

Abstract
Mycosis fungoides is usually an indolent disease that, after a variable period of time in a stable phase, evolves into a tumoral form with aggressive behavior. The molecular events that mark this progression remain essentially unknown to date, and this prompted us to investigate the hypothetical role of p16INK4a silencing in mycosis fungoides progression. We analyzed the three most frequent mechanisms of inactivation of the p16INK4a gene (deletion, promoter hypermethylation, and mutation) in nine cases with patch/plaque and tumoral lesions of mycosis fungoides. The existence of alterations was investigated by microsatellite analysis, methylation-specific polymerase chain reaction, and direct sequencing. Alterations of the p16INK4a gene were found in four of nine of the plaque lesions (hypermethylation in three samples and allelic loss in one sample) and seven of nine in the tumor lesions (hypermethylation in five samples and allelic loss in three samples). No case presented point mutations. Although a higher incidence of p16INK4a hypermethylation was observed in the cases that failed to achieve a complete remission, the limited size of our series prompted us to evaluate this finding cautiously. The results of this study therefore showed a common genetic alteration that is found more frequently in tumoral lesions than it is in plaque lesions of mycosis fungoides. It also offers data that could suggest a relationship between p16INK4a hypermethylation and unfavorable clinical outcome. Broader studies are needed to confirm both relationships.
Lymphoid malignancies are a heterogeneous group of diseases with different clinical, morphological, immunophenotypical, and genetic characteristics. Mycosis fungoides (MF), the most common subtype of cutaneous T-cell lymphoma, characteristically evolves through several clinical stages. 1 Usually MF has an indolent clinical course with slow progression over years or decades, from a premycotic cutaneous appearance that may resemble that of other common dermatological disorders, to patches and more infiltrated plaques. After a variable period of time it may develop aggressive behavior, with cutaneous tumors and spreading to lymph nodes and visceral sites. The rapidity of progression is variable and unpredictable, 2 and some cases may show an aggressive course from the beginning.
Histopathologically it consists of epidermotropic band-like infiltrates containing small or medium-sized mononuclear cells with cerebriform nuclei. Colonization of the lower layers of the epidermis by simple or small groups of neoplastic cells is a characteristic finding. Epidermotropism is usually lost with progression to tumor stage, when the dermal infiltrate becomes more diffuse and the tumor cells increase in number and size, while there is a concomitant decrease in the number of admixed reactive inflammatory cells.
In lymphomas, as in other tumors, tumoral progression has been found to be the consequence of multiple cell cycle alterations, with loss of some of the negative regulatory pathways. Cell cycle progression is regulated by complexes formed between cyclins and cyclin-dependent kinases (CDKs). CDK4 and CDK6 bind to D-type cyclins in the G1 phase of the cell cycle and control G1/S transition through the phosphorylation of retinoblastoma protein (pRb). The activity of cyclin D-CDK4/6 complexes is subject to additional levels of regulation, including the association with CDK inhibitors (CDKIs). The p16INK4a gene is located in the short arm of chromosome 9, region 9p21, encoding for a nuclear protein that can block cell cycle progression by effectively inhibiting the kinase activity of CDK4/6, thereby exerting a negative control on cell proliferation. 3
The p16INK4a gene has been found to be altered in a high percentage of cell lines (75%) and various primary human tumors, 4-6 such as breast and colon cancer, lymphoma, and leukemia. In these tumors, the loss of protein expression, secondary to 5′CpG island methylation, homozygous deletion, and, less frequently, p16INK4a gene mutations, is usually associated with tumoral progression. 7-15
The molecular mechanisms by which advanced cases of MF undergo large cell transformation and develop an aggressive behavior are essentially unknown. Several studies have failed to identify the genes involved in this transformation, and thus the data obtained in the analysis of p53, lyt10, c-myc, bcl-1, and bcl-2 genes have shown a low frequency of genetic alterations associated with MF. 16-20 A large body of evidence has recently suggested that p16INK4a gene inactivation is a key event in the development and progression of hematological malignancies. 21 However, the data on p16INK4a status in MF remain sparse and are less than compelling. 22-27
The aim of this study was to investigate the hypothetical pathogenic role of p16INK4a inactivation in a representative panel of MF with patch/plaque and tumoral sequential samples. To determine the possible implication of p16INK4a gene alterations in the development and progression of MF, we analyzed the three most frequent mechanisms of inactivation of the p16INK4a gene: deletion, promoter hypermethylation, and mutation. As previous data obtained in the study of other lymphoma types have shown that p16INK4a inactivation is characteristically associated with advanced tumoral stages, we restricted analysis to cases concurrently showing both early and advanced stages of MF, thus making it possible to compare the results in biopsies that are representative of both phases of tumoral progression.
Materials and Methods
Case Selection
A group of MF patients was selected from the medical records of the 12 de Octubre Hospital, Madrid, and the Virgen de la Salud Hospital, Toledo (both of which are in the center of Spain). Diagnosis was based on generally accepted clinical-pathological criteria. 2 Patch or plaque lesions were erythematous, slightly scaling macules or plaques with well-defined borders. Histopathologically they presented a characteristic infiltrate of epidermotropic atypical T cells with convoluted cerebriform nuclear contours. Tumors were defined as nodules, whether they were ulcerated or not, when histopathological examination showed a majority of large cells (Figure 1) ▶ .
Figure 1.
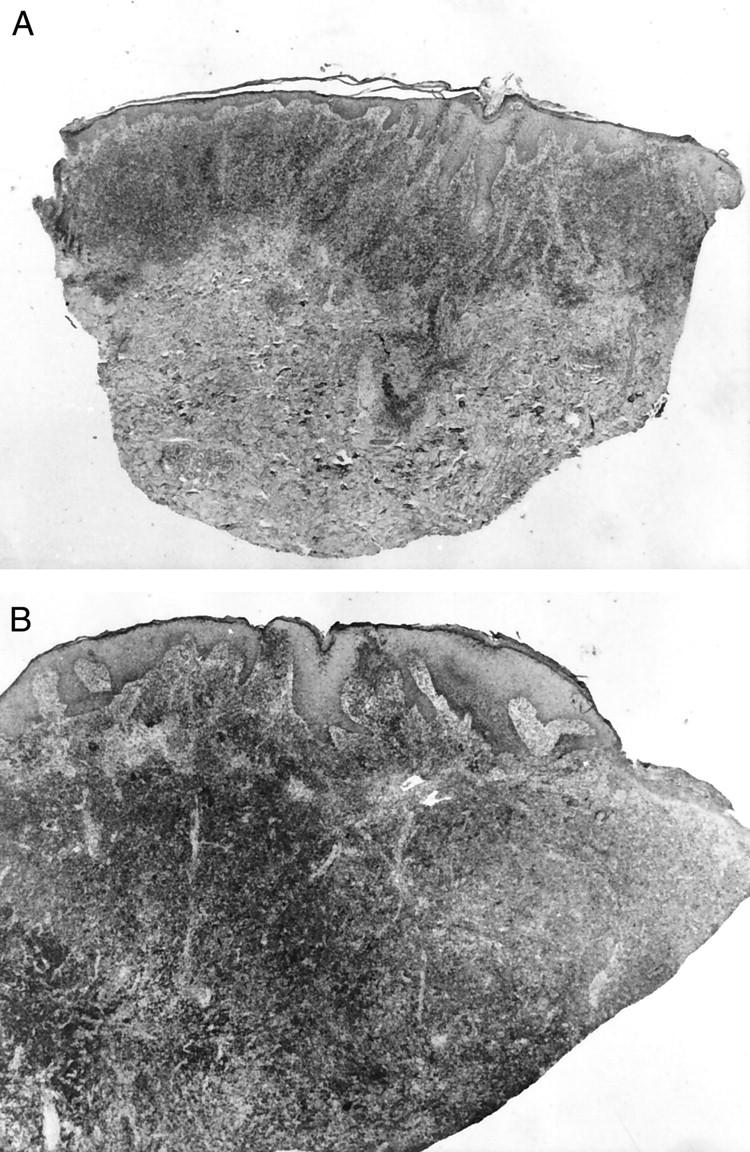
A: Patient MF78. Panoramic view of a plaque lesion. H&E. Original magnification, ×50. B: Patient MF78. Panoramic view of MF, tumor stage. H&E. Original magnification, ×65.
We identified nine cases from which we could obtain formalin-fixed paraffin-embedded tissue or fresh frozen material from patch/plaque as well as tumoral lesions. Some patients had patch/plaque and tumor lesions present simultaneously on different areas on their skin. All of the samples analyzed were studied in parallel for routine hematoxylin-eosin examination.
Complete remission (CR) is defined as no evidence of residual disease after a follow-up of at least 3 months. The TNM staging system was used, as defined by the MF Cooperative Group for MF/SS. 2
Molecular Studies
DNA Extraction
Genomic DNA from the cases selected was obtained from frozen or paraffin-embedded tissue from patch/plaque and tumoral lesions. To provide a negative control in methylation studies, DNA was obtained from different samples of reactive lymphoid tissue (12 cases of peripheral blood lymphocytes from healthy donors, three reactive lymph nodes, and one normal thymus) and six cases of inflammatory dermatosis (pytiriasis lichenoides). The Raji cell line was used as the positive control. For analysis of allelic loss nontumoral DNA was obtained from neutrophils (seven cases), oral swabs (one case), or nontumoral paraffin-embedded tissue (one case).
Allelic Loss Assays at the 9p21 Locus
Tumoral and normal DNA was analyzed for loss of heterozygosity (LOH) or homozygous deletion (HD) by amplification of dinucleotide repeats containing sequence microsatellite markers, 28 under conditions previously described by Villuendas et al. 9 These markers are at the 9p21 region, surrounding the p16INK4a gene (IFNα, D9S736, D9S1749, D9S1747, D9S974, D9S1748, D9S1752, D9S171). Briefly, 100 ng of DNA was amplified in a volume of 25 μl with 1× polymerase chain reaction (PCR) buffer, 200 μmol/L deoxynucleoside triphosphates, 10 pmol of each primer, 1 μCi of [32P]dCTP, and 1 U of Taq polymerase. Amplified products were separated by electrophoresis by denaturing 7 mol/L urea on 6% acrylamide gels, followed by autoradiography. Allelic loss was determined if the intensity of the signal from any one allele was significantly reduced in the tumor DNA when compared with normal DNA by direct visualization.
The presence of homozygous deletions was also assessed in all cases by comparative multiplex PCR assay, using two primer sets from loci outside the 9p21 region (D9S934 and D7S1824, in chromosomes 9q and 7q, respectively).
Methylation Studies
Methylation-specific PCR (MSP) assays were performed to analyze the methylation status of CpG islands of the first exon of p16INK4a, as described by Herman et al.. 29 Briefly, 1 μg of denatured genomic DNA was modified by reaction with sodium bisulfite under conditions that convert all unmethylated cytosine residues, except 5-methylcytosine, to uracil by deamination. Modified DNA was purified using the Wizard DNA clean-up system (Promega, Madison, WI). Modification was completed by NaOH 0.3 mol/L treatment for 5 minutes at room temperature, followed by ethanol precipitation.
Bisulfite-modified DNA was amplified using p16INK4a unmethylated-specific primers (U), methylated-specific primers (M), and unmodified or wild-type primers (W). One hundred nanograms of bisulfite-modified DNA was amplified using 1 U of AmpliTaq Gold (PE Applied Biosystems, Weiterstadt, Germany) under the following conditions: 30 seconds at 94°C, 30 seconds at 60°C, and 30 seconds at 72°C, for 35 cycles. Controls without DNA and positive controls for U and M reactions were performed for each set of PCRs. The PCR product was visualized in agarose gels stained with ethidium bromide under UV illumination.
If a methylation-specific PCR product was detected, the whole procedure using sodium bisulfite and MSP was performed again to minimize the possible influence of contamination or incomplete bisulfite treatment.
DNA methylation was determined by the presence of a 150-pb fragment in those samples amplified with the M primers.
The PCR products obtained with p16-M and p16-U primers of two cases were sequenced with an Automated DNA Sequencer ABI PRISM 310 Genetic Analyzer (PE Applied Biosystems) according to the manufacturer’s instructions.
Mutation Study
DNA from seven frozen tumoral stage samples were analyzed for mutations in exons 1 and 2 of the p16INK4a gene, comprising 97% of the coding sequences, with primers described previously. 30 Briefly, 200 ng of DNA was amplified with 25 pmol of each primer, 200 μmol/L of each deoxynucleoside triphosphate, 5% dimethylsulfoxide, and 1 U Taq polymerase, under amplification conditions described previously by Villuendas et al. 9 Direct sequencing of amplified products was performed with an Automated DNA Sequencer ABI PRISM 310 Genetic Analyzer (PE Applied Biosystems) according to the manufacturer’s procedures.
Data Analysis
Statistical analysis was performed by means of Epilnfo6.0 or SAS2.0 software. The following characteristics were evaluated using Fisher’s exact test or the χ 2 test, with Yates correction when necessary: treatment previous to the biopsy (with α-interferon, psolaven and ultraviolet A light (PUVA), sunbathing, radiotherapy, chemotherapy, topical bis-chlorethylnitrosourea (BCNU), topical mechloretamine, or topical corticosteroids) versus genetic alteration (p16INK4a hypermethylation or allelic loss or both). The same tests were used to analyze the relationship between clinical outcome (long complete remission or not) versus genetic alteration (p16INK4a hypermethylation, allelic loss, or both). The Wilcoxon and median score tests were used to evaluate the influence of the variable interval of time between the onset of the disease and biopsy examination and the existence and type of p16INK4a genetic alteration.
Results
Clinical Data
The clinical data of our patients are summarized in Table 1 ▶ . Sex distribution was 2:1, M/F; mean age at the beginning of MF was 44.7 years (range 21–70 years); mean delay to diagnosis was 75 months (range 3–288 months). The mean time until the appearance of tumors was 93.1 months (range 0–298 months). Two patients developed tumors and patch/plaques simultaneously. In another patient tumors appeared within 6 months of the beginning of patch/plaque lesions.
Table 1.
Main Clinical Data of the Patients
| Patient | Age/sex. onset of MF | Plaque lesions | Tumoral lesions | Status (June 99) | |||
|---|---|---|---|---|---|---|---|
| Date of sampling* | Previous treatment† | Onset of tumors | Date of sampling* | Previous treatment† | |||
| MF36 | 39/M 1993 | +36 (March-96) | Topical corticosteroids | 0 (March-93) | +36 (March-96) | Same | Alive, CR |
| MF39 | 67/M 1985 | +127 (April-96) | α-IFN | +118 (July-95) | +120 (Sept-95) | None | Alive t2NOMO |
| MF113 | 36/M 1984 | +168 (June-98) | Sunbathing Topical corticosteroids | +178 (April-99) | +180 (May-99) | Sunbathing, PUVA α-IFN | Alive T2N1MO |
| MF90 | 34/F 1986 | +36 (Sept 89) | None | +100 (July-94) | +130 (Oct-97) | PUVA, RePUVA, α-IFN, Electron-bean, C-MOPP, ABMT, Etoposide | Dead (lymphoma) |
| MF91 | 21/F 1973 | +288 (June 97) | Topical corticosteroids | +298 (March-98) | +298 (March-98) | Topical corticosteroids, PUVA | Alive T2N0M0 |
| MF73 | 43/M 1988 | +72 (Nov-94) | RePUVA α-IFN | +48 (Nov 92) | +48 (Nov 92) | BCNU | Aive T3N1M1 |
| Sunbathing, PUVA | |||||||
| MF78 | 27/M 1982 | +172 (July 96) | RePUVA, α-IFN RT (electrons) Topical mecloretamine, Photopheresis | +90 (Sept 89) | +184 (July 97) | Same | Alive T2N0M0 |
| MF82 | 66/F June-88 | +18 (Jan-90) | Sunbathing | +6 | +18 (Jan-90) | Same | Alive, CR |
| MF122 | 70/M 1995 | +6 (Feb 96) | Topical corticosteroids | 0 | 6+ (Feb 96) | Same | Dead (Oat-cell carcinoma) |
*Time from the onset of MF until molecular study was performed (months).
†Treatments performed before the date of sampling.
M, male; F, female; IFN, interferon; PUVA, photochemotherapy; RT, radiotherapy; ABMT, autologous bone marrow transplantation; T2, patches or plaques covering more than 10% of the body surface; T3, tumoral lesions; N0, no palpable lymph nodes; N1, palpable lymph nodes, histopathologically negative; M0, no visceral involvement; M1, visceral involvement; CR, complete remission.
In four of nine patients, the patch/plaque samples analyzed were taken 10–94 months before the sampling of tumor lesions. In two of nine patients patch/plaque samples were taken seven and 24 months after the tumor lesion. In three patients, the two kinds of samples were taken simultaneously.
Two of the patients achieved complete remission, and they remain free of disease after 39 and 99 months of follow-up, respectively. Only two of the seven remaining patients underwent complete remission during their medical history: MF73 and MF78, 6 and 11 months, respectively, early in the course of their disease.
Molecular Studies
A table showing the results obtained in the analysis of p16INK4a is included (Table 2) ▶ . Alterations of the p16INK4a gene were found in four of nine samples in plaque lesions and in seven of nine samples in tumoral samples (including both deletion and methylation analysis).
Table 2.
Summary of 9p21 Region Alterations in the Plaque and Tumoral Stages of MF
| Cases | Plaque stage | Tumoral stage | |||
|---|---|---|---|---|---|
| Methylation p16INK4a | Allelic loss | Methylation p16INK4a | Allelic loss | Mutation p16INK4a | |
| MF36 | ND | − | − | HD | wt* |
| MF39 | + | − | + | LOH | ND |
| MF113 | ND | LOH | ND | LOH | wt |
| MF90 | ND | − | ND | − | ND |
| MF91 | + | − | + | − | wt |
| MF73 | − | ND | + | − | wt |
| MF78 | − | ND | + | − | wt |
| MF82 | − | − | − | − | wt |
| MF122 | + | − | + | − | wt |
| 3/6 | 1/7 | 5/7 | 3/9 | 0/7 | |
| Total alterations | 4/9 | 7/9 |
HD, homozygous deletion; LOH, loss of heterozygosity; ND, not done; +, positive; −, negative.
*wt due to reactive cells associated with the tumor as it presents HD.
Allelic Loss Studies
Nine cases of MF for which DNA from nontumoral tissue was available were studied using markers for microsatellites surrounding the p16INK4a gene in the 9p21 region. The results of allelic loss analysis are shown in Figure 2 ▶ , and representative cases are illustrated in Figure 3 ▶ .
Figure 2.

Summary of allelic loss data for the 9p21 region in MF. Graphic representation of the eight microsatellite markers. All cases studied are illustrated in plaque (PL) and tumoral (T) stages. Noninformative cases marked with an asterisk indicate a signal markedly diminished in comparison with normal DNA.
Figure 3.

Representative examples of allelic loss in chromosome 9p21. The presence of allelic losses in tumor (T) and plaque (PL) in comparison with matched normal (N) DNA is shown by arrows. Case MF36 shows allelic loss in informative loci D9S1749 and D9S974. A band markedly diminished in tumoral DNA in comparison with normal DNA is shown in the noninformative locus D9S1748. This case showed retention of both alleles for locus D9S1747. Case MF39 shows LOH for informative loci D9S1749 and D9S1747 and retention of both alleles for locus D9S1748.
Only one of nine cases showed allelic loss in plaque lesion MF (MF113). In contrast to this finding, three of nine cases showed genetic deletion in the tumoral phase (Table 2) ▶ . Deletions were most commonly observed at marker D9S1749.
Case MF36 (Figure 3) ▶ showed LOH in one informative marker in the sample corresponding to the tumoral phase. Homozygous deletion was identified by multiplex PCR at the D9S974 marker, close to the p16INK4a gene. Noninformative loci D9S1748 and D9S1752 also showed a band in tumoral DNA that was markedly diminished in comparison with normal DNA. This case also showed retention of heterozygosity at the D9S1747 locus. This apparent retention of heterozygosity at a single locus within a large deletion supports the hypothesis of the existence of a homozygous deletion, including the p16INK4a gene, rather than two separate regions of loss at 9p, as the signal from the deleted alleles in the tumor lane probably arises from normal cell contamination of the tumor biopsy.
In case MF39 (Figure 3) ▶ , LOH was detected in only two markers.
Case MF113 displayed LOH, involving four different markers in plaque and tumoral samples. The existence of LOH in markers D9S974 and D9S1748, which flank the region occupied by the p16INK4a gene, indicates hemizygous deletion of this gene.
Methylation at the CpG Island of the p16INK4a Gene Assays
The MSP technique was used to identify the hypermethylation status of the CpG island in the promoter region of p16INK4a gene. Hypermethylation of the p16INK4a gene was found in five of seven tumoral lesions and three of six plaque lesions (Table 2) ▶ . In these three early cases, methylation of the p16INK4a gene was also detected in the sample corresponding to the tumoral phase. Two cases only presented hypermethylation of the p16INK4a gene in advanced-stage biopsy. Representative cases are illustrated in Figure 4 ▶ .
Figure 4.

Methylation-specific PCR assay of the p16INK4a gene. Agarose gel electrophoresis of two representative cases (MF78, MF91) is shown at the plaque (PL) and tumoral (T) stages. Primer sets used for amplification are designated as unmethylated (U) or methylated (M). The Raji cell line and peripheral blood lymphocytes (PBLs) serve as positive controls for methylated (M) and unmethylated (U) p16 alleles, respectively.
Tumoral and plaque samples from case MF90 and the plaque sample from MF36 were not amplified with the p16-U and p16-M primers, probably because of the extreme fragmentation of DNA from these samples preserved in paraffin. Material from patient MF113 was not available for methylation study.
All 22 controls included (skin, lymphoid tissue in different reactive conditions, and peripheral blood lymphocytes) showed amplification with unmethylated-specific primers (U) but not with methylated-specific primers (M). DNA from the Raji cell line was used as a positive control for the amplification reaction with p16-M primers, and no amplification was seen in the PCR reaction with p16-U primers (Figure 4) ▶ . PCR reactions with wild-type primers (W) were used as a control of efficiency of chemical modification.
p16-M and p16-U amplification products from the plaque and tumoral stages of cases MF91 and MF122 were sequenced. The sequence of p16-M products did not show any change from C to T in any of the CpG sites, in the plaque or the tumoral samples, thus confirming the presence of methylation in all of the CpG sites (Figure 5A) ▶ . However, changes from C to T (indicating the absence of methylation) were observed in the p16-U product sequence (Figure 5B) ▶ . p16-W PCR was performed using non-bisulfite-treated DNA.
Figure 5.
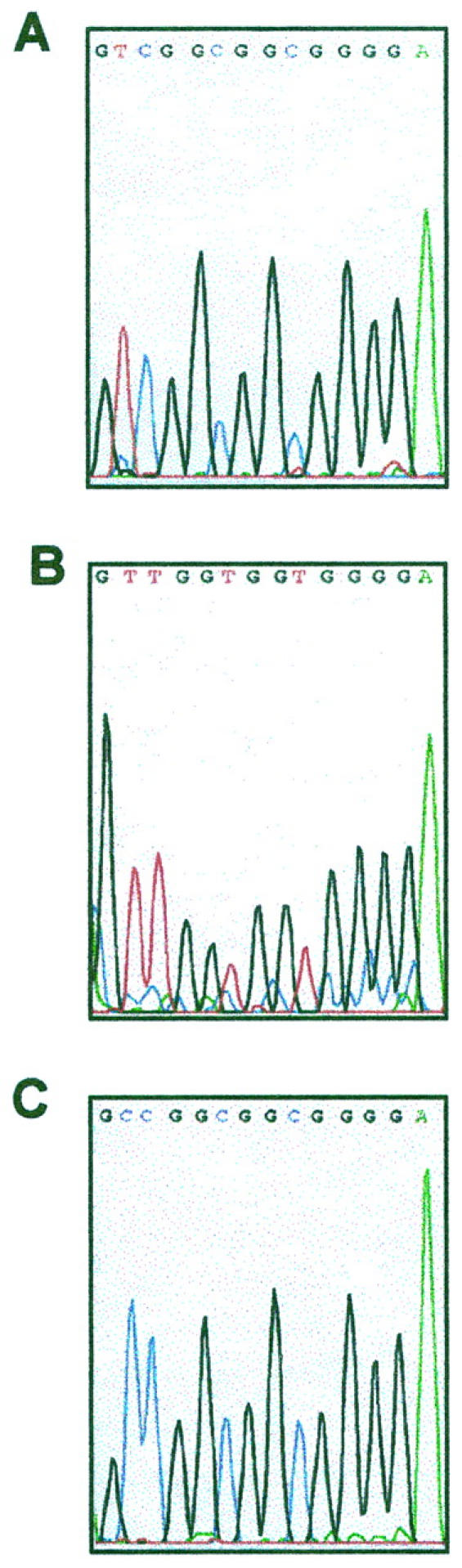
Direct sequencing of the amplified products with p16-M (A), p16-U (B), and p16-W (C) primers in case MF91. All cytosines (C) in the unmethylated have been converted to thymidines (T), whereas all C in methylated CpG dinucleotides remain as C, indicating the presence of methylation in all of the CpG sites.
The sequence of p16-W products is shown in Figure 5C ▶ .
Mutational Analysis of the p16INK4a Gene
To determine whether mutations of the p16INK4a gene were present, exons 1 and 2 of the gene were analyzed by direct sequencing. None of the seven cases analyzed showed mutations.
Correlation between Clinical and Molecular Data
The type of molecular alteration identified in this study (methylation or gene deletion) was independent of previous treatment received by the patient (P > 0.05).
The existence of molecular alteration was also independent of the length of the evolution of the disease previous to the biopsy analyzed. For plaque lesions, the mean times were 147 months for hypermethylated and 74.5 months for nonhypermethylated plaques. The difference between these two groups is not significant (Wilcoxon and Median score tests). For tumoral lesions, the mean time interval between the first clinical manifestations of the disease and the date of the biopsy on which molecular study was performed was 124.5 months for the cases with hypermethylation or allelic loss and 74 months for cases with none. These results were not significantly different.
To determine whether clinical outcome is related to p16INK4a alteration or to some specific molecular abnormality (methylation or gene deletion), a comparison was made between patients in terms of their response to treatment. Five of the patients in whom complete remission was not achieved had p16INK4a hypermethylation in either the tumor or both the tumor and plaque stages, whereas neither of the two patients (MF36 and MF82) with long complete remission had p16INK4a hypermethylation in plaque or tumor lesions. However, patient MF36 showed homozygous deletion, thus making methylation unnecessary.
Discussion
Our series includes a representative group of MF patients (nine cases) showing tumoral progression, in which patch/plaque and tumoral lesions were analyzed. Their clinical features are not different from those of similar previously published series.
This series allowed us to investigate some molecular mechanisms underlying tumoral progression, which in other lymphoma types have been found to be associated with inactivation of some of the more common suppresser pathways, p14/ARF-p53 and p16INK4a-Rb. We decided to focus on p16INK4a, because previous works have shown there is a low frequency of p53 inactivation in advanced forms of MF. 18-19
The tumor suppresser gene p16INK4a is now recognized as the one that is most often subject to genetic alteration in different types of tumor. The frequency and type of alterations depend on tumoral type, because it has been established that gene deletions, promoter hypermethylation, or point mutation may lead to the silencing of the p16INK4a gene. 4-8,31 In non-Hodgkin’s lymphomas, p16INK4a inactivation has been found to be very characteristically associated with tumoral progression, this being caused most often by promoter hypermethylation and gene deletion. The highest frequencies of deletion of the p16INK4a gene are found in acute lymphoblastic leukemias (ALLs), mainly of the T-cell precursor type. 13,24 Rates of hypermethylation of the 5′ CpG island of p16INK4a are more strongly associated with a higher rate of tumor progression in Burkitt’s lymphoma, multiple myeloma, 32-33 and large B-cell lymphoma than they are in stable forms of indolent B-cell lymphomas, such as B-chronic lymphocytic leukemia, follicle center lymphoma, mantle cell lymphoma, and mucosa-associated lymphoid tissue lymphoma. 9-12 Information about the frequency and mechanisms of p16INK4a inactivation in MF is scarce, although recently the loss of p16INK4a expression in a significant proportion of cases of MF has been demonstrated. 27
This study showed that p16INK4a alterations take place in the MF lesions of patients, at both the plaque and tumoral stages. Tumoral samples of MF showed a higher frequency (seven of nine cases) of p16INK4a gene alterations (including both deletion and methylation analysis), in contrast with a lower frequency (four of nine samples) in plaque samples. Comparing these data with those obtained in the analysis of B-cell lymphomas, the relatively higher frequency of p16INK4a alterations in early phases of the disease in this series of MF is remarkable. The higher frequency of p16INK4a alterations in tumoral lesions could suggest a role for p16INK4a in the progression of MF from the plaque to the tumoral stage. Screening for these abnormalities could hypothetically identify patients with a higher probability of transformation to aggressive forms of the disease, although we cannot exclude the possibility that the p16INK4a alteration could be a hallmark of MF that is frequently present from the beginning of the disease. A more comprehensive study of MF cases without progression would be needed to confirm the significance of this alteration. If additional studies confirm that p16INK4a inactivation is a marker of tumoral progression in MF, this could be used in the routine evaluation of these patients, thus making it possible to identify cases that would be candidates for different therapeutic schemes. Only one case (MF39), which shows methylation in both the plaque and tumoral phases of the disease, additionally acquired LOH in the tumoral sample, suggesting that in at least this case gene deletion is subsequent to methylation. Indeed, comparative analysis of the molecular results of this series suggests that the differences between plaques and tumors seem to be more closely related to allelic loss than they are to p16INK4a hypermethylation. However, we cannot discount the possibility that these differences could be due to technical difficulties associated with samples having a low percentage of tumoral cells, which may lead to a significant underestimation of allelic loss.
None of our cases presented mutations in tumoral DNA. Consistent with other investigations, 27 our findings suggest that mutations of the p16INK4a gene are not an important mechanism in the tumoral progression of MF.
Furthermore, the clinical implications of the molecular data obtained in this study have been analyzed. Because most of our patients had undergone different forms of treatment over long periods of time, we hypothesized that some of these treatments could have selected cell subpopulations with molecular alterations that would give a survival advantage. None of the treatments that our patients had received before the biopsies analyzed seemed to influence the molecular results.
Second, we attempted to determine whether the acquisition of p16INK4a alterations could be a result of a longer period of disease development before biopsy. Although the mean duration of the disease until biopsy of the lesions without molecular alterations was lower than that in the case of samples with p16INK4a alterations, the small size of the series made it impossible for us to reach significant conclusions on this point. Finally, although p16INK4a hypermethylation was more frequent in those cases that failed to achieve complete remission, the small size of our series and the existence of a homozygous deletion in one of the cases without methylation forced us to make a cautious interpretation of this finding.
If these results were reproduced in a larger series, the existence of p16INK4a hypermethylation could be used as a predictor of aggressive disease and would be useful in identifying patients who could benefit from different treatments.
Acknowledgments
We thank Dras. Margarita Sánchez-Beato and M-Sol Mateo for their helpful comments and valuable discussion; Dr. Agustín Gómez for his useful help with statistical analysis, and Pablo Morales for his kind contribution of patient samples.
Footnotes
Address reprint requests to Dra. Patrocinio Algara, Department of Genetics, Hospital Nacional de Parapléjicos, Finca La Peraleda s/n, 45071 Toledo, Spain. E-mail: p.martinez@mx2.redestb.es.
Drs. Navas and Ortiz-Romero contributed equally to this work.
References
- 1.Edelson RL: Cutaneous T-cell lymphoma: mycosis fungoides, Sezary syndrome and other variants. J Am Acad Dermatol 1980, 2:89-106 [DOI] [PubMed] [Google Scholar]
- 2.Diamandidou E, Cohen PR, Kurzrock R: Mycosis fungoides and Sezary syndrome. Blood 1996, 88:2385-2409 [PubMed] [Google Scholar]
- 3.Serrano M: The tumour suppresser protein p16INK4a. Exp Cell Res 1997, 237:7-13 [DOI] [PubMed] [Google Scholar]
- 4.Nobori T, Miura K, Wu DJ, Lois A, Takabayashi K, Carson DA: Deletions of the cyclin-dependent kinase-4 inhibitor gene in multiple human cancers. Nature 1994, 368:753-756 [DOI] [PubMed] [Google Scholar]
- 5.Cairns P, Mao L, Merlo A, Lee DJ, Schwab D, Eby Y, Tokino K, van der Riet P, Blaugrund JE, Sidransky D: Rates of p16 (MTS1) mutations in primary tumors with 9p loss. Science 1994, 265:415-417 [DOI] [PubMed] [Google Scholar]
- 6.Liggett WH, Jr, Sidransky D: Role of the p16 tumour suppresser gene in cancer. J Clin Oncol 1998, 16:1197-1206 [DOI] [PubMed] [Google Scholar]
- 7.Herman JG, Merlo A, Mao L, Lapidus RG, Issa JP, Davidson NE, Sidransky D, Baylin SB: Inactivation of the CDKN2/p16/MTS1 gene is frequently associated with aberrant DNA methylation in all common human cancers. Cancer Res 1995, 20:4525-4530 [PubMed] [Google Scholar]
- 8.Merlo A, Herman JG, Mao L, Lee DJ, Grabielson E, Burger PC, Baylin SB, Sidransky D: 5′CpG island methylation is associated with transcriptional silencing of the tumour suppressor p16/CDKN2/MTS1 in human cancers. Nature Med 1995, 1:686-692 [DOI] [PubMed] [Google Scholar]
- 9.Villuendas R, Sánchez-Beato M, Martínez JC, Sáez AI, Martínez-Delgado B, García JF, Mateo MS, Sánchez-Verde L, Benítez J, Martínez P, Piris MA: Loss of p16/INK4A protein expression in non-Hodgkin’s lymphomas is a frequent finding associated with tumor progression. Am J Pathol 1998, 153:887-897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Elenitoba-Johnson KS, Gascoyne RD, Lim MS, Chhanabai M, Jaffe ES, Raffeld M: Homozygous deletions at chromosome 9p21 involving p16 and p15 are associated with histologic progression in follicle centre lymphoma. Blood 1998, 91:4677-4685 [PubMed] [Google Scholar]
- 11.Pinyol M, Cobo F, Bea S, Jares P, Nayach I, Fernández PL, Montserrat E, Cardesa A, Campo E: p16INK4a gene inactivation by deletions, mutations and hypermethylation is associated with transformed and aggressive variants of non-Hodgkin’s lymphomas. Blood 1998, 91:2977-2984 [PubMed] [Google Scholar]
- 12.Martínez-Delgado B, Fernandez-Piqueras J, García MJ, Arranz E, Gallego J, Rivas C, Robledo M, Benitez J: Hypermethylation of a 5′CpG island of p16 is a frequent event in non-Hodgkin’s lymphoma. Leukemia 1997, 11:425-428 [DOI] [PubMed] [Google Scholar]
- 13.Uchida T, Kinoshita T, Murate T, Saito H, Hotta T: CDKN2 (MTS1/p16INK4a) gene alterations in adult T-cell leukemia/lymphoma. Leuk Lymphoma 1998, 29:27-35 [DOI] [PubMed] [Google Scholar]
- 14.Uchida T, Kinoshita T, Saito H, Hotta T: CDKN2 (MTS1/p16INK4a) gene alterations in hematological malignancies. Leuk Lymphoma 1997, 24:449-461 [DOI] [PubMed] [Google Scholar]
- 15.Baur AS, Shaw P, Burri N, Delacrétaz F, Bosman FT, Chaubert P: Frequent methylation silencing of p15INK4b (MTS2) and p16INK4a (MTS1) in B-cell and T-cell lymphomas. Blood 1999, 94:1773-1781 [PubMed] [Google Scholar]
- 16.Beylot-Barry M, Vergier B, DeMascarel A, Beylot C, Merlio JP: p53 oncoprotein expression in cutaneous lymphoproliferations. Arch Dermatol 1995, 131:1019-1024 [PubMed] [Google Scholar]
- 17.McGregor JM, Dublin EA, Levison DA, MacDonald DM, Smith NP, Whittaker S: p53 immunoreactivity is uncommon in primary cutaneous lymphoma. Br J Dermatol 1995, 132:353-358 [DOI] [PubMed] [Google Scholar]
- 18.de Misa RF, Azana JM, Harto A, Bellas C, Ledo A: Infrequent expression of protein p53 in epidermotropic variants of cutaneous T-cell lymphomas. J Dermatol 1995, 22:524-526 [DOI] [PubMed] [Google Scholar]
- 19.Garatti SA, Roscetti E, Trecca E, Fracchiolla NS, Neri A, Berti E: bcl1, bcl2, p53, c-myc and lyt10 analysis in cutaneous lymphomas. Recent Results Cancer Res 1995, 139:249-261 [DOI] [PubMed] [Google Scholar]
- 20.Li G, Chooback L, Wolfe JT, Rook AH, Felix CA, Lessin SR, Salhani KE: Overexpression of p53 protein in cutaneous T-cell lymphoma: relationship to large cell transformation and disease progression. J Invest Dermatol 1998, 110:767-770 [DOI] [PubMed] [Google Scholar]
- 21.Drexler HG: Review of alterations of the cyclin dependent kinase inhibitor INK4 family genes p15, p16, p18 and p19 in human leukemia-lymphoma cells. Leukemia 1998, 12:845-849 [DOI] [PubMed] [Google Scholar]
- 22.Dreyling MH, Roulston D, Bohlander SK, Vardiman J, Olopade OI: Codeletion of CDKN2 and MTAP genes in a subset of non-Hodgkin lymphomas may be associated with histologic transformation from low-grade to diffuse large-cell lymphoma. Genes Chromosom Cancer 1998, 22:72-78 [PubMed] [Google Scholar]
- 23.Stranks G, Height SE, Mitchell P, Jadayel D, Yuille MA, De Lord C, Clutterbuck RD, Treleaven JG, Powles RL, Nacheva E, Oscier DG, Karpas A, Lenoir GM, Smith SD, Millar JL, Catovsky D, Dyer MJS: Deletions and rearrangement of CDKN2 in lymphoid malignancy. Blood 1995, 85:893-901 [PubMed] [Google Scholar]
- 24.Ogawa S, Hangaishi A, Miyawaki S, Hirosawa S, Miura Y, Takeyama K, Kamada N, Ohtake S, Uike N, Shimazaki Ch, Toyama K, Hirano M, Mizoguchi H, Kobayashi Y, Furusawa S, Saito M, Emi N, Yazaki Y, Ueda R, Hirai H: Loss of the cyclin-dependent kinase 4-inhibitor (p16;MTS1) gene is frequent in and highly specific to lymphoid tumors in primary human hematopoietic malignancies. Blood 1995, 86:1548-1556 [PubMed] [Google Scholar]
- 25.Gombart AF, Morosetti R, Miller CW, Said JW, Koeffler HP: Deletions of the cyclin-dependent kinase inhibitor genes p16INK4A and p15INK4B in non-Hodgkin’s lymphomas. Blood 1995, 86:1534-1539 [PubMed] [Google Scholar]
- 26.Siebert R, Willers CP, Schramm A, Fossa A, Dresen IM, Uppenkamp M, Nowrousian MR, Seeber S, Opalka B: Homozygous loss of the MTS1/p16 and MTS2/p15 genes in lymphoma and lymphoblastic leukaemia cell lines. Br J Haematol 1995, 91:350-354 [DOI] [PubMed] [Google Scholar]
- 27.Peris K, Stanta G, Fargnoli MC, Bonin S, Felli A, Amantea A, Chimenti S: Reduced expression of CDKN2a/p16INK4a in mycosis fungoides. Arch Dermatol Res 1999, 291:207-211 [DOI] [PubMed] [Google Scholar]
- 28.Cairns P, Polascik TJ, Eby Y, Tokino K, Califano J, Merlo AA, Mao L, Herath J, Jenkins R, Westra W, Rutter JL, Buckler A, Gabrielson E, Tockman M, Cho KR, Hedrick L, Bova GS, Isaacs W, Koch W, Schwab D, Sidransky D: Frequency of homozygous deletion at p16/CDKN2 in primary human tumours. Nature Genet 1995, 11:210-212 [DOI] [PubMed] [Google Scholar]
- 29.Herman JG, Graff JR, Myohanen S, Nelkin BD, Baylin SB: Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci USA 1996, 93:9821-9826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kamb A, Gruis NA, Weaver-Feldhaus J, Liu Q, Harshman K, Tavtigian SV, Stockert E, Day RS, Johnson BE, Skolnick MH: A cell cycle regulator potentially involved in genesis of many type of tumors. Science 1994, 264:436-440 [DOI] [PubMed] [Google Scholar]
- 31.Gonzalez-Zulueta M, Bender CM, Yang AS, Nguyen T, Beart RW, Van Tornout JM, Jones PA: Methylation of the 5′CpG island of the p16/CDKN2 tumour suppresser gene in normal and transformed human tissues correlates with gene silencing. Cancer Res 1995, 55:4531-4535 [PubMed] [Google Scholar]
- 32.Herman JG, Civin CI, Issa J-PJ, Collector MI, Sharkis SJ, Baylin SB: Distinct patterns of inactivation of p15INK4b and p16INK4a characterize the major types of hematological malignancies. Cancer Res 1997, 57:837-841 [PubMed] [Google Scholar]
- 33.Ng MHL, Chung YF, Lo KW, Wickham NWR, Lee JCK, Huang DP: Frequent hypermethylation of p16 and p15 genes in multiple myeloma. Blood 1997, 89:2500-2506 [PubMed] [Google Scholar]