

Abstract
Vascular endothelial growth factor (VEGF or vascular permeability factor) is an important angiogenic factor that is up-regulated in numerous benign and malignant disorders, including angiosarcoma, hemangiomas, and solid tumors. To determine the functional role of VEGF in the development of endothelial tumors, we expressed primate VEGF 121 in an endothelial cell line, MS1, derived from primary murine cells by immortalization with a temperature-sensitive SV40 large T antigen. This cell line expresses the VEGFR-2 (Flk-1/Kdr) receptor for VEGF. Expression of VEGF 121 led to the development of slowly growing endothelial tumors, which were histologically well-differentiated angiosarcomas. The angiosarcomas generated from MS1 VEGF cells demonstrated up-regulation of the VEGF receptors VEGFR-2 and VEGFR-1 (Flt-1) in vivo compared with benign hemangiomas generated from MS1 cells. Treatment of these cells with the VEGFR-2 tyrosine kinase inhibitor SU 1498 led to decreased expression of ets-1, a transcription factor which has been shown to be stimulated by VEGF. These results suggest that high level expression of VEGF in endothelial cells may result in malignant transformation. This transformation process likely involves both autocrine and paracrine pathways.
Endothelial tumors constitute a group of tumors responsible for significant morbidity and mortality, but the pathogenesis of these tumors is not well understood. 1 Hemangiomas constitute the most common tumor of childhood and, though benign, may require systemic therapy to alleviate complications, such as compression of vital structures, coagulopathy (Kasabach-Merritt syndrome), and high output heart failure. 2-4 Endothelial malignancies are difficult to treat surgically, because of multifocal growth and potential for distant spread, the latter often arising after successful resection of a primary tumor. 5,6 Thus, increased understanding of the pathogenesis of angiosarcoma is required and may lead to improved therapy.
In humans, angiosarcoma may arise as a result of exposures to various toxins. The most commonly implicated causative agents are vinyl chloride and thorotrast, both of which are associated with hepatic angiosarcomas. 7,8 These angiosarcomas, and experimental angiosarcomas arising in rodents from exposure to vinyl chloride, have been shown to have activated Ras mutations in a number of cases. 8-10 Radiation of hemangiomas has been shown to result in the later development of angiosarcoma. 11 Finally, lymphedema from a variety of causes, including postsurgical (Stewart-Treves syndrome), congenital, and even post-traumatic lymphedema is associated with the development of angiosarcoma. 12-14
Animal models have proven useful in determining molecular events necessary for the development of angiosarcoma. We have previously generated a model of rapidly growing angiosarcoma through the sequential introduction of a temperature-sensitive SV40 large T antigen and activated H-Ras into murine endothelial cells. 15 Upon injection into nude mice, endothelial cells containing SV40 large T antigen alone, MS1 cells, develop dormant hemangiomas that do not grow in size. Endothelial cells (SVR) with two oncogenes, SV40 large T antigen and H-ras, form rapidly growing angiosarcomas that cause death of mice. 15 To evaluate whether overexpression of VEGF in endothelial cells that expressed the VEGF receptors VEGFR-1 and VEGFR-2 would lead to malignant transformation, we overexpressed primate VEGF using a retrovirus in MS1 cells. The resulting cells, MS1 VEGF cells, form slowly growing angiosarcomas upon injection into mice. These tumors display invasiveness and continued growth in vivo. These MS1 VEGF angiosarcomas, like the angiosarcomas that express large T antigen and Ras, show high level expression of VEGFR-1 and VEGFR-2 in vivo. Our studies suggest that expression of VEGF in cells that express receptors for VEGF is a transforming event in vivo.
Materials and Methods
Generation of Cell Lines
MS1 murine endothelial cells 15 (no. 2279, American Type Culture Collection, Manassas, VA) were infected with a retrovirus encoding primate VEGF and puromycin resistance, or puromycin resistance alone, and selected in 1 μg/ml puromycin. MS1 cells are murine endothelial cells immortalized with a temperature-sensitive SV40 large T antigen, and SVR cells were derived by transforming MS1 cells with oncogenic H-ras. 15 Clones were pooled, and supernatants were assayed for the presence for human VEGF using an enzyme-linked immunosorbent assay specific for VEGF (R&D Systems, Minneapolis, MN). The cells containing VEGF or puromycin (Sigma Chemical Co., St. Louis, MO) resistance alone were named MS1 VEGF and MS1puro, respectively.
Receptor Cross-Linking Studies
MS1 and SVR endothelial cells were grown to confluence in 5-cm plates. Medium was aspirated, plates were washed with 10 ml cold phosphate-buffered saline (PBS) and 1.2 ml binding buffer (DMEM 20 mmol/L, HEPES, pH 7.4, 0.1% gelatin), and radio-iodinated VEGF (10 ng/ml) was added according to the method of Soker et al. 16 Cells were shaken for 2 hours at 4°C. Plates were washed twice with 8 ml cold PBS, and 20 μl of 20 mmol/L disuccinimidyl suberate in dimethylsulfoxide (DMSO) were added and the plates incubated 15 minutes at room temperature. Two hundred microliters of stop buffer (10 mmol/L Tris-HCl, pH 7.5, 20 mmol/L glycine, 2 mmol/L EDTA) were added, and the plates were incubated for 1 hour, then washed twice with 8 ml PBS.
The cells on the plate were lysed by the addition of 700 μl scrape buffer consisting of 20 ml PBS, 1.5 mmol/L EDTA, and 2 mmol/L phenylmethylsulfonyl fluoride (PMSF) twice, followed by scraping with a rubber policeman. The lysed cells were spun at the maximum speed of a microfuge for 10 minutes. The supernatant was removed, and the pellet was suspended in 40 μl lysis buffer (10 mmol/L Tris-HCl, pH 7.0, 1% Nonidet P-40, 1 mmol/L EDTA, 2 mmol/L PMSF). An equal volume of sample buffer (0.125 mol/L Tris-HCl, 4% sodium dodecyl sulfate (SDS), 20% glycerol, 10% β-mercaptoethanol/bromphenol blue) was added, and the samples were boiled for 3 minutes. The samples were loaded on a 6% acrylamide gel and assayed by autoradiography.
In Vivo Tumorigenesis
One million cells were injected subcutaneously into the right flank of nude male mice (Massachusetts General Hospital, Boston, MA) 5 to 6 weeks old. Three mice were injected per cell line. Tumor size was measured by Vernier caliper, and tumor volume was calculated by the formula (w2 × l) × 0.52, where w (width) represents the smallest diameter of the tumor. 15 Tumors were excised at 5 months, fixed in formalin, and stained with hematoxylin and eosin.
In Situ Hybridization (ISH)
ISH was performed on 4-mm-thick sections of formalin-fixed, paraffin-embedded tissue. Details of ISH have been reported previously. 17-19 Briefly, slides were passaged through xylene and graded alcohols; 0.2 mol/L HCl; Tris/EDTA with 3 μg/ml proteinase K/0.2% glycine, 4% paraformaldehyde in PBS, pH 7.4; 0.1 mol/L triethanolamine containing 1/200 (v/v) acetic anhydride; and 2× SSC. Slides were hybridized overnight at 50°C with 35S-labeled riboprobes in the following mixture: 0.3 mol/L NaCl, 0.01 mol/L Tris, pH 7.6, 5 mmol/L EDTA, 0.02% w/v Ficoll, 0.02% w/v polyvinylpyrollidone, 0.02% w/v bovine serum albumin fraction V/50% formamide, 10% dextran sulfate, 0.1 mg/ml yeast tRNA, and 0.01 mol/L dithiothreitol (DTT). Post-hybridization washes included 2× standard saline citrate (SSC)/50% formamide and 10 mmol/L dithiothreitol at 50°C, 4× SSC, 10 mmol/L Tris, and 1 mmol/L EDTA with 20 μg/ml ribonuclease at 37°C; and 2× SSC, 50% formamide, 10 mmol/L EDTA at 65°C and 2× SSC. Slides were then dehydrated through graded alcohols containing 0.3 mol/L ammonium acetate, dried, coated with Kodak NTB 2 emulsion, and stored in the dark at 4°C for 2 weeks. The emulsion was developed with Kodak D19 developer and the slides were counterstained with hematoxylin. Antisense single-stranded 35S-labeled human VPF/VEGF RNA probe and its sense control have been described previously. 18 The antisense probe hybridizes specifically with a region of VPF/VEGF RNA mRNA common to all known VPF/VEGF splice variants. 35S-labeled single-stranded antisense and sense RNA probes for mouse VPF/VEGF mRNA and the mouse VPF/VEGF receptors VEGFR-1 and VEGFR-2 mRNAs have been described previously. 19
In Vitro Kinase Assay
In vitro kinase assays were performed according to the method of Waltenberger et al. 20 Subconfluent cell cultures were starved overnight in DMEM (Life Technologies, Inc.) containing 0.1% fetal calf serum (Sigma, St. Louis, MO) and then stimulated or not with VEGF 165 (100 ng/ml; Peprotech, Rocky Hill, NJ) for 7 minutes at 37°C. The cells were rinsed with ice-cold PBS containing 100 μmol/L Na3VO4 and lysed for 10 minutes on ice in NP-40 lysis buffer (20 mmol/L Hepes, pH 7.5, 150 mmol/L NaCl, 1% NP-40, 10% glycerol, 300 μmol/L Na3VO4, 1% aprotinin, 1 mmol/L PMSF).
Lysates were clarified at 10,000 × g for 15 minutes at 4°C, and the supernatants were incubated with antibodies against either VEGFR-2 (RS-2) or phosphotyrosine (4G10, Upstate Biotechnology, Waltham, MA) for 1 hour at 4°C, followed by a final incubation for 45 minutes with immobilized protein A (Immunosorb; EC Diagnostics, Uppsala, Sweden). The precipitates were washed three times in NP-40 lysis buffer and twice in PBS containing 100 μmol/L Na3VO4, then incubated for 15 minutes at room temperature in 40 μl kinase buffer (20 mmol/L Hepes, pH 7.5, 10 mmol/L MgCl2, 2 mmol/L MnCl2, 0.05% Triton X-100, 1 mmol/L DTT) containing 5 μCi [γ-32P]ATP (Amersham). The kinase reactions were terminated by addition of 40 μl 2× sample buffer (8% SDS, 0.4 mol/L Tris/HCl, pH 8.8, 1 mol/L sucrose, 10 mmol/L EDTA, 0.02% bromphenol blue, 4% β-mercaptoethanol) and the samples were boiled at 95°C for 4 minutes and analyzed by SDS-PAGE in a 10% SDS-polyacrylamide gel. After fixation in methanol/acetic acid, the gel was treated with 1 mol/L KOH for 1 hour at 55°C, fixed again, dried, and analyzed by autoradiography.
Metabolic Labeling
Semiconfluent MS1 and MS1 VEGF cultures were washed with methionine- and cysteine-deficient DMEM (Life Technologies) and subsequently incubated for 2 hours at 37°C in the same medium, complemented with 100 μCi/ml [35S]methionine/cysteine mix (Amersham Pharmacia Biotech, Uppsala, Sweden). The cultures were rinsed once with ice-cold PBS to terminate the labeling and then lysed in 0.5% NP 40/20 mmol/L Tris-HCl, pH 7.5, 150 mmol/L NaCl, 10 mmol/L EDTA, 1% aprotinin, and 0.2 mmol/L PMSF. Cell lysates were immunoprecipitated with a rabbit antiserum raised against a 25-amino acid residue peptide corresponding to a part of the kinase insert in VEGFR-2, 20 followed by incubation with immobilized protein A. Control lysates were incubated with protein A-Sepharose alone. Samples were analyzed by SDS-PAGE. The gel was treated with Amplify (Amersham Pharmacia Biotech), dried, and analyzed using a BioImager (Fuji, Tokyo, Japan).
Western Blot Analysis of c-Ets-1 Expression
MS1 VEGF cells were treated with 5 μg/ml SU-1498 (Calbiochem, La Jolla, CA) in DMSO or with DMSO alone for 24 hours. Cells were lysed in lysis buffer containing 50 mmol/L PBS, pH 7.2, 5 mmol/L EDTA, 5% glycerol, and 0.3 mmol/L PMSF. The protein concentration was determined by Protein Assay (Bio-Rad, Hercules, CA) using bovine serum albumin as a standard. Samples were mixed with Laemmli sample buffer and heated at 95°C for 5 minutes before SDS-PAGE (Bio-Rad MiniVertical Electrophoresis standard protocol) and transferred to PVDF membrane (Bio-Rad). The membrane were blocked with blocking buffer (5% dry milk, 10 mmol/L Tris-HCl, pH 7.5, 100 mmol/L NaCl, 0.1% Tween-20) and subsequently incubated with the appropriate antibodies for immunoblotting. Anti-Ets-1 polyclonal antibody 21 0.2 μg/ml and peroxidase 0.4 μg/ml conjugated anti-Rabbit lgG were from Santa Cruz Biotechnology (Santa Cruz, CA). 21
Assay of Anchorage-Independent Growth
Anchorage-independent growth was determined by adding equal numbers of viable cells to a plate containing 0.35% Noble agar in 5% fetal bovine serum-containing media at a density of 5 × 10 5 cells per 6-cm 2 plate. The number of colonies formed in agar was recorded 2 weeks after plating the cells.
Results
MS1 murine endothelial cells were generated by expression of a temperature-sensitive SV40 large T antigen in primary murine microvascular endothelial cells. SVR cells were generated by the introduction of oncogenic H-Ras into MS1 cells. 15 MS1 and SVR cells were found to bind radio-iodinated VEGF 165, yielding a band consistent with the combined molecular weight of VEGF and VEGFR-2 after cross-linking (data not shown). The presence of VEGFR-2 was confirmed by Northern blot analysis of MS1 and SVR cells (Figure 1) ▶ . VEGFR-2 levels were not altered by various stimuli in vitro, including Ras activation and hypoxia.
Figure 1.

Northern blot analysis of VEGFR-2 expression in MS1 and SVR cells. Expression of flk-1 is present, but is not altered by ras transformation, phorbol ester, hypoxia, or free radicals. Lanes 1 under both MS1 and SVR represent cells exposed to free radicals, lanes 2 represent cells after exposure to phorbol ester, lanes 3 represent cells exposed to hypoxia (3.5% oxygen), and lane 4 represents normoxia controls.
MS1 cells were infected with a retrovirus encoding primate VEGF 121 and selected with puromycin. As a vector control, MS1 cells were also infected with retrovirus encoding the plasmid pBABEpuro. After selection in puromycin, supernatant from each group was assayed for the presence of VEGF using an enzyme-linked immunosorbent assay kit that detects human VEGF. Supernatants from MS1 VEGF cells showed high levels of VEGF expression (approximately 1000 pg/ml), compared with undetectable levels of VEGF in MS1puro cells. Expression of primate VEGF was confirmed by Northern blot analysis (Figure 2) ▶ . The human probe used in the Northern blot analysis does not cross-hybridize with endogenous murine VEGF.
Figure 2.

Northern blot analysis of VEGF expression in MS1puro and MS1VEGF cells using a human VEGF probe. This probe shows no hybridization with endogenous mouse VEGF
One million cells of each type were injected subcutaneously into the flank of nude mice. MS1 cells, which do not overexpress VEGF, formed benign hemangiomas, which did not grow beyond 3 to 4 mm in diameter throughout the observation period of up to 5 months. In contrast, MS1 VEGF cells formed slowly growing tumors which reached 1 cm in diameter at 5 months (Figure 3, A and B) ▶ . Tumors derived from MS1 and MS1puro cells resembled hemangiomas. They consisted of well-circumscribed tumor masses within the subcutaneous tissue. Fibrous septa divided the tumor masses into coarse nodules similar to the lobular pattern seen in many capillary hemangiomas. Each nodule consisted of well-formed, discrete capillary vessels lined by differentiated endothelium. The vessels did not intercommunicate nor dissect through the subcutaneous tissues as one sees in angiosarcoma (Figure 4A) ▶ . On the other hand, those tumors derived from MS1 VEGF cells formed large masses occupying both the dermis and subcutaneous tissue with poorly circumscribed borders. The proliferating endothelial cells formed a ramifying sinusoidal network of vessels, which dissected through collagen and around adnexal structures (Figure 4B) ▶ . Despite this aggressive pattern of growth the tumors consisted entirely of areas in which vasoformative features were identified and lacked solid or undifferentiated areas.
Figure 3.
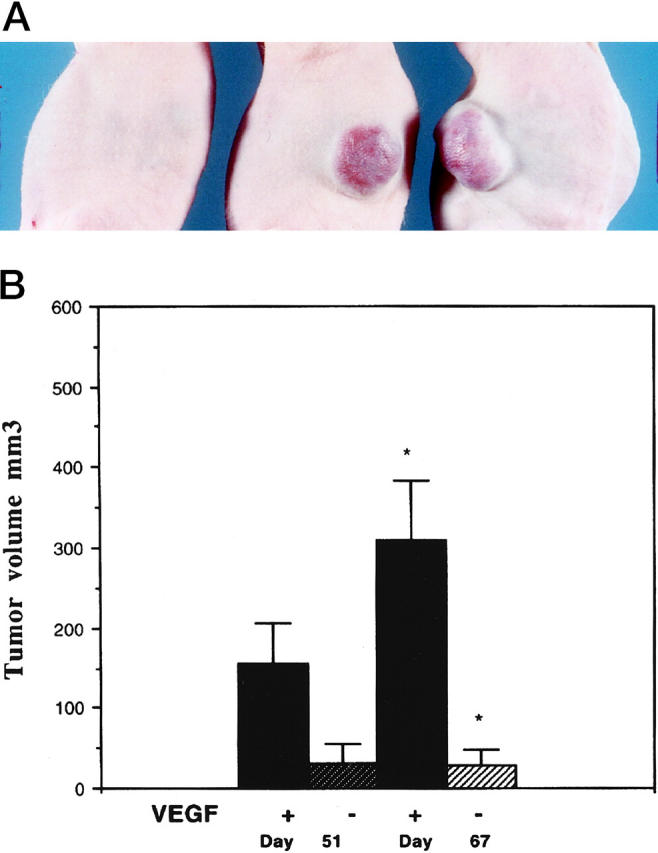
A: Gross appearance of tumors overexpressing VEGF 121. The mouse on the left was injected with MS1puro cells (MS1 cells with vector control), and the two mice on the right were injected with MS1 VEGF cells. This picture was taken 5 months after inoculation of cells. B: Growth of tumors in the presence of absence of VEGF overexpression. The differences in tumor volume between VEGF overexpressing and control cells is significant at P < 0.05. Four mice were injected with each cell line.
Figure 4.
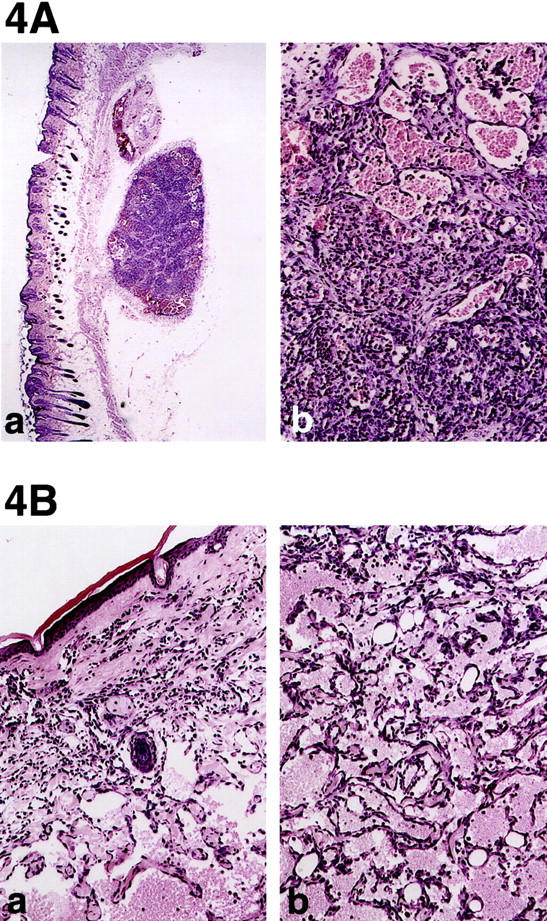
A. a: Hemangioma induced by MS1 cells which do not overexpress VEGF. Parental MS1 tumor cells are histologically identical to MS1puro cells. Tumor consists of two well circumscribed masses in the subcutaneous tissue (original magnification, ×40). In the larger mass the tumor is subdivided into lobules by fibrous bands similar the pattern seen in capillary hemangiomas in humans. b: Higher power of one tumor lobule shows mixture of small vessels with tiny lumina (left) to larger vessels with distended lumina filled with erythrocytes (original magnification, ×200). Note that the vascular channels do not intercommunicate with one another as they do in angiosarcoma. B: Angiosarcoma induced by MS1 VEGF cells. a: Irregular endothelial lined channels dissect through the dermal collagen isolating individual adnexal structures (center, original magnification, ×40). b: Higher power showing intercommunicating vascular channels dissecting around fat cells of subcutaneous tissue (original magnification, ×200). Solid or undifferentiated areas were not observed, however.
In situ hybridization was performed on tissue sections of MS1puro, MS1 VEGF, and SVR cells. High level of expression was seen using a human VEGF probe in MS1 VEGF tumors. Tumors derived from MS1 VEGF and SVR cells demonstrated high level of expression of VEGFR-1 and VEGFR-2 message, whereas MS1 hemangiomas, which do not overexpress VEGF tumors, showed little hybridization of either receptor (Figure 5A) ▶ .
Figure 5.
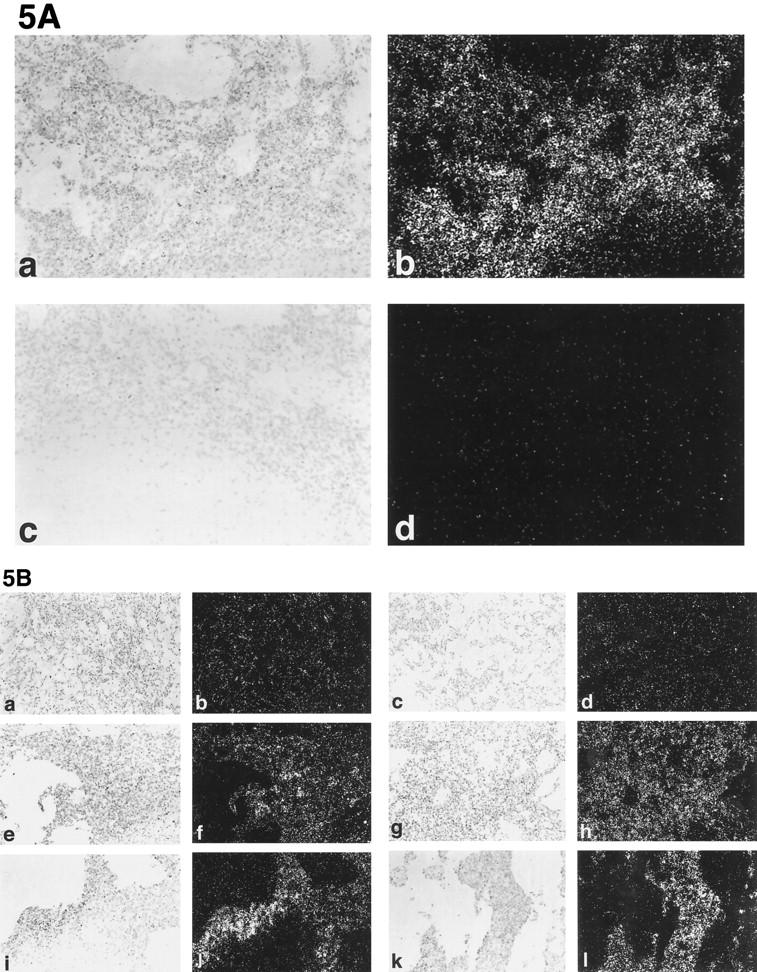
In situ hybridization of MS1, MS1 VEGF, and SVR tumors. A: Expression of VEGF is maintained in vivo. Frames a and b represent antisense human VEGF probe, whereas frames c and d represent sense VEGF probe control. Frames a and c are brightfield; frames b and d are darkfield. B: Expression of VEGFR-1 and VEGFR-2 in endothelial tumors. Frames a−d represent MS1 tumors, frames e−h represent MS1 VEGF tumors, and frames i−l represent SVR tumors. The leftmost column represents brightfield images of VEGFR-1 hybridization, the second column from the left represents darkfield images of VEGFR-1 hybridization, the second column from the right represents brightfield images of VEGFR-2 hybridization, and the rightmost column represents darkfield images of VEGFR-2 hybridization. Original magnifications, ×200.
To show that the VEGF 121 secreted from MS1 VEGF cells is biologically active, we performed in vitro kinase assays on intact cells. These assays examined phosphorylation of VEGFR-2 on intact cells in response to exogenously added VEGF. Control MS1 cells demonstrated appropriate phosphorylation of VEGFR-2 in response to exogenous VEGF. In contrast, no phosphorylation was seen in MS1 VEGF cells. These results are consistent with blockade of receptor by endogenously produced VEGF and with increased receptor turnover due to chronic VEGF stimulation of VEGFR-2 (Figure 6A) ▶ .
Figure 6.

A: MS1 cells and MS1 cells overexpressing VEGF were analyzed for induction of kinase activity using an in vitro kinase assay. Lanes 1–4 show the presence of the VEGFR-2 in both MS1 cells (lanes 1 and 2), and in MS1 VEGF cells which overexpress VEGF 121 (lanes 3 and 4). Cells were treated (+) or left untreated (−) withVEGF (100 ng/ml) for 7 minutes at 37°C, lysed, and immunoprecipitated with antibodies against VEGFR-2 (RS-2) or phosphotyrosine (4G10), and subjected to an in vitro kinase assay. Samples were analyzed by SDS-PAGE and autoradiography. VEGFR-2 is marked in the figure with an arrow. Demonstration of phsophorylation of VEGFR-2 is seen in lane 6. B: MS1 and MS1 VEGF cells express VEGFR-2. MS1 and MS1 VEGF cells were metabolically labeled with [35S]methionine/cysteine, lysed and immunoprecipitated with a rabbit antiserum to VEGFR-2 (+), followed by incubation with protein A Sepharose. Parallel samples were incubated with protein A Sepharose alone (−). Migration positions of marker proteins are indicated to the left; migration positions of VEGFR-2 intracellular precursor and cell surface mature forms are indicated to the right.
To confirm that MS1 VEGF cells express VEGFR-2, MS1 and MS1 VEGF cells were metabolically labeled with [35S]methionine/cysteine, lysed, and immunoprecipitated using an anti-VEGFR-2 antiserum. As seen in Figure 6B ▶ , the VEGFR-2 precursor and mature forms of 200 and 220 kd, respectively, 22 were immunoprecipitated both from MS1 and MS1 VEGF cells. It is noteworthy that the mature form was less pronounced in the MS1 VEGF cells than in the MS1 cells, in accordance with a rapid turnover of the receptor due to binding of the endogenously produced VEGF121 in the MS1 VEGF cells.
A downstream target of VEGF-VEGFR interactions is the transcription factor Ets-1. 21 To demonstrate the possibility of an autocrine loop between exogenously produced VEGF and VEGFR-2, we incubated MS1 VEGF cells with SU1498, a specific inhibitor of VEGFR-2 receptor phosphorylation. This blocks signal transduction from VEGFR-2. In the presence of SU1498, levels of Ets-1 were decreased, suggesting that VEGF-VEGFR-2 interactions contributed to baseline levels of Ets-1 expression, and interruption of this autocrine interaction with SU1498 led to decreased expression of Ets-1 (Figure 7) ▶ .
Figure 7.

Western blot analysis of c-Ets-1 expression in the presence or absence of SU-1498. The left lane represents c-Ets-1 expression in the presence of vehicle alone. The right lane represents expression in the presence of SU-1498. Representative lanes are shown. Experiments were performed in triplicate.
Discussion
Vascular endothelial growth factor (VEGF) is a potent angiogenesis factor 23-27 that can be induced by hypoxia, 28 glucose deprivation, 29 dominant oncogenes, and inflammatory stimuli. 15,25-27,29-32 VEGF is a potent endothelial chemoattractant, but has also been shown to promote endothelial proliferation and resistance to apoptosis. 33,34 VEGF has two major receptors, VEGFR-1 and VEGFR-2. 35,36 Of these receptors, VEGFR-2 appears to play a major role in VEGF-induced mitogenesis and chemoattraction. 37 Therapeutically, VEGF has been shown to augment revascularization of ischemic limbs in animals and humans. 38
In humans, VEGF protein and mRNA have been demonstrated in a variety of endothelial tumors, including benign hemangiomas of infancy and angiosarcomas. 39-41 Because these tumors have also been demonstrated to express VEGF receptors, both autocrine and paracrine roles for VEGF in endothelial tumorigenesis have been implied. VEGF has been shown to have autocrine activity in retinal pigment epithelial cells in vitro, 42 but this is the first report of transforming activity of VEGF in vivo. We demonstrate that overexpression of human VEGF in immortalized endothelial cells is sufficient to convert them from benign hemangiomas to malignant angiosarcomas. These angiosarcomas differ from those derived from sequential introduction of SV40 large T antigen and Ras in terms of their slower rate of in vivo growth. Angiosarcomas derived from endothelial cells containing both large T antigen and H-Ras reach a volume of 1 cm 3 in approximately 3 to 4 weeks, 15 whereas angiosarcomas containing large T antigen and VEGF 121 require approximately 5 months to reach the same size. MS1VEGF cells do not form colonies in soft agar, thus demonstrating that in this system, growth in vivo does not correlate with soft agar growth.
The VEGF secreted by these tumors is biologically active, as judged from the fact that MS1 VEGF cells fail to respond to exogenous VEGF 165 with an increase in autophosphorylation of VEGFR-2 as compared with the response in the control MS1 cells. The lack of increased phosphorylation of VEGFR-2 is likely to be due to an intracellular binding of endogenously produced VEGF 121. Binding of ligand to the receptor could take place in any intracellular component where VEGF 121 would have access to extracellular domains of VEGFR-2, such as the endoplasmic reticulum or the Golgi apparatus. Activation of the kinase activity of receptor tyrosine kinases is known to lead to rapid degradation, via either via lysosomal degradation or ubiquitination and proteosome-mediated degradation. It is, therefore, expected that this type of chronic stimulation would not lead to a detectable increase in the level of autophosphorylated receptors. Furthermore, if the level of endogenously produced ligand is sufficiently high to saturate receptor binding sites, exogenously added growth factor would not have access to receptors. Similar effects of intracrine stimulation in cells expressing a growth factor and its cognate receptor have been described before. 43 In addition, the tumors derived from MS1 VEGF cells demonstrate continued synthesis of primate MS1 VEGF in vivo.
Because VEGFR-2 protein could be identified in immunoprecipitates from metabolically labeled MS1 VEGF cells, the failure to induce kinase activity with exogenous VEGF strongly indicates that VEGF produced by MS1 VEGF cells interacts with endogenous VEGFR-2. To further demonstrate the presence of an autocrine VEGF loop in these cells, MS1 VEGF cells were incubated with SU1498, 44 a specific inhibitor of flk-1 tyrosine kinase. Incubation of cells with this inhibitor decreased levels of c-ets-1, a transcription factor that has been shown to be up-regulated by VEGF 121.
A large number of angiosarcomas have alterations in the activity of the p53 tumor suppressor gene. 9,10,45 This may be a result of loss of p53 through mutation or deletion, or amplification of the MDM2 gene, which functionally inactivates p53. 40 p53-null mice also develop angiosarcoma at a high frequency. 46 Our immortalized endothelial cells express SV40 large T antigen, which functionally impairs p53 activity and may serve as a model of an angiosarcoma precursor. Chemically induced angiosarcomas in rodents and humans have been shown to have mutations in activated Ras, leading to constitutive Ras activity, suggesting that p53 inactivation and Ras activation may be synergistic in rapidly growing angiosarcomas. Little is known of the molecular pathogenesis of more slowly growing endothelial malignancies. We show that overexpression of primate VEGF in immortalized endothelial cells that express VEGFR-1 and VEGFR-2 leads to malignant transformation, but to a novel, slowly growing tumor, in contrast to the vast majority of tumors that can be propagated in animals. This model may shed light on the pathogenesis of slow growing malignancies and establishes VEGF as a oncogenic factor in endothelial cells.
Acknowledgments
We thank Dr. Judah Folkman for his suggestions and support, and Susan Connors and Karen Keough for assistance with VEGF RIA.
Footnotes
Address reprint requests to Jack L. Arbiser, Department of Dermatology, Emory University School of Medicine, WMB 5309, 1639 Pierce Drive, Atlanta, GA 30322. E-mail: jarbise@emory.edu.
Funded in part by grants from the KAO Corporation, the Dermatology Foundation, the American Skin Association, the Emory Skin Disease Research Core Center P30 AR 42687 (National Institutes of Health), and the National Institute of Arthritis and Musculoskeletal and Skin Diseases KO8 AR02096–01 and RO3 AR44947–03 (all to J. L. A.).
References
- 1.Pratt GA: Birthmarks in infants. Arch Dermatol 1953, 67:302-305 [DOI] [PubMed] [Google Scholar]
- 2.Vin-Christian K, McCalmont TH, Frieden IJ: Kaposiform hemangioendothelioma. A locally invasive vascular tumor that can mimic hemangioma of infancy. Arch Dermatol 1997, 133:1573-1578 [DOI] [PubMed] [Google Scholar]
- 3.Morrison WH, Byers RM, Garden AS, Evans HL, Ang KK, Peters LJ: Cutaneous angiosarcoma of the head and neck: a therapeutic dilemma. Cancer 1995, 76:319-327 [DOI] [PubMed] [Google Scholar]
- 4.Blei F, Karp N, Rofski N, Rosen R, Greco MA: Successful multimodal therapy for kaposiform hemangioendothelioma complicated by Kasabach-Merritt phenomenon: case report and review of the literature. Pediat Hematol Oncol 1998, 15:295-305 [DOI] [PubMed] [Google Scholar]
- 5.Schmitz-Rixen T, Horsch S, Arnold G, Peters PE: Angiosarcoma in primary lymphedema of the lower extremity-Stewart-Treves syndrome. Lymphology 1984, 17:50-53 [PubMed] [Google Scholar]
- 6.Brady MS, Garfein CF, Petrek JA, Brennan MF: Post-treatment sarcoma in breast cancer patients. Ann Surg Oncol 1994, 1:66-72 [DOI] [PubMed] [Google Scholar]
- 7.Infante PF, Wagoner JK, McMichael AJ, Waxweiler RJ, Falk H: Genetic risks of vinyl chloride. Lancet 1976, 1:1289-1290 [DOI] [PubMed] [Google Scholar]
- 8.Przygodzki RM, Finkelstein SD, Keohavong P, et al: Sporadic and thorotrast induced hepatic angiosarcomas of the liver manifest frequent and multiple point mutations in K-ras-2. Lab Invest 1997, 76:153-159 [PubMed] [Google Scholar]
- 9.Barbin A, Froment O, Boivin S, Marion MJ, Belopggi F, Maltoni C, Montesano R: p53 gene mutation pattern in rat liver tumors induced by vinyl chloride. Cancer Res 1997, 57:1695-1698 [PubMed] [Google Scholar]
- 10.Barbin A, Froment O, Boivin S, Marion MJ, Maltoni C, Montesano R: p53 gene mutation pattern in rat liver tumors induced by vinyl chloride. Cancer Res 1997, 57:1695-1698 [PubMed] [Google Scholar]
- 11.Caldwell JB, Ryan MT, Benson PM, James WD: Cutaneous angiosarcoma arising in the radiation site of a congenital hemangioma. J Am Acad Dermatol 1995, 33:865-870 [DOI] [PubMed] [Google Scholar]
- 12.Stewart NJ, Pritchard DJ, Nascimento AG, Kang YK: Lymphangiosarcoma following mastectomy. Clin Orthop Rel Res 1995, 320:135-141 [PubMed] [Google Scholar]
- 13.Zukerberg LR, Nickoloff BJ, Weiss SW: Kaposiform hemangioendothelioma of infancy and childhood: an aggressive neoplasm associated with Kasabach-Merritt syndrome and lymphangiomatosis. Am J Surg Pathol 1993, 17:321-328 [DOI] [PubMed] [Google Scholar]
- 14.Kirova YM, Feuilhade F, Calitchi E, Otmezguine Y, Belembaogo E, Le Bourgeois JP: Radiation-induced sarcoma after breast cancer: apropos of 8 cases and review of the literature. Cancer Radiother 1998, 2:381-386 [DOI] [PubMed] [Google Scholar]
- 15.Arbiser JL, Moses MA, Fernandez CA, Ghiso N, Cao Y, Klauber N, Frank D, Brownlee M, Flynn E, Parangi S, Byers HR, Folkman J: Oncogenic H-Ras stimulates tumor angiogenesis by two distinct pathways. Proc Natl Acad Sci USA 1997, 94:861-866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Soker S, Takashima S, Miao HQ, Neufeld G, Klagsbrun M: Neuropilin-1 is expressed by endothelial and tumor cells as an isoform-specific receptor for vascular endothelial growth factor. Cell 1998, 92:735-745 [DOI] [PubMed] [Google Scholar]
- 17.Offrench-Constant C, Van De Water L, Dvorak HF, Hynes RO: Reappearance of an embryonic pattern of fibronectin splicing during wound healing in the adult rat. J Cell Biol 1989, 109:903-914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brown LF, Berse B, Tognazzi K, Manseau EJ, Van De Water L, Senger D, Dvorak H, Rosen S: Vascular permebility factor mRNA and protein expression in human kidney. Kidney Int 1992, 42:1457-1461 [DOI] [PubMed] [Google Scholar]
- 19.Claffey KP, Brown LF, del Aguila LF, Tognazzi K, Yeo KT, Manseau EJ, Dvorak HF: Expression of vascular permeability factor/vascular endothelial growth factor by melanoma cells increases tumor growth, angiogenesis, and experimental metastasis. Cancer Res 1996, 56:172-181 [PubMed] [Google Scholar]
- 20.Waltenberger J, Claesson-Welsh L, Siegbahn A, Shibuya M, Heldin CH: Different signal transduction properties of KDR and Flt1, two receptors for vascular endothelial growth factor. J Biol Chem 1994, 269:26988-26995 [PubMed] [Google Scholar]
- 21.Iwasaka C, Tanaka K, Abe M, Sato Y: Ets-1 regulates angiogenesis by inducing the expression of urokinase-type plasminogen activator and matrix metalloproteinase-1 and the migration of vascular endothelial cells. J Cell Physiol 1996, 169:522-531 [DOI] [PubMed] [Google Scholar]
- 22.Shibuya M, Ito N, Claesson-Welsh L: Structure and function of vascular endothelial growth factor receptor-1 and -2. Curr Top Microbiol Immunol 1999, 237:59-83 [DOI] [PubMed] [Google Scholar]
- 23.Senger DR, Galli SJ, Dvorak AM, Perruzzi CA, Harvey VS, Dvorak HF: Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science 1983, 219:983-985 [DOI] [PubMed] [Google Scholar]
- 24.Ferrara N, Davis-Smyth T: The biology of vascular endothelial growth factor. Endocr Rev 1997, 18:4-25 [DOI] [PubMed] [Google Scholar]
- 25.Rak J, Mitsuhashi Y, Bayko L, Filmus J, Shirasawa S, Sasazuki T, Kerbel RS: Mutant ras oncogenes upregulate VEGF/VPF expression: Implications for induction and inhibition of tumor angiogenesis. Cancer Res 1995, 55:4575-4580 [PubMed] [Google Scholar]
- 26.Grugel S, Finkenzeller G, Weindel K, Barleon B, Marme D: Both v-Ha-ras and v-raf stimulate expression of the vascular endothelial growth factor in NIH3T3 cells. J Biol Chem 1995, 270:25915-25919 [DOI] [PubMed] [Google Scholar]
- 27.Mukhopadhyay D, Tsiokas L, Zhou XM, Foster D, Brugge JS, Sukhatme VP: Hypoxic induction of human vascular endothelial growth factor expression through c-src activation. Nature 1995, 375:577-81 [DOI] [PubMed] [Google Scholar]
- 28.Levy AP, Levy NS, Goldberg MA: Post-translational regulation of vascular endothelial growth factor by hypoxia. J Biol Chem 1996, 271:2746-2753 [DOI] [PubMed] [Google Scholar]
- 29.Shweiki D, Neeman M, Itin A, Keshet E: Induction of vascular endothelial growth factor expression by hypoxia and by glucose deficiency in multicell spehroids: implications for tumor angiogenesis. Proc Natl Acad Sci USA 1995, 92:768-772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mukhopadhyay D, Tsiokas L, Sukhatme VP: Wild type p53 and v-src exert opposing influences on human vascular endothelial growth factor gene expression. Cancer Res 1995, 55:6161-6165 [PubMed] [Google Scholar]
- 31.Kerbel RS, Viloria-Petit A, Okada F, Rak J: Establishing a link between oncogenes and tumor angiogenesis. Mol Med 1998, 4:286-295 [PMC free article] [PubMed] [Google Scholar]
- 32.Petit AM, Rak J, Hung MC, Rockwell P, Goldstein N, Fendly B, Kerbel RS: Neutralizing antibodies against epidermal growth factor and ErbB-2/neu receptor tyrosine kinases down-regulate vascular endothelial growth factor production by tumor cells in vitro and in vivo: angiogenic implications for signal transduction therapy of solid tumors. Amer J Pathol 1997, 151:1523-1530 [PMC free article] [PubMed] [Google Scholar]
- 33.Gerber HP, McMurtrey A, Kowalski J, Yan M, Keyt BA, Dixit V, Ferrara N: Vascular endothelial growth factor regulates endothelial cell survival through the phosphatidylinositol-3-kinase/Akt signal transduction pathway: requirement for flk-1/kdr activation. J Biol Chem 1998, 273:30336-30343 [DOI] [PubMed] [Google Scholar]
- 34.Yoshida A, Anand-Apte B, Zetter BR: Differential endothelial migration and proliferation to basic fibroblast growth factor and vascular endothelial growth factor. Growth Factors 1996, 13:57-64 [DOI] [PubMed] [Google Scholar]
- 35.Fong GH, Rossant J, Gertsenstein M, Breitman ML: Role of the flt-1 receptor tyrosine kinase in regulating the assembly of vascular endothelium. Nature 1995, 376:66-70 [DOI] [PubMed] [Google Scholar]
- 36.Shalaby F, Rossant J, Yamaguchi TP, Gertsenstein M, Wu XF, Breitman ML, Schuh AC: Failure of blood-island formation and vasculogenesis in flk-1 deficient mice. Nature 1995, 376:62-66 [DOI] [PubMed] [Google Scholar]
- 37.Landgren E, Schiller P, Cao Y, Claesson-Welsh L: Placenta growth factor stimulates MAP kinase and mitogenicity but not phospholipase c-gamma and migration of endothelial cells expressing flt-1. Oncogene 1998, 16:359-367 [DOI] [PubMed] [Google Scholar]
- 38.Isner JM, Pieczek A, Schainfeld R, Blair R, Haley L, Asahara T, Rosenfield K, Razvi S, Walsh K, Symes JF: Clinical evidence of angiogenesis after arterial gene transfer of phVEGF 165 in patient with ischaemic limb. Lancet 1996, 348:370-374 [DOI] [PubMed] [Google Scholar]
- 39.Takahashi K, Mulliken JB, Kozakewich HP, Rogers RA, Folkman J, Ezekowitz RA: Cellular markers that distinguish the phases of hemangioma during infancy and childhood. J Clin Invest 1994, 93:2357-2364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zietz C, Rossle M, Haas C, Sendelhofert A, Hirschmann A, Sturzl M, Lohrs U: MDM-2 Oncoprotein overexpression, p53 gene mutation, and VEGF up-regulation in angiosarcomas. Am J Pathol 1998, 153:1425-1433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brown LF, Tognazzi K, Dvorak HF, Harrist TJ: Strong expression of kinase insert domain-containing receptor, a vascular permeability factor/vascular endothelial growth factor receptor in AIDS associated Kaposi’s sarcoma and cutaneous angiosarcoma. Am J Pathol 1996, 148:1065-1074 [PMC free article] [PubMed] [Google Scholar]
- 42.Guerrin M, Scotet E, Malecaze F, Houssaint E, Plouet J: Overexpression of vascular endothelial growth factor induces cell transformation in cooperation with fibroblast growth factor 2. Oncogene 1997, 14:463-471 [DOI] [PubMed] [Google Scholar]
- 43.Valgeirsdottir S, Eriksson A, Nister M, Heldin CH, Westermark B, Claesson-Welsh L: Compartmentalization of autocrine signal transduction pathways in Sis transformed NIH 3T3 cells. J Biol Chem 1995, 270:10161-10170 [DOI] [PubMed] [Google Scholar]
- 44.Sun L, Tran N, Tang F, App H, Hirth P, McMahon G, Tang C: Synthesis and biologic evaluation s of 3-substituted indolin-2-ones: a novel class of tyrosine kinase inhibitors that exhibit selectivity toward particular receptor tyrosine kinases. J Med Chem 1998, 41:2588-2603 [DOI] [PubMed] [Google Scholar]
- 45.Soini Y, Welsh JA, Ishak KG, Bennett WP: P53 mutations at AT base pairs in primary hepatic angiosarcomas not associated with vinyl chloride exposure. Carcinogenesis 1995, 16:2879-2881 [DOI] [PubMed] [Google Scholar]
- 46.Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA, Jr, Butelk JS, Bradley A: Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 1992, 356:215-221 [DOI] [PubMed] [Google Scholar]