 human small-cell lung cancer cells. Am J Respir Cell Mol Biol 1999, 20:541-549 [DOI] [PubMed] [Google Scholar]
- 41.Mandell JW, Hussani IM, Zecevic M, Weber MJ, VandenBerg SR: In situ visualization of intratumor growth factor signaling: immunohistochemical localization of activated ERK/MAP kinase in glial neoplasms. Am J Pathol 1998, 153:1411-1423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hardie WD, Kerlakian CB, Bruno MD, Huelsman KM, Wert SE, Glasser SW, Whitsett JA, Korfhagen TR: Reversal of lung lesions in transgenic transforming growth factor α mice by expression of mutant epidermal growth factor receptor. Am J Respir Cell Mol Biol 1996, 15:499-508 [DOI] [PubMed] [Google Scholar]