

Abstract
The heterogeneity of tumor cells is frequently observed in lung cancer, but the clonality of these cells has not yet been established. The distinct components of 12 lung adenosquamous carcinomas were compared by genetic alterations of p53 and K-ras, chromosomal abnormalities at 9p21 and 9q31–32, and immunohistochemical reactions. The immunoreactivity of p53 was consistent in both adenocarcinomatous and squamous cell carcinomatous components as well as in the transitional areas, retaining the morphological characteristics of the distinct components. The same p53 mutation was found in both components of each tumor with p53 overexpression. No K-ras mutations were detected in any of the tumors examined. Three of the four tumors with chromosomal abnormalities detected, one at 9p21 and two at 9q31–32, had coincident abnormalities between the distinct components, whereas one tumor deleted homozygously at 9p21 (D9S259) in the adenocarcinomatous component with loss of heterozygosity in the other component. The expression of squamous cell carcinoma-related antigen in adenocarcinomatous components was significantly higher than that of lung adenocarcinomas (57 ± 5.8% vs. 1.0 ± 0.5%, P < 0.0001), whereas Mucin 1 expression is less in these components (9.0 ± 4.9% vs. 55 ± 8.2%, P = 0.003). These results suggest monoclonal transition from squamous cell carcinoma to adenocarcinoma in lung adenosquamous carcinoma.
Although tumor heterogeneity is a fundamental feature of carcinogenesis and cancer progression, 1-3 the mechanism by which it occurs remains largely unknown. 4,5 The heterogeneity of tumor cells is frequently observed in lung cancer. 1,6-8 The main pattern consists of a regional distribution of individual cells, similar to their immediate neighbors, whereas more distant cells tend to be similar to their immediate neighbors. 6-9 According to fundamental concepts concerning the carcinogenesis of lung cancer, field cancerization, and the multistep carcinogenic process, the pathogenesis of these heterogeneous tumor cells would be explained by various carcinogenic stages involving monoclonal or polyclonal expansion. 5,10 Direct evidence of the clonality of lung cancer cells, however, still remains quite limited, though such information would be essential not only for explaining the process of carcinogenesis and cancer progression, but also for designing successful treatment modalities for heterogeneous tumors. 2,3,9
Adenosquamous carcinoma of the lung, one of the rather rare subtypes of lung cancer, 11-13 is a typical example of the heterogeneous tumors, containing two distinct components that are either adenocarcinomatous or squamous carcinomatous. The diagnosis of adenosquamous carcinoma should be restricted to carcinomas that show unequivocal squamous differentiation in the form of keratin or intracellular bridges, and unequivocal glandular differentiation in the form of acini, tubules, or papillary structures. 14 According to the most recent World Health Organization classification of lung tumors, a definitive diagnosis of lung adenosquamous carcinoma requires a minimum of 10% of each component in the whole tumor. 15 Although morphological characterizations of both components of adenosquamous carcinoma have been made, no genetic approach to the clonality of adenosquamous carcinoma has yet been attempted. In this study, we performed topographic genotyping and immunohistochemical analysis on 12 adenosquamous carcinoma tumors with a definitive diagnosis, free from either anti-cancer chemotherapy or irradiation therapy before sampling, that would cause genetic changes to the cancer cells. As topographic genotyping, comparative DNA sequences of p53, K-ras, and chromosomal analysis at 9p21 and 9q31–32 were performed on the distinct components in each tumor. p53 mutations occur in approximately one-half of non-small-cell carcinomas at various points within hot spots, 16 supposed as a good marker for detecting the clonality of cancer cells. 17,18 K-ras mutations have been reported in 27 to 46% of adenocarcinomas of the lung, and less or not at all in squamous cell carcinoma. 19-21 9p21 22,23 and 9q31–32 24 are frequent abnormal loci in squamous cell carcinoma, but less in adenocarcinoma of the lung. The immunolocalization of p53 overexpression was compared with the p53 mutations between the distinct components in each tumor. In addition, statistical difference of immunohistochemical expression of the tumor-associated antigens was examined, including the antigens predominantly expressed in adenocarcinomas, such as carcino-embryonic antigen (CEA), 25,26 cancer antigen 19-9 (CA19-9), 26-28 and Mucin 1 (MUC1). 29,30 Squamous cell carcinoma-related antigen (SCC) was also examined, because its rather selective expression in squamous cell carcinomas. 31-33 The expressions of vascular endothelial growth factor (VEGF), 34,35 proliferating cell nuclear antigen (PCNA), 36,37 and p21WAF1/Cip1 (WAF1) 38,39 were also examined to characterize the two components of these tumors. The degrees of immunoreactivity were quantified according to the distribution of immunoreactive tumor cells among whole tumor cells according to our previous study. 36
The main purposes of this study were to clarify the clonality of adenosquamous carcinoma of the lung as a typical model of heterogeneous tumors and to elucidate the process of carcinogenesis and cancer progression of these tumors. Such information may suggest successful therapies for heterogeneous tumors. 1-3
Materials and Methods
Tumors Examined
The tumors of 12 patients with adenosquamous carcinoma of the lung were obtained at the Hospital of the Institute of Development, Aging and Cancer, Tohoku University (cases 1–8), and the Central Hospital of Aomori Prefecture (cases 9–12). Eight of these tumors (cases 1–4 and 9–12) were resected by surgical operation; the others (cases 5–8) were autopsied (Table 1) ▶ . Two patients (cases 10 and 11) are without smoking habit; the rest are current or former smokers (Table 1) ▶ with the range of Brinkman index for cigarette smoking from 690 to 1400 (958 ± 104, mean ± SEM). Informed consent for genetic analysis of these tumors was obtained from each patient or their families. None of these patients was treated by anti-cancer chemotherapy or irradiation before the tumors were sampled. Metastatic lesions were available in three cases (cases 3, 5, and 8), and were examined by microscopic observation. A pathological diagnosis of adenosquamous carcinoma was based on the WHO Histological Typing of Lung Tumors 15 and checked by at least three independent pathologists. Three to five sampled specimens of each tumor were formalin-fixed and paraffin-embedded, and 10 serial sections 3 μm thick were cut from each block. The first sections were stained with hematoxylin-eosin (H&E), the second sections for periodic acid-Schiff (PAS)-alcian blue staining, and the remaining eight sections were used for immunohistochemical study. Three additional sections 8 μm thick were cut and used for microdissection of the two distinct components of each tumor to extract DNA as described below. Ten primary tumors of lung adenocarcinoma and ten primary tumors of lung squamous cell carcinoma, surgically resected in our hospital, were also used in our pilot study to get control reactions by immunohistochemistry, especially for non-commercially available antibodies against human squamous cell carcinoma-related antigen (F2H7C) and against human MUC1 (MUSEII).
Table 1.
Characteristics of Patients and Adenosquamous Carcinomas Examined, and p53 Status in Each Component
| Case no. | Age (y)/gender | Smoking | Stage | Sampling | Predominant p53 component | IHC AD/SQ | p53 mutation AD/SQ |
|---|---|---|---|---|---|---|---|
| 1 | 72 /M | former | I | operated | SQ (80%) | N /N | WT/WT |
| 2 | 65 /M | former | II | operated | SQ (80%) | N /N | WT/WT |
| 3 | 68 /M | current | IIIA | operated | SQ (70%) | N /N | WT/WT |
| 4 | 68 /F | current | IIIA | operated | AD (80%) | P /P | 167Gln to Arg/167Gln to Arg |
| 5 | 89 /M | former | IV | autopsied | AD (60%) | P /P | 297His to Tyr/297His to Tyr |
| 6 | 68 /M | current | IV | autopsied | SQ (60%) | N /N | WT/WT |
| 7 | 78 /M | current | IV | autopsied | SQ (80%) | P /P | 210Asn to Asp/210Asn to Asp |
| 8 | 74 /M | current | IV | autopsied | AD (70%) | N /N | WT/WT |
| 9 | 68 /M | current | II | operated | SQ (60%) | P /P | ND |
| 10 | 63 /F | no | I | operated | AD (60%) | P /P | ND |
| 11 | 60 /F | no | I | operated | SQ (80%) | N /N | WT/WT |
| 12 | 65 /M | current | IIIA | operated | SQ (80%) | N /N | WT/WT |
M, male; F, female; AD, adenocarcinomatous component; SQ, squamous carcinomatous component; IHC, immunohistochemistry; P, positive; N, negative; WT, wild-type; ND, not determined; 167Gln to Arg, mutation from glutamine to arginine at codon 167; 297His to Tyr, mutation from histidine to tyrosine at codon 297; 210Asn to Asp, mutation from asparagine to asparatic acid at codon 210; cdns, codons.
Microdissection and DNA Extraction
The two different foci in each tumor, determined by subsequent H&E-stained sections, were scraped off individually from three deparaffinized thick sections under microscopic observation. To avoid the contamination of each sample from different foci in one section, the tissue sections were dried up before sampling, and only the foci to be scraped off were wetted using microcapillary tips. The tissue fragments were collected into microtubes and digested as previously described. 40 For the analysis of loss of heterozygosity at 9p21 and 9q31–32, additional DNA samples were extracted from tumor-free lung tissues, which were available in cases 1 to 8 and used for control DNA for each tumor.
Detecting p53 Mutations
For detecting p53 mutations within exons 5 to 8, the isolated DNA samples from the two different components of each tumor were amplified using primer sets as described previously. 41 The amplified polymerase chain reaction (PCR) products were cloned into a pCRII vector (Invitrogen Corp., Carlsbad, CA) and purified through Qiagen Mini Columns (Qiagen Inc., Chatsworth, CA). The mixtures of 20 clones of each DNA sample were sequenced by a dsDNA cycle sequencing system (Gibco BRL, Rockville, MD) using primers end-labeled with [γ-32P] ATP (Amersham Corp., Arlington, IL) according to the manufacturers’ manuals. Mutations were accepted when the genetic alterations were detected in the independent PCR products of the same DNA samples.
Detecting K-ras Mutations
The isolated DNA samples from the two different two components in each tumor were also analyzed for K-ras mutations at codons 12, 13, and 61 using PCR-restriction fragment length polymorphisms (RFLP) analysis, and direct sequencing. As for codon 12, PCR-primer introduced restriction with enrichment for mutant alleles (PIREMA) was used as described previously. 21 Briefly, the PCR products (192 bp) were digested with BstNI (TOYOBO, Osaka, Japan), and the digested products were amplified again in the same protocol with the first PCR. As positive controls of the K-ras mutations at these sites, the extracted DNA samples of the cell lines NCI-H358 (TGT) and NCI-H727 (GTT) 42 were used, in addition to human genomic DNA (Clontech Laboratories, Inc., Palo Alto, CA) as a normal control (GGT). Direct sequencing of DNA samples was also performed for detecting other possible mutations at codon 12, 13, and 61 using a double-strand DNA cycle sequencing system (Gibco BRL) with the primers end-labeled with [γ-32P]ATP (Amersham Corp.) according to a previous study. 40
Microsatellite Analysis at 9p21 and 9q31–32
Three different DNA samples from each tumor were used for microsatellite analysis, collected independently from the adenocarcinomatous components, squamous carcinomatous components, and tumor-free sites. Microsatellite analysis was omitted in cases 9 to 12, which lacked tumor-free materials. Six microsatellite markers were used in this study to distinguish the chromosomal regions at 9p21 (D9S265, D9S126, D9S259) and 9q31–32 (KM9.1, D9S177) using primer sequences reported before. 22,24 The primers were end-labeled with 0.2 μCi each of [γ-32P] ATP and 0.07 U of T4 polynucleotide kinase (Gibco BRL). PCR amplifications were performed in 10 μl reaction volumes, including 50 ng of genomic DNA, 0.6 pg of labeled primer, 50 μmol/L of each dNTP (Takara), and 0.5 U of Taq DNA polymerase (Takara). After initial denaturation at 94°C for 3 minutes, 40 cycles of PCR (30 seconds at 94°C, 60 seconds at 50°C, and 60 seconds for 72°C) were performed, with final extension for 10 minutes at 72°C. PCR products were mixed with loading buffer, heat denatured, and electrophoresed in 8% polyacrylamide-urea-formamide gel. The intensity of the radioactivity of the signal was measured using Bio-Imaging Analyzer system (Fuji Film Co., Minami-Ashigara, Japan), and loss of heterozygosity was defined as a reduction in the intensity of one allele in the tumor DNA by at least 50% as compared with the corresponding normal DNA. 22,24 When both alleles were deleted, the results were confirmed at least twice.
Immunohistochemistry
The mouse monoclonal antibody antiserums used in this study were as follows: anti-human CEA (ZC23, Histofine, Tokyo), anti-human CA19-9 (1116 NS 19-9, CIS bio International, Tokyo), anti-human SCC (F2H7C, kindly provided by Dr. Kato, Yamaguchi University School of Medicine, Yamaguchi, Japan), anti-human VEGF (Santa Cruz Biotechnology, Santa Cruz, CA), anti-human MUC1 (MUSEII, kindly provided by Dr. Imai, Sapporo University, Sapporo, Japan), anti-human p53 (DO-7, Dako Corporation, Carpinteria, CA), anti-human PCNA (PC-10, Dako), and anti-human p21Waf1/Cip1 (Ab-1, Oncogene Science). The optimal dilution of each immunohistochemical reaction was determined by pilot studies as follows: antibodies against CEA and CA19-9 were diluted according to the manufacturer’s recommendations, anti-SCC at 1:100, anti-VEGF at 1:500, anti-MUC1 at 1:50, anti-p53 at 1:20, anti-PCNA at 1:20, and anti-p21Waf1/Cip1 at 1:10. Primary incubation was performed overnight at 4°C with these primary antibodies, and staining was performed using the avidin-biotinylated peroxidase complex technique using Vectastain kits (Vector Laboratories, Burlingame, CA) according to our previous study. 36 For the reactions with the antibodies against p53 and Waf1, microwave pretreatment was performed using 0.01 mol/L sodium citrate buffer at pH 6.0.
Quantification and Statistical Analysis
To quantify the immunoreactive tumor cells, five random sites each per one adenocarcinomatous and one squamous carcinomatous component of the same tumor were color-photographed and observed at 1000× as the final magnification using parallel sampling lines (one nucleus once). The degrees of immunoreactivity of each component were determined by percentage of the positive tumor cells among total tumor cells, positive and negative (approximately 300), according to the quantification methods. 36 Statistical analysis was performed by χ 2 test. The immunoreactivity of cases 9 to 12 was quantified only for the reaction by anti-p53 to determine the overexpression of p53 when more than 10% of the whole tumor cells in each component were positive. 36
Results
Light Microscopic Observation
All of the primary tumors examined were found to have both of the well-differentiated adenocarcinomatous and squamous carcinomatous components. The approximate percentages of the areas occupied by the predominant components are shown in Table 1 ▶ . Each component of these tumors showed a distribution of more than 20% of the whole tumor area, thereby confirming the diagnosis of adenosquamous carcinoma (Figures 1 and 2) ▶ ▶ . The adenocarcinomatous components were often, but not always, positive for PAS-alcian blue staining, with little or no reaction in the squamous components of these tumors (Figure 1b) ▶ . The tumor cells distributed in the boundary of the two components were observed to have the cytologic features of squamous cell carcinoma, despite forming tubular or papillomatous structures, the characteristic features of adenocarcinoma (Figures 1 and 2) ▶ ▶ . Although such intervening areas, referred to as “transitional areas,” were limited in size, they were found in all of the tumors of the twelve cases. Metastatic lesions were available in three cases (3, 5, and 8). Case 3 had lymph node metastasis, which was infiltrated with adenocarcinoma of moderate differentiation (Figure 2f) ▶ . The autopsied Case 5 had metastasis to multiple organs including the liver, adrenal glands, and lymph nodes, which were infiltrated by adenocarcinomatous components. The metastases to the liver and lymph nodes of case 8, another autopsied case, were also composed only of adenocarcinomatous components (Figure 2g) ▶ . These metastatic lesions were occupied by homogeneous tumor cells, in contrast to the heterogeneous primary tumors of the lungs.
Figure 1.
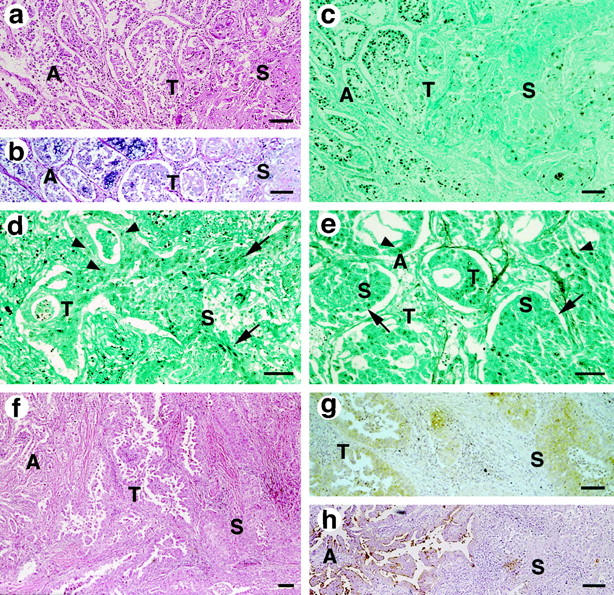
Photomicrographs of the adenosquamous carcinomas of case 10 (a-c), case 5 (d), case 7 (e), and case 2 patients (f-h) Bar, 100 μm. a: H&E staining shows the transitional areas (T) between the tumor composed of two well-defined components comprising well-differentiated adenocarcinoma growing in a bronchioloalveolar fashion (A) and nests of well-differentiated squamous cell carcinoma with increased cytoplasm (S). b: Heterogeneous staining in the adenocarcinomatous components (A) with PAS-alcian blue stain, without reactions in transitional areas (T), and in the squamous carcinomatous components (S). c−e: The degrees of p53 overexpression are consistent in both of the components, adenocarcinomatous (A, arrowheads) and squamous cell carcinomatous (S, arrows), and in the transitional area (T) as well. f: Transitional area (T) distributed between the two distinct components, adenocarcinomatous (A) and squamous carcinomatous (S). g: SCC immunoreactivity was revealed in the cells composing the transitional area (T), those with the cytologic features of squamous cell carcinoma (S), and those forming tubular and papillomatous structures, characteristic of adenocarcinoma. h: Higher immunoreactivity to CA19-9 was shown in the adenocarcinomatous components (A), but it was lower in the cells of the transitional areas and in the squamous carcinomatous components (S).
Figure 2.
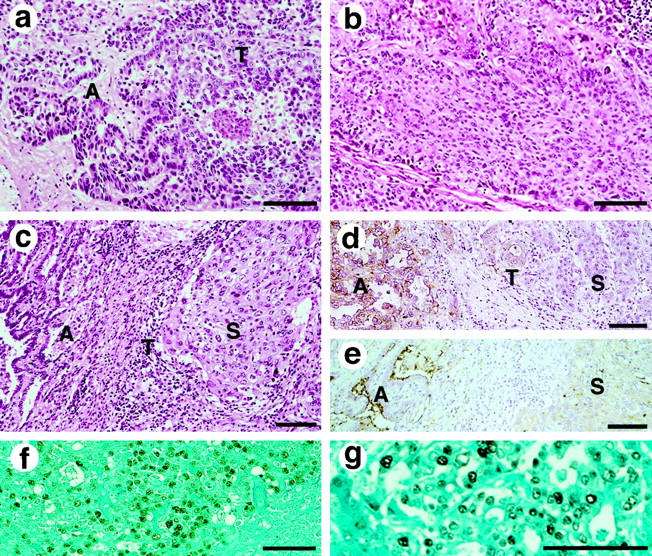
Photomicrographs of the adenosquamous carcinomas of case 8 (a, b, g), and Case 3 (c−f) (bar, 100 μm). a: H&E staining. The adenocarcinomatous component (A) showed greater cellular intensity of eosinophilic staining and nuclear chromatin density than the transitional component (T). b: Squamous carcinomatous components are shown with differentiation (H&E). c: The transitional area (T) was limited in this intervening area of case 3 (H&E). d: Higher expression of CEA was revealed in the adenocarcinomatous components (A), and less prominent in the squamous carcinomatous components (S). Cells with the characteristic features found in the transitional area (T) were distributed apart from the adenocarcinomatous components with expression of CEA. e: The immunohistochemical staining of MUC1 in the adenocarcinomatous compartment (A) is significantly more intense than in the other component (S). f and g: The metastatic tumors showing adenocarcinomatous features overexpressed p53, though both components of the primary sites of these tumors were negative for p53 immunoreactivity. p53 overexpression was found in the lymph nodes with metastasis (f, case 3) and in the liver metastasis (g, case 8), which were invaded by adenocarcinomatous components with moderate differentiation.
p53 Immunoreactivity and p53 Gene Status
Nuclear staining was observed only in the cells immunoreactive for anti-p53 antibody. p53 overexpression was assumed only when more than 10% of the tumor cells were immunoreactive, according to our previous study. 36 Five out of twelve tumors were found to have p53 immunoreactivity. It is of interest that both components of these five tumors exhibited overexpression of p53 protein (Table ▶ 1 and Figure 1, c ▶ −e). No significant difference was found between the degrees of p53 overexpression in the distinct areas in each tumor (Figure 5) ▶ . Sequencing revealed p53 mutations in the distinct components of the tumor with p53 overexpression; those of cases 4, 5, and 7 were consistent throughout each tumor (Table 1) ▶ . Both components of case 4 contained the same p53 mutation from CAG (glutamine) to CGG (arginine) in exon 5 (at codon 167, Figure 3a ▶ ). In Case 5, CAC (histidine) was changed to TAC (tyrosine) in exon 8 (at codon 297), and in Case 7, AAC (asparagine) was changed to GAC (aspartic acid) in exon 6 (at codon 210). p53 mutations were not determined in cases 9 and 10 because of the poor quality of DNA samples. Interestingly, no expression of p53 protein was observed and no p53 mutations were detected in either of the components of cases 3 and 8, although the metastatic lesions of these cases overexpressed p53 protein homogeneously and intensively, showing almost all tumor cells positive for p53 protein (Figure 2, f and g) ▶ .
Figure 5.
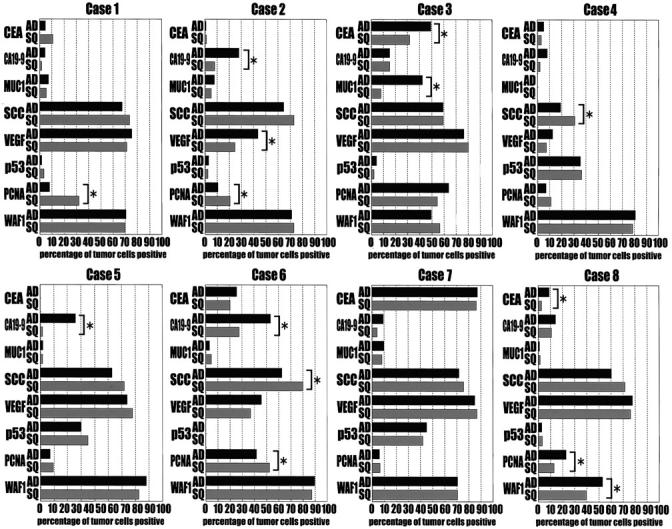
The panel of immunoreactivity of each component of eight tumors to the antibodies against CEA, CA19-9, MUC1, SCC, VEGF, p53 PCNA, and WAF1. The percentage of immunoreactive tumor cells in the whole tumor cells was calculated as the index of immunoreactivity. The results were compared between the two distinct components of each tumor, adenocarcinomatous (AD) and squamous carcinomatous (SQ), and the difference was determined to be significant when the P value was <0.001 (*).
Figure 3.

Comparison of genetic alterations of p53 and K-ras between the two distinct components of adenosquamous carcinomas. a: Both components of case 4, adenocarcinomatous (AD) and squamous carcinomatous (SQ), contained the same p53 mutation from CAG (glutamine) to CGG (arginine) at codon 167, compared with tumor-free tissues (N). b: Three tumors contained adenocarcinoma as predominant component: cases 4 (80%), 5 (60%), and 8 (70%). Complete digestion by BstNI of 192-bp PCR products to 163- and 29-bp fragments showed no mutations were included in these tumors as in the normal control DNA (GGT). The PCR products of the cell lines, NCI-H358 (TGT) and NCI-H727 (GTT), as positive controls of the K-ras mutations, were retained uncut.
K-ras Mutations
The DNA samples extracted from different foci of the twelve adenosquamous carcinomas were examined for K-ras mutations. No K-ras mutations were detected in either the adenocarcinomatous or squamous carcinomatous portions of each tumor at codons 12 using PCR-RFLP. 21 Figure 3b ▶ shows the results of three cases with predominantly adenocarcinomatous components: 80% in case 4, 60% in case 5, and 70% in case 8. Neither component of these cases was digested by BstNI, indicating that no K-ras mutations at codon 12 were included in these tissues, whereas the positive controls (NCI-H358 and NCI-H727) were clearly digested. Direct sequencing of DNA samples extracted from both of the components of these 12 tumors was also performed at codons 12, 13, and 61 without detecting any mutations.
Microsatellite Analysis of Chromosome 9
The microsatellite regions at 9p21, marked by D9S265, D9S126, and D9S259, 22 and at 9q31–32, marked by KM9.1 and D9S177, 24 were compared between the different components of each tumor. Control DNA samples taken from a tumor-free locus are required to show chromosomal alterations, but were available only in cases 1 to 8. Four of these eight cases contained heterozygous or homozygous deletions detected by the five markers examined (Table 2 ▶ , Figure 4 ▶ ). The deletions occurred more frequently in 9q31–32 (4/28, 14.3%) than in 9p21 (4/44, 9.1%). The identical abnormality (heterogeneous deletion) was found in the two components in three cases. Only one case, Case 8, was found to have different abnormalities in the two distinct components. In the adenocarcinomatous component of Case 8, both alleles were lost (homozygous deletion) at D9S259, whereas only a heterozygous deletion was detected in the squamous carcinomatous component (Figure 4c) ▶ . These abnormalities were confirmed by independent PCR reactions at least twice.
Table 2.
Chromosomal Status at 9p21 and 9q31-32 in Each Component of Adenosquamous Carcinomas
| Case no. | 9p21 | 9q31-32 | |||
|---|---|---|---|---|---|
| D9S265 AD/SQ | D9S126 AD/SQ | D9S259 AD/SQ | KM9.1 AD/SQ | D9S177 AD/SQ | |
| 1 | R /R | NI /NI | R /R | R /R | R /R |
| 2 | R /R | R /R | R /R | L /L | R /R |
| 3 | R /R | R /R | R /R | NI /NI | R /R |
| 4 | L /L | R /R | NI /NI | R /R | NI /NI |
| 5 | R /R | R /R | R /R | R /R | R /R |
| 6 | R /R | R /R | R /R | R /R | L /L |
| 7 | R /R | R /R | R /R | R /R | R /R |
| 8 | R /R | R /R | HD /L | R /R | R /R |
AD, adenocarcinomatous component; SQ, squamous carcinomatous component; R, retention of both alleles; L, loss of heterozygosity; HD, homozygous deletion; NI, not informative.
Figure 4.

Chromosomal abnormalities at 9p21 and 9q31–32 revealed by microsatellite analysis using five markers. D9S265 (a), D9S126 (b), and D9S259 (c) detected chromosomal abnormalities at 9p21, whereas KM9.1 (d), and D9S177 (e) detected 9q31–32. DNA samples extracted from three parts of the lung were used: tumor-free lung tissues (N), adenocarcinomatous components (AD) and squamous carcinomatous components (SQ). Loss of heterozygosity (L), homozygous deletion (HD) or retention (R) of both alleles was determined by densitometry of the polymorphic bands noted by arrows. These results are summarized in Table 2 ▶ . Case 8 (c) had different abnormalities between the adenocarcinomatous (homozygous deletion) and squamous carcinomatous lesions (heterozygous deletion), whereas both alleles were clearly retained in tumor-free foci.
Quantitative Analysis of Immunohistochemistry
The distributions of immunoreactive tumor cells among the whole tumor cells were quantified by calculating the percentage of positive tumor cells in each component, and analyzed by χ 2 test. The similarities between the different components of each tumor were revealed to be 79.7% (51/64) in coincident immunoreactivity. Higher immunoreactivity in the adenocarcinomatous components was revealed by the expression of CEA in cases 3 (Figure 2d) ▶ and 8, CA19-9 in cases 2 (Figure 1h) ▶ , 5, and 6, MUC1 in case 3 (Figure 2e) ▶ , and VEGF in case 2.
In 7 of 8 cases (87.5%), more than 50% positive tumor cells in the adenocarcinomatous component were found to have squamous cell carcinoma-related antigen (SCC). The SCC immunoreactivity in the adenocarcinomatous components was much higher than the average of SCC immunoreactivity in 10 lung adenocarcinomas by our pilot study (57.2 ± 5.8% versus 1.0 ± 0.5%, mean ± SEM, P < 0.0001), close to the SCC expressions in squamous components of these tumors (57.0 ± 9.7%, P = 0.988) and in 10 lung squamous cell carcinomas (43.4 ± 9.0%, P = 0.241). The intervening tumor cells in the transitional areas between the two distinct components were also reactive with anti-SCC antigen, as shown in Figure 1g ▶ . In contrast, the expression of MUC1, which is 55.3 ± 8.2% in 10 lung adenocarcinomas and 1.25 ± 0.71% in 10 lung squamous cell carcinomas by our pilot study, was very low (<10% positive) in 7 adenocarcinomatous components. The mean MUC1 expression of 8 adenocarcinomatous components was 9.04 ± 4.9%, significantly lower than that of 10 lung adenocarcinomas (P = 0.003). The MUC1 expression of squamous carcinomatous components was 4.30 ± 1.08%. Higher PCNA immunoreactivity was seen in the squamous cell carcinomatous components in three cases (1, 2, and 6), but only in the adenocarcinomatous lesions in one case, case 8.
Discussion
It is widely accepted that cancer results from the accumulation of mutations in the genes that directly control cell birth or cell death. 43 Such genetic instabilities of tumor cells are partly induced by the selective conditions in the tumor environment, including aberrant humoral, cell-substratum, and cell-cell interactions, that give rise to clonal expansion, allowing the tumor cell with the mutation to overtake its sister cells. 43 In this context, heterogeneity is a frequent event in cancer, which may happen by clonal selection 44 of the appropriate cells in the tumor environment.
Concerning the histogenesis of adenosquamous carcinoma, several possible pathways have been proposed, divided into monoclonal or polyclonal pathways. Monoclonality consists of a transformation from one component to the other, and the polyclonal pathway is assumed to result from a collision of two tumors, each encountering the other as an independent component. 13,45 Our two major findings, the similarities of the two distinct components by genetic and immunohistochemical analysis and the major characteristics of squamous cell carcinomas, were detected even in the adenocarcinomatous components, strongly suggesting a transition from the squamous cell carcinomatous components. The transitional areas, distributed between the two distinct components in all of the tumors examined in this study, were occupied by tumor cells retaining the characteristics of both adenocarcinoma and squamous cell carcinoma. The existence of transitional areas in the intervening region between distinct components was partly suggested by morphological studies. 46,47 Hammond et al described the frequent concurrence of adenocarcinoma and squamous cell carcinoma arising separately in the small lesions in their hamster lung cancer models, but they also suggested the possibility that these cells arose from bipotential cells manifesting two separate histological patterns. 47 Such previous studies were based mainly on morphological observations and would not, therefore, address the problem of the clonality of heterogeneous cells.
In this study, we revealed the genetic similarities of the different components of each tumor. The immunoreactivity of p53 was detected in five of twelve tumors (42%) examined, and no statistically significant difference was found in the degrees of intensity or distribution of p53 expression between each component of these tumors. The same point mutation was detected in the distinct components of each tumor of the three cases with p53 overexpression, suggesting the early occurrence of the p53 mutations in these tumors. We also found a coincidence between the two components in the gene status of K-ras (100%) at codons 12, 13, and 61, and the chromosomal status except only one case at one locus. These findings strongly support the hypothesis of a monoclonal expansion of a single mutant progenitor cell clone, which expands over time to populate widespread areas of the respiratory tract.
We sought to determine the possible existence of a progenitor component in the adenosquamous carcinomas. No K-ras mutations were detected at codons 12, 13, and 61 in any of the components of the 12 tumors examined in this study, and there are also previous reports that showed no K-ras mutations in the adenosquamous carcinomas of the lung. 19,20 This lack of K-ras mutations even in the adenocarcinomatous components conflicts with the numerous reports showing frequent K-ras mutations in usual adenocarcinomas of the lung. 19-21,43 By immunohistochemical comparison, moreover, we found higher expression levels of SCC and lower expression of MUC1 in these adenocarcinomatous compartments than in usual adenocarcinomas of the lung. 30,31,33 The absence of K-ras mutations and the immunohistochemical characteristics of the adenocarcinomatous components strongly suggested that the squamous compartment was the original lesion of these adenosquamous carcinomas. The progressed abnormality found in the adenocarcinomatous component of case 8 at 9p21, D9S259, may also support our hypothesis of a transition from the squamous carcinomatous component to the adenocarcinomatous component, caused by subsequent genetic alterations.
Clinicopathological studies showing a poorer prognosis for adenosquamous carcinomas of the lung than adenocarcinoma and squamous cell carcinomas 48-50 suggest that adenosquamous carcinomas may originate from progenitor cells different from those of homogeneous tumor cells, acquiring bipotential differentiation. The most likely precursor for squamous cell carcinoma of the lung is squamous metaplasia originating from basal cell hyperplasia. 10,51 McDowell et al 52 proposed the mucous secretory cell as the major proliferating component, with proliferation of these cells leading ultimately to invasive carcinomas that can become differentiated into either squamous cell carcinoma, adenocarcinoma, or combined adenosquamous carcinoma. Considering that atypical adenomatous hyperplasia is now more widely accepted as the precursor lesion of peripheral adenocarcinomas 10,53-55 and in which K-ras mutations are detected as in adenocarcinomas, 55 it is unlikely that a transition from adenocarcinoma to squamous cell carcinoma occurs. The biological meaning of differentiation into distinct components from bipotential stem cells might be explained by the hypothesis of Mabry et al, according to which admixtures of the phenotypes of lung cancer may mimic the transitions of normal cellular differentiation in bronchial mucosa. 1
The three metastatic tumors were consisted of adenocarcinoma with a higher grade of differentiation, which is consistent with previous studies showing higher frequencies of adenocarcinoma invading metastatic lesions of adenosquamous carcinomas. 51 These results also support the notion of adenocarcinomatous components acquiring invasive potency. The evidence revealed in the present study that the metastatic lesions of two tumors without p53 overexpression acquired intense expression of p53 protein may be one example of accumulating abnormalities.
Higher immunoreactivity of CEA, CA19-9, MUC1, and VEGF, shown in some adenocarcinomatous components of these tumors, does not imply the origin of the heterogeneous tumor cells because transcriptional changes, either gain or loss, could happen during differentiation. Although the expression of PCNA was significantly higher in three squamous components and in one adenocarcinomatous component, higher expression of PCNA is not always correlated with excessive proliferation in non-small-cell lung cancer 36 because of abnormal cell-cycle regulation 56 and/or prolonged half life of PCNA. 57
In conclusion, we investigated the clonal expansion in the carcinogenic process of adenosquamous carcinoma, and the results suggest that the consistent genetic abnormalities that are retained within both components may be targeted for successful treatment.
Footnotes
Address reprint requests to Masahito Ebina, M.D., Ph.D., Department of Respiratory Oncology and Molecular Medicine, Institute of Development, Aging and Cancer, Tohoku University, 4–1, Seiryo, Aoba-ku, Sendai 980-8575, Japan. E-mail: ebinam@idac.tohoku.ac.jp.
Supported in part by a grant from Ministry of Education, Science, Sports and Culture of Japan (No. 08670647 to M. E.).
References
- 1.Mabry M, Nelkin B, Falco JP, Barr LF, Baylin SB: Transitions between lung cancer phenotypes-implications for tumor progression. Cancer Cells 1991, 3:53-58 [PubMed] [Google Scholar]
- 2.Kern SE: Clonality: more than just a tumor-progression model. J Natl Cancer Inst 1993, 85:1020-1021 [DOI] [PubMed] [Google Scholar]
- 3.Godfrey M: Molecular heterogeneity: a clinical dilemma; clinical heterogeneity: a molecular dilemma. Am J Hum Genet 1993, 53:22-25 [PMC free article] [PubMed] [Google Scholar]
- 4.Heppner GH: Tumor heterogeneity. Cancer Res 1984, 44:2259-2265 [PubMed] [Google Scholar]
- 5.Williams GT, Wynford-Thomas D: How many clonality be assessed in human tumours? Histopathology 1994, 24:287-292 [DOI] [PubMed] [Google Scholar]
- 6.Ashley DJ, Davies HD: Cancer of the lung: histology and biological behavior. Cancer 1967, 20:165-174 [DOI] [PubMed] [Google Scholar]
- 7.Roggli VL, Vollmer RT, Greenberg SD, McGavran MH, Spjut HJ, Yesner R: Lung cancer heterogeneity: a blinded and randomized study of 100 consecutive cases. Hum Pathol 1985, 16:569-579 [DOI] [PubMed] [Google Scholar]
- 8.Dunnill MS, Gatter KC: Cellular heterogeneity in lung cancer. Histopathology 1986, 10:461-475 [DOI] [PubMed] [Google Scholar]
- 9.Coons SW, Johnson PC, Shapiro JR: Cytogenetic and flow cytometry DNA analysis of regional heterogeneity in a low grade human glioma. Cancer Res 1995, 55:1569-1577 [PubMed] [Google Scholar]
- 10.Lee SJ, Mao L, Hong WK: Biology of preneoplastic lesions. 2nd ed. Roth JA Cox JD Hong WK eds. Lung Cancer, 1998, :pp 25-55 Blackwell Science Inc, Malden, MA, [Google Scholar]
- 11.Naunheim KS, Taylor JR, Skosey C, Hoffman PC, Ferguson MK: Adenosquamous lung carcinoma: clinical characteristics, treatment, and prognosis. Ann Thorac Surg 1987, 44:462-466 [DOI] [PubMed] [Google Scholar]
- 12.Stridhar K, Raub W, Duncan R, Hilsenbeck S: The increasing recognition of adenosquamous lung carcinoma. Am J Clin Oncol 1992, 15:356-362 [DOI] [PubMed] [Google Scholar]
- 13.Ishida T, Kaneko S, Yokoyama H, Inoue T, Sugio K, Sugimachi K: Adenosquamous carcinoma of the lung. Clinicopathologic and immnohistochemical features. Am J Clin Pathol 1992, 97:678-685 [DOI] [PubMed] [Google Scholar]
- 14.Colby TV, Koss MN, Travis WD: Tumors of the lower respiratory tract. Atlas of Tumor Pathology, Third series, Fascicle 13. 1995:pp 279-286 Armed Forces Institute of Pathology, Washington, DC,
- 15.Travis WD, Colby TV, Corrin B, Shimosato Y, Brambilla E: Histological Typing of Lung and Pleural Tumors, 3rd ed. 1999, Springer-Verlag, Berlin
- 16.Hollstein M, Sidransky D, Vogelstein B, Harris C: p53 mutations in human cancer. Science 1991, 253:49-53 [DOI] [PubMed] [Google Scholar]
- 17.Sidransky D, Mikkelsen T, Schwechheimer K, Rosenblum ML, Cavanee W, Vogelstein B: Clonal expansion of p53 mutant cells is associated with brain tumour progression. Nature 1992, 355:846-847 [DOI] [PubMed] [Google Scholar]
- 18.Holst VA, Finkelstein S, Colby TV, Myers JL, Yousem S: P53, and K-ras mutational genotyping in pulmonary carcinoma, spindle cell carcinoma, and pulmonary blastoma: implications for histogenesis. Am J Surg Pathol 1997, 21:801-811 [DOI] [PubMed] [Google Scholar]
- 19.Rodenhuis S, Slebos RJC, Boot AJM, Everts SG, Mooi WJ, Wagenaar SS, van Bodegom PC, Bos JL: Incidence and possible clinical significance of K-ras oncogene activation in adenocarcinoma of the human lung. Cancer Res 1998, 48:5738-5741 [PubMed] [Google Scholar]
- 20.Suzuki Y, Orita M, Shiraishi M, Hayashi K, Sekiya T: Detection of ras gene mutations in human lung cancers by single-strand conformation polymorphism analysis of polymerase chain reaction products. Oncogene 1990, 5:1037-1043 [PubMed] [Google Scholar]
- 21.Mills NE, Fishman CL, Rom WN, Dubin N, Jacobson DR: Increased prevalence of K-ras oncogene mutaions in lung adenocarcinoma. Cancer Res 1995, 55:1444-1447 [PubMed] [Google Scholar]
- 22.Wiest JS, Franklin WA, Otstot JT, Forbey K, Varella-Garcia M, Rao K, Drabkin H, Gemmill R, Ahrent S, Sidransky D, Saccomanno G, Fountain JW, Anderson MW: Identification of a novel region of homozygous deletion on chromosome 9p in squamous cell carcinoma of the lung: the location of a putative tumor suppressor gene. Cancer Res 1997, 57:1-6 [PubMed] [Google Scholar]
- 23.Lukeis R, Irving L, Garson M, Hasthorpe S: Cytogenetics of non-small cell lung cancer: analysis of consistent non-random abnormalities. Genes Chromosomes Cancer 1990, 2:116-124 [DOI] [PubMed] [Google Scholar]
- 24.Miura K, Suzuki K, Tokino T, Isomura M, Inazawa J, Matsuno S, Nakamura Y: Detailed deletion mapping in squamous cell carcinomas of the esophagus narrows a region containing a putative tumor suppressor gene to about 200 kilobases on distal chromosome 9q. Cancer Res 1996, 56:1629-1634 [PubMed] [Google Scholar]
- 25.Travis WD, Linnoila RI, Tsokos MG, Hitchcock CL, Cutler GB, Jr, Nieman L, Chrousos G, Pass H, Doppman J: Neuroendocrine tumors of the lung with proposed criteria for large-cell neuroendocrine carcinoma. An ultrastructural, immunohistochemical, and flow cytometric study of 35 cases. Am J Surg Pathol 1991, 15:529-553 [DOI] [PubMed] [Google Scholar]
- 26.Boilleau G, Pujol JL, Ychou M, Faurous P, Marty-Ane C, Michel FB, Godard P: Detection of lymph node metastases in lung cancer: comparison of 131I-anti-CEA-anti-CA 19-9 immunoscintigraphy versus computed tomography. Lung Cancer 1994, 11:209-219 [DOI] [PubMed] [Google Scholar]
- 27.Ohshio G, Yamaki K, Imamura T, Suwa H, Chang CY, Wada H, Sueno Y, Imamura M: Distribution of the carbohydrate antigens, DU-PAN-2 and CA19-9, in tumors of the lung. Tumori 1995, 81:67-73 [DOI] [PubMed] [Google Scholar]
- 28.Narimatsu H, Iwasaki H, Nishihara S, Kimura H, Kudo T, Yamauchi Y, Hirohashi S: Genetic evidence for the Lewis enzyme, which synthesizes type-1 Lewis antigens in colon tissue, and intracellular localization of the enzyme. Cancer Res 1996, 56:330-338 [PubMed] [Google Scholar]
- 29.Ban T, Imai K, Yachi A: Immunohistological and immunohistochemical characterization of a nobel pancreatic cancer-associated antigen MUSE11. Cancer Res 1989, 49:7141-7146 [PubMed] [Google Scholar]
- 30.Jarrard JA, Linnoila RI, Lee H, Steinberg SM, Witschi H, Szabo E: MUC1 is a novel marker for the type II pneumocyte lineage during lung carcinogenesis. Cancer Res 1998, 58:5582-5589 [PubMed] [Google Scholar]
- 31.Kato H, Torigoe T: Radioimmunoassay for tumor antigen of human cervical squamous cell carcinoma. Cancer 1977, 40:1621-1628 [DOI] [PubMed] [Google Scholar]
- 32.Kato H, Tamai K, Morioka H, Nagai M, Nagaya T, Torigoe T: Tumor-antigen TA-4 in the detection of recurrence in cervical squamous cell carcinoma. Cancer 1984, 54:1544-1546 [DOI] [PubMed] [Google Scholar]
- 33.Mino N, Iio A, Hamamoto K: Availability of tumor-antigen 4 as a marker of squamous cell carcinoma of the lung and other organs. Cancer 1988, 62:730-734 [DOI] [PubMed] [Google Scholar]
- 34.Mattern J, Koomagi R, Volm M: Association of vascular endothelial growth factor expression with intratumoral microvessel density and tumour cell proliferation in human epidermoid lung carcinoma. Br J Cancer 1996, 73:931-934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shoji M, Hancock WW, Abe K, Micko C, Casper KA, Baine RM, Wilcox JN, Danave I, Dillehay DL, Matthews E, Contrino J, Morrissey JH, Gordon S, Edgington TS, Kudryk B, Kreutzer DL, Rickles FR: Activation of coagulation and angiogenesis in cancer: immunohistochemical localization in situ of clotting proteins and vascular endothelial growth factor in human cancer. Am J Pathol 1998, 152:399-411 [PMC free article] [PubMed] [Google Scholar]
- 36.Ebina M, Steiberg SM, Mulshine JL, Linnoila RI: Relationship of p53 overexpression and up-regulation of proliferating cell nuclear antigen with the clinical course of non-small cell lung cancer. Cancer Res 1994, 54:2496-2503 [PubMed] [Google Scholar]
- 37.Fontanini G, Macchiarini P, Pepe S, Ruggiero A, Hardin M, Bigini D, Vignati S, Pingitore R, Angeletti CA: The expression of proliferating nuclear cell antigen in paraffin sections of peripheral, node-negative non-small cell lung cancer. Cancer 1992, 70:1520-1527 [DOI] [PubMed] [Google Scholar]
- 38.El-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW, Vogelstein B: WAF1, a potential mediator of p53 tumor suppression. Cell 1993, 75:817-825 [DOI] [PubMed] [Google Scholar]
- 39.Hayashi H, Miyamoto H, Ito T, Kameda Y, Nakamura N, Kubota Y, Kitamura H: Analysis of p21Waf1/Cip1 expression in normal, premalignant, and malignant cells during the development of human lung adenocarcinoma. Am J Pathol 1997, 151:461-470 [PMC free article] [PubMed] [Google Scholar]
- 40.Teneriello MG, Ebina M, Linnoila RI, Henry M, Nash JD, Park RC, Birrer MJ: p53 and Ki-ras gene mutations in epithelial ovarian neoplasms. Cancer Res 1993, 53:3103-3108 [PubMed] [Google Scholar]
- 41.Hall KL, Teneriello MG, Taylor RR, Lemon S, Ebina M, Linnoila RI, Norris JH, Park RC, Birrer MJ: Analysis of Ki-ras, p53, and MDM2 genes in uterine leiomyomas and leiomyosarcomas. Gynecol Oncol 1997, 65:330-335 [DOI] [PubMed] [Google Scholar]
- 42.Mitsudomi T, Viallet J, Mulshine JL, Linnoila RI, Minna JD, Gazdar AF: Mutations of ras genes distinguish a subset of non-small-cell lung cancer cell lines from small-cell lung cancer cell lines. Oncogene 1991, 6:1353-1362 [PubMed] [Google Scholar]
- 43.Lengauer C, Kinzler KW, Vogelstein B: Genetics instabilities in human cancers. Nature 1998, 396:643-649 [DOI] [PubMed] [Google Scholar]
- 44.Nowell PC: The clonal evolution of tumor cell populations. Science 1976, 194:23-28 [DOI] [PubMed] [Google Scholar]
- 45.Takamori S, Noguchi M, Morinaga S, Goya T, Tsugane S: Clinicopathologic characteristics of adenosquamous carcinoma of the lung. Cancer 1991, 67:649-654 [DOI] [PubMed] [Google Scholar]
- 46.Myer R: Beitrag zur Verstamdigung uber die Namengebung in der Geschwulstlehre. Zentralbl Allg Pathol 1919, 30:291-296 [Google Scholar]
- 47.Hammond WG, Tesluk H, Benfield JR: Histogenesis of adenosquamous bronchogenic carcinoma. Cancer Lett 1995, 96:163-168 [DOI] [PubMed] [Google Scholar]
- 48.Hofmann H-S, Knolle J, Neef H: The adenosquamous lung carcinoma: clinical and pathological characteristics. J Cardiovasc Surg 1994, 35:543-547 [PubMed] [Google Scholar]
- 49.Shimizu J, Oda M, Hayashi Y, Nonomura A, Watanabe Y: A clinicopathologic study of resected cases of adenosquamous carcinoma of the lung. Chest 1996, 109:989-994 [DOI] [PubMed] [Google Scholar]
- 50.Kamiyoshihara M, Hirai T, Kawashima O, Ishikawa S, Morishita Y, Maeshima A: A clinicopathologic study of the resected cases of adenosquamous carcinoma of the lung. Oncol Rep 1998, 5:861-865 [DOI] [PubMed] [Google Scholar]
- 51.Melamed MR, Zaman MB: Pathogenesis of epidermoid carcinoma of lung. Shimosato Y Melamed MR Nettesheim P eds. Pathogenesis of Lung Cancer. 1982, :pp 7-64 FL, CRC Press, Boca Raton [Google Scholar]
- 52.McDowell EM: Bronchogenic carcinomas. McDowell EM eds. Lung Carcinomas. 1987, :pp 255-258 Churchill Livingstone, New York [Google Scholar]
- 53.Kerr KM, Carey FA, King G, Lamb D: Atypical alveolar hyperplasia: relationship with pulmonary adenocarcinoma, p53, and c-erbB-2 expression. J Pathol 1994, 174:249-256 [DOI] [PubMed] [Google Scholar]
- 54.Mori M, Tezuka F, Chiba R, Funae Y, Watanabe M, Nukiwa T, Takahashi T: Atypical adenomatous hyperplasia and adenocarcinoma of the human lung: their heterology in form and analogy in immunohistochemical characteristics. Cancer 1996, 77:665-674 [PubMed] [Google Scholar]
- 55.Westra WH, Baas IO, Hruban RH, Askin FB, Wilson K, Offerhaus GJ, Slebos RJ: K-ras oncogene activation in atypical alveolar hyperplasias of the human lung. Cancer Res 1996, 56:2224-2228 [PubMed] [Google Scholar]
- 56.Celis JE, Celis A: Cell cycle-dependent variation in the distribution of the nuclear protein cycline proliferating cell nuclear antigen in cultured cells: subdivision of S phase. Proc Natl Acad Sci USA 1985, 82:3262-3266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.van Dierendonck JH, Wijsman JH, Keijzer R, van de Velde CJ, Cornelisse CJ: Cell-cycle-related staining patterns of anti-proliferating cell nuclear antigen monoclonal antibodies. Comparison with BrdU labeling and Ki-67. Am J Pathol 1991, 138:1165–1172 [PMC free article] [PubMed]