

Abstract
Medullary carcinomas of the pancreas are a recently described, histologically distinct subset of poorly differentiated adenocarcinomas that may have a unique pathogenesis and clinical course. To further evaluate these neoplasms, we studied genetic, pathological, and clinical features of 13 newly identified medullary carcinomas of the pancreas. Nine (69%) of these had wild-type K-ras genes, and one had microsatellite instability (MSI). This MSI medullary carcinoma, along with three previously reported MSI medullary carcinomas, were examined immunohistochemically for Mlh1 and Msh2 expression, and all four expressed Msh2 but did not express Mlh1. In contrast, all of the medullary carcinomas without MSI expressed both Msh2 and Mlh1. Remarkably, the MSI medullary carcinoma of the pancreas in the present series arose in a patient with a synchronous but histologically distinct cecal carcinoma that also had MSI and did not express Mlh1. The synchronous occurrence of two MSI carcinomas suggests an inherited basis for the development of these carcinomas. Indeed, the medullary phenotype, irrespective of MSI, was highly associated with a family history of cancer in first-degree relatives (P < 0.001). Finally, one medullary carcinoma with lymphoepithelioma-like features contained Epstein-Barr virus-encoded RNA-1 by in situ hybridization. Therefore, because of medullary carcinoma’s special genetic, immunohistochemical, and clinical features, recognition of the medullary variant of pancreatic adenocarcinoma is important. Only by classifying medullary carcinoma as special subset of adenocarcinoma can we hope to further elucidate its unique pathogenesis.
Most, but not all, poorly differentiated carcinomas arising in the pancreas are conventional ductal adenocarcinomas. 1-3 Some poorly differentiated pancreatic carcinomas, however, are medullary carcinomas. 4 Although historically grouped with poorly differentiated ductal adenocarcinomas, medullary carcinomas are histologically distinct. As first described in 1998, they have poorly defined cellular boundaries (syncytial growth pattern), expanding tumor borders, and extensive necrosis, features similar to their histological counterparts arising in the colorectum (Figure 1) ▶ . 4-8
Figure 1.
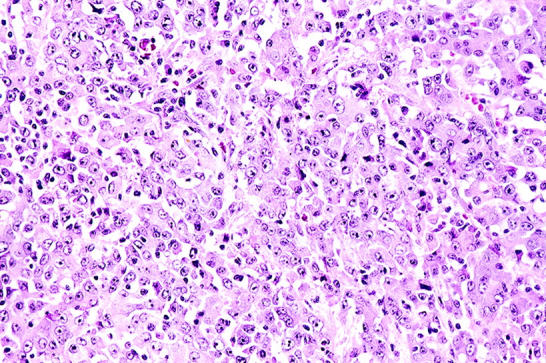
Histology of medullary carcinoma of the pancreas. The tumor has a syncytial growth pattern of the cells.
Medullary carcinomas may also be genetically distinct from conventional ductal adenocarcinomas of the pancreas. Indeed, of five medullary carcinomas first described by Goggins et al, 4 three (60%) had both microsatellite instability (MSI) and a wild-type K-ras gene. (One of the six medullary carcinomas reported by Goggins et al 4 was later determined to be a metastatic renal cell carcinoma.) These data are atypical for nonmedullary ductal adenocarcinomas of the pancreas, which nearly universally harbor K-ras gene mutations and seldom if ever have MSI. 4,9-13
Although these preliminary findings favor a distinct pathogenetic course for medullary pancreatic carcinomas, the small number of cases studied so far has required a cautious view. To further elucidate the clinical and genetic features associated with the medullary morphology, we studied a larger series of medullary pancreatic carcinomas. Specifically, our goals were to determine 1) the frequency of wild-type K-ras genes and MSI among medullary carcinomas; 2) whether immunohistochemistry for the DNA-repair gene products Mlh1 and Msh2 could identify the medullary carcinomas with MSI; and 3) whether the medullary carcinomas were associated with any other immunohistochemical, pathological, or clinical variables, including latent Epstein-Barr (EBV) infection or family history of cancer. Answering these three questions would help us to gauge the utility of classifying medullary carcinomas as a special subset of ductal adenocarcinomas of the pancreas.
Materials and Methods
Case Selection
The surgical pathology files of The Johns Hopkins Hospital and the Academic Medical Center (University of Amsterdam) were screened for pancreatic carcinomas with the medullary histological pattern. 4 A histological review of 450 randomly chosen pancreatic cancers revealed 18 (4.0%) possible medullary carcinomas, each originally diagnosed as a poorly differentiated ductal adenocarcinoma. Five of these cases were those originally identified by Goggins et al. 4 One of the medullary carcinomas also contained adenosquamous carcinoma comprising approximately half of the neoplasm. All 17 of the remaining medullary carcinomas were pure medullary carcinomas.
The 18 medullary carcinomas were then pooled by a reference pathologist (REW) with 17 other (nonmedullary) pancreatic carcinomas. The 17 nonmedullary carcinomas included 10 poorly differentiated ductal adenocarcinomas, four moderately differentiated ductal adenocarcinomas, two mucinous adenocarcinomas, and one adenosquamous carcinoma. One slide from each case was coded, and the set of slides was reviewed by three other pathologists (GJAO, RHH, and NVA), one of whom (NVA) was from an institution not associated with our original study on medullary carcinomas. 4 Each pathologist graded the cases according to criteria originally proposed by Goggins et al 4 (gland formation, invasion pattern, syncytial growth pattern, necrosis). Each neoplasm then received an overall grade from 1 (most similar) to 5 (least similar), based on its similarity to the index medullary carcinoma case identified in the original study by Goggins et al. 4
A neoplasm was considered a medullary carcinoma only when it met the following two criteria: first, an average grade less than 3; and second, at least two of the three pathologists providing a grade of 2 or lower. The three pathologists were said to disagree when any of the pathologists gave a score that was more than one point away from either of the other two pathologists.
Histological Analysis for Pancreatic Intraepithelial Neoplasias
When available, sections containing nonneoplastic pancreatic tissue surrounding each medullary carcinoma were histologically analyzed by the reference pathologist (REW) for associated pancreatic intraepithelial neoplasias (PanINs), the putative precursors of pancreatic ductal adenocarcinoma. 14-16 PanINs were identified and graded according to accepted criteria established at a National Cancer Institute-sponsored Pancreas Cancer Think Tank, held September, 1999, in Park City, Utah. These criteria are available on the Worldwide Web (http://pathology.jhu.edu/pancreas/panin).
In Situ Hybridization for Epstein-Barr Virus
Because they share some features with EBV-containing lymphoepitheliomas of the nasopharynx, all 18 medullary carcinomas were assayed by in situ hybridization for Epstein-Barr virus-encoded RNA-1. In addition, five of the 17 nonmedullary carcinomas were also studied for latent EBV infection. These five nonmedullary carcinomas included two poorly differentiated conventional adenocarcinomas, one moderately differentiated conventional adenocarcinoma, and two mucinous adenocarcinomas.
Formalin-fixed, paraffin-embedded tissue sections were deparaffinized and hydrated in graded ethanol. Sections were washed in water, digested with 10 μg/ml of proteinase K in 50 mmol/L Tris-HCl (pH 7.5) for 30 minutes at 37°C, and washed again in water. Sections were hybridized at 37°C for 16 to 18 hours with 20 μl of fluorescein isothiocyanate-labeled Epstein-Barr virus-encoded RNA riboprobe in hybridization solution (Novocastra, Newcastle on Tyne, UK). A parallel section was hybridized with a negative control probe. The sections were washed in Tris-buffered saline (pH 8.0) with 0.05% Tween 20 (TBS-T; Sigma Chemical Co., St. Louis, MO) and then immersed in 0.2× sodium chloride-sodium citrate/0.1% sodium dodecyl sulfate solution at 52°C for 10 minutes.
Sections were incubated in 10% normal rabbit serum for 30 minutes and then in a 1:200 dilution of anti-fluorescein isothiocyanate-alkaline phosphatase conjugate (In Situ Hybridization Detection Kit, Novocastra) in Tris-buffered saline (pH 8.0) with 0.05% Tween 20 for 30 minutes. After three washes, sections were placed in 1× alkaline-phosphatase buffer (100 mmol/L Tris-HCl, pH 9.5, 100 mmol/L NaCl, 50 mmol/L MgCl2) for 5 minutes. Chromogenic detection was then performed with nitroblue tetrazolium and X-phosphate. Adequate color development occurred within 2 hours of incubation in the chromogen solution. The slides were rinsed in water, counterstained with hematoxylin, and mounted with aqueous mounting medium (GlycerGel; DAKO, Carpinteria, CA). An intense bluish-purple color indicated a positive hybridization signal.
Microdissection and DNA Extraction
The five medullary carcinomas originally identified by Goggins et al 4 had already been analyzed genetically. Therefore, 13 genetically unanalyzed medullary carcinomas remained. We also genetically analyzed five of the 17 nonmedullary pancreatic carcinomas with the same technique to ensure a sensitivity adequate to detect single base-pair genetic alterations. These five nonmedullary carcinomas included those also studied for EBV latent infection by in situ hybridization. Therefore, we microdissected and genetically analyzed 18 carcinomas (13 medullary and five nonmedullary) in this study.
Each of the 18 carcinomas to be genetically analyzed were microdissected from unstained 10-μm sections cut from archived paraffin blocks. The microdissected tissue was deparaffinized, digested with proteinase K, purified by phenol-chloroform extraction, and precipitated, as previously described. 9,17 The final product was reconstituted in 50 μl LoTE (3 mmol/L Tris, 0.1 mmol/L ethylenediaminetetraacetic acid, pH 7.5) and stored at −20°C. Because of the solid growth pattern of medullary carcinomas and microdissection with avoidance of lymphocytes, relatively pure samples of carcinoma were obtained. Samples containing at least 200 neoplastic cells were used for each polymerase chain reaction (PCR) below. Both the medullary and adenosquamous components of the mixed medullary carcinoma were separately microdissected.
K-ras Gene Analysis
Portions of exons 1 and 2 (encompassing codons 12, 13, and 61) of the K-ras gene (KRAS2) were amplified by PCR, as previously described. 9 Direct cycle sequencing (SequiTherm Excel II, Epicentre Technologies, Madison, WI) was performed using internal primers at an annealing temperature of 60°C. Products were separated on a 6% polyacrylamide gel and subjected to autoradiography.
Unusual K-ras genotypes were confirmed by direct sequencing of an independent PCR product and by sequencing of cloned PCR products (TOPO TA, Invitrogen, Carlsbad, CA). The N- and H-ras genes were not studied in this series, as we found previously that mutations in these two genes do not occur in pancreatic carcinomas, including medullary carcinomas. 18,19
Microsatellite Instability Analysis
Evidence of MSI was sought in each of the 18 microdissected samples by: 1) length analysis of BAT 25 and BAT 26 markers; and by 2) direct sequencing of the polythymidine tract of the TGFBR2 gene. 4,20-22 BAT 25 and BAT 26 sequences were amplified by PCR incorporating [α]-32P-dCTP, and the products were resolved with 6% denaturing polyacrylamide gels, as previously described. 4 A portion of the TGFBR2 gene was amplified by PCR, and the products were cycle sequenced and similarly resolved. 4 The three MSI carcinomas from the previous study of Goggins et al 4 were used as positive controls.
For each of these three loci, gain or loss of at least one nucleotide was regarded as a shift. Each carcinoma tested was found to have either shifts in at least two of the three loci (designated MSI), or shifts at no locus (designated microsatellite stable, MSS). Because carcinomas lacking a mononucleotide shift in at least one of these three markers generally do not have shifts in additional dinucleotide markers, additional markers were not used. 4,23
Immunohistochemistry for Mlh1 and Msh2
Immunohistochemical labeling for the products of the MLH1 and MSH2 genes was performed on the current series of 13 medullary and five nonmedullary carcinomas, as well as on three MSI medullary carcinomas identified in the previous series collected by Goggins et al. 4 Unstained 6-μm sections were deparaffinized and rehydrated with xylene and graded alcohols. The slides were treated with sodium citrate buffer (10 mmol/L, pH 6.0) and then submitted to microwave antigen retrieval. Endogenous peroxidase was blocked by 20% hydrogen peroxide, and nonspecific binding was blocked by 20% Protein Blocker (Signet Laboratories, Dedham, MA) in buffered saline. After cooling for 5 minutes, the slides were labeled with mouse monoclonal antibodies to Mlh1 (1:50 dilution, clone G168–728; PharMingen, San Diego, CA) or to Msh2 (1:100 dilution, clone FE11; Oncogene Research Products, Cambridge, MA) and incubated overnight at room temperature. Each primary antibody was detected by biotinylated secondary antibodies, avidin-biotin complex, and 3,3′-diaminobenzidine. Sections were counterstained with hematoxylin. 24
Cancers interpreted to lack Mlh1 or Msh2 expression required a complete absence of labeling in the nuclei of neoplastic cells. Labeling of nonneoplastic epithelium, stromal cells, or lymphocytes served as an internal positive control in each of the sections.
Germline MLH1 Analysis
One patient in this study had synchronous pancreatic and colonic carcinomas. This patient’s pancreatic carcinoma had a medullary phenotype with microglandular features and MSI, and his colonic carcinoma was an adenocarcinoma that also had MSI. In addition, as will be discussed later, neither carcinoma had immunodetectable MLH1 gene product. Therefore, informed consent was obtained for germline MLH1 analysis under a protocol approved by the institutional review board of The Johns Hopkins Hospital. Conformation-sensitive gel electrophoresis was performed under clinical laboratory conditions at the University of Pennsylvania (Philadelphia, PA). 25-28 The results of the analysis were discussed with the patient, and he is currently undergoing genetic counseling.
Clinical and Pathological Data Collection
Clinical and pathological data for each of the patients were obtained from various sources, including the medical records of The Johns Hopkins Hospital and the Academic Medical Center, the surgical pathology databases of The Johns Hopkins Hospital and Academic Medical Center, and The Johns Hopkins Oncology Center’s clinical information system. Specifically, family history was obtained from extensive review of these sources. Fourteen (78%) of the 18 patients with medullary carcinomas and 69 (90%) of the 77 patients with nonmedullary carcinomas had familial cancer pedigrees available. Data collected included tumor size, presence of lymph node metastases at presentation, age, gender, race, smoking history, alcohol history, comorbidities, presenting symptoms, and family history of cancer in a first-degree relative. Comorbidities included a history of myocardial infarction, peptic ulcer disease, peripheral vascular disease, hypertension, chronic or acute pancreatitis, and inflammatory bowel disease. Presenting symptoms included weight loss, abdominal pain, jaundice, nausea/vomiting, and fever/chills.
Statistical Methods
Data from the 18 microdissected tumors first described in this study and 77 xenografted tumors, many previously studied by Goggins et al 4 and by others in our laboratory, 9 were analyzed. Means were compared with a t-test or Wilcoxon rank sum test when the assumption of normality was not valid. Cross-tabulations were performed using Fisher exact tests.
The statistical endpoint of this study was survival status. Event-time distributions for this endpoint were estimated with the method of Kaplan and Meier 29 and compared using the log-rank statistic 30 or the proportional hazards regression model. 31 The simultaneous effect of two or more factors was studied using the multivariate proportional hazards regression model. Covariates that were marginally significant (P < 0.19) in univariate analyses were entered into the multivariate regression model, and nonsignificant effects were removed in a stepwise fashion.
All P values are two-sided. Computations were performed using the Statistical Analysis System 32 or EGRET. 33,34
Results
Histology of the Medullary Carcinomas
The reference pathologist selected 35 cases for histological study. These included 18 cases classified as medullary by the reference pathologist and 17 cases classified as nonmedullary. The three other reviewing pathologists verified the diagnosis of medullary carcinoma in each of the 18 cases selected by the reference pathologist. The distribution of average grades for the similarity of the case in question to the index medullary carcinoma case was as follows: 1.0 (3 carcinomas); 1.2 (2 carcinomas); 1.8 (2 carcinomas); 2.0 (4 carcinomas); 2.1 (1 carcinoma); 2.2 (3 carcinomas); 2.3 (1 carcinoma); and 2.7 (2 carcinomas) (Table 1) ▶ . The medullary component from the case also containing adenosquamous carcinoma received an average grade of 1.8. The distribution of average scores for the 17 nonmedullary carcinomas was as follows: 5.0 (16 carcinomas) and 4.5 (1 carcinoma).
Table 1.
Selected Histological, Clinical, and Genetic Characteristics of 18 Medullary Carcinomas of the Pancreas
| Case* | Average grade† | PanINs | EBV in situ | Special histologic features | Gender | Race | Age | Family history of cancer | Survival status | Survival (months) | K-ras codon 12 | MMR phenotype |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2.0 | n/a | Neg | Squamoid features | M | W | 72 | Pos | Dec | 0 | CGT | MSS |
| 2 | 2.2 | n/a | Neg | F | W | 74 | Unk | Dec | 8 | Wt | MSS | |
| 3 | 2.0 | n/a | Neg | M | W | 79 | Pos | Dec | 5 | GAT | MSS | |
| 4 | 2.2 | Pres | Neg | F | W | 53 | Pos | Dec | 45 | Wt | MSS | |
| 5 | 2.3 | Pres | Neg | Clear-cell features | M | W | 44 | Unk | Dec | 11 | Wt | MSS |
| 6 | 2.2 | Not | Neg | M | W | 49 | Unk | Dec | 15 | Wt | MSS | |
| 7 | 1.0 | Pres | Neg | M | W | 74 | Pos | Dec | 12 | Wt | MSS | |
| 8 | 2.7 | Pres | Neg | F | W | 74 | Unk | Dec | 12 | Wt | MSS | |
| 9 | 1.8 | Not | Neg | Microglandular features | M | W | 34 | Pos | Alive | 13 | Wt | MSI |
| 10 | 1.2 | Not | Pos | Lymphoepithelioma-like features | M | W | 33 | Neg | Alive | 126 | Wt | MSS |
| 11 | 2.0 | Pres | Neg | F | W | 67 | Pos | Dec | 15 | TTT | MSS | |
| 12 | 2.1 | Not | Neg | Clear-cell features | M | W | 67 | Pos | Dec | 4 | GAT | MSS |
| 13 | 1.8 | Not | Neg | Mixed adenosquamous carcinoma | F | AA | 66 | Pos | Dec | 7 | Wt | MSS |
| 14 | 1.0 | Not | Neg | M | W | 71 | Pos | Alive | 67 | Wt | MSI | |
| 15 | 1.2 | Not | Neg | F | W | 84 | Pos | Dec | 4 | Wt | MSI | |
| 16 | 1.0 | Not | Neg | M | W | 72 | Pos | Alive | 24 | Wt | MSI | |
| 17 | 2.0 | n/a | Neg | M | W | 85 | Pos | Dec | 40 | GAT | MSS | |
| 18 | 2.7 | n/a | Neg | M | W | 65 | Pos | Dec | 9 | GCT | MSS |
Abbreviations: PanINs, pancreatic intraepithelial neoplasias; Pres, present; Not, not present; n/a, slides of non-neoplastic pancreas surrounding the medullary carcinoma were not available for review; EBV, Epstein-Barr Virus; Pos, positive; Neg, negative; M, male; F, female; W, white; AA, African-American; Unk, unknown family history; Dec, deceased; MMR, mismatch repair; MSS, microsatellite stable; MSI, microsatellite unstable.
*Cases 14 through 18 were first identified in the original study by Goggins et al. 4 Cases 1 through 13 were first identified in the present series.
†Average grade indicates similarity of the case to the index medullary carcinoma.
Among the three pathologists, there was disagreement in only three (two medullary carcinomas and one nonmedullary carcinoma) of the 35 cases analyzed histologically. For each medullary carcinoma, two pathologists gave a score of 2, and one gave a score of 4 (average score, 2.7). For the nonmedullary carcinoma, two pathologists gave a score of 5, and one pathologist gave a score of 3.5 (average score, 4.5).
Whereas all of the medullary carcinomas had syncytial growth patterns, expanding tumor borders, and extensive necrosis, some had additional special features. For example, two medullary carcinomas had foci of clear cells, one had a focal microglandular growth pattern, and one had areas of squamoid differentiation (Table 1) ▶ . In addition, one medullary carcinoma had lymphoepithelioma-like features, with extensive intraepithelial and stromal infiltration by lymphocytes (Figure 2A) ▶ . In general, pancreatic medullary carcinomas do not have a large number of intratumoral lymphocytes, as was seen in this latter neoplasm. 4
Figure 2.
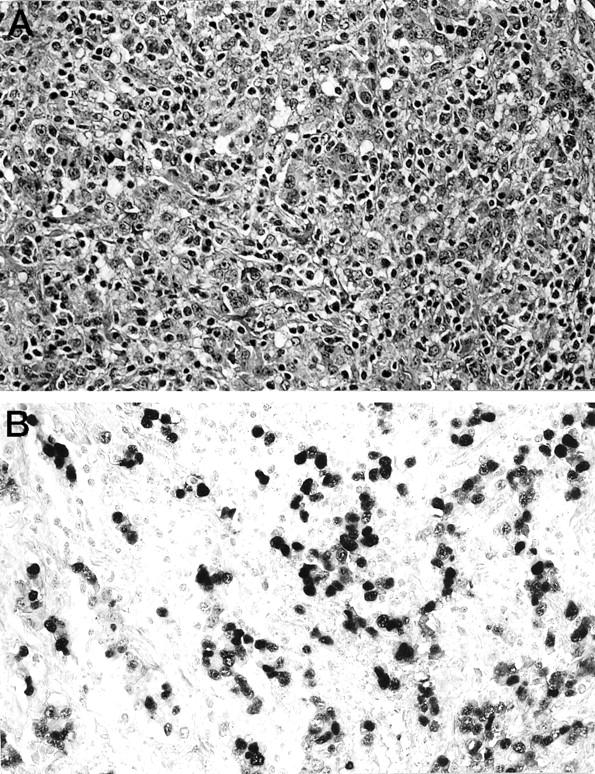
Histology of a medullary carcinoma of the pancreas with lymphoepithelioma-like features. A: The neoplasm has the characteristics of medullary carcinoma with the additional finding of an intense intratumoral lymphocytic infiltrate. B: In situ hybridization for Epstein-Barr virus-encoded RNA-1 is positive in the nuclei of the neoplastic cells.
Identification of Pancreatic Intraepithelial Neoplasia in Pancreata with Medullary Carcinoma
When available, slides of pancreatic parenchyma surrounding each of the medullary carcinomas were searched for PanIN. Slides of noncancerous tissue were available from 11 (61%) of the 18 medullary carcinomas (average of 7.2 slides per case; range, 2 to 25 slides). Five (45%) of these 11 cases had PanINs: case 4 had two PanINs-1B, case 5 had one PanIN-1B, case 7 had four PanINs-1A, case 8 had one PanIN-1B, and case 11 had two PanINs-1A and seven PanINs-1B. No higher grade duct lesions (PanINs-2 or -3) were identified.
In Situ Hybridization for Epstein-Barr Virus RNA
All 18 medullary carcinomas and five nonmedullary carcinomas were probed for latent EBV infection by in situ hybridization against Epstein-Barr virus-encoded RNA-1. The nuclei of the neoplastic cells in one (6%) of the 18 medullary carcinomas strongly labeled for the virus-encoded RNA (Figure 2B) ▶ . The neoplasm demonstrating EBV infection was the one with lymphoepithelioma-like features. None of the other 17 medullary carcinomas and none of the five nonmedullary carcinomas contained EBV RNA.
Prevalence of K-ras Mutations
Each of the 18 microdissected carcinomas produced amplifiable K-ras PCR products. Of the 13 medullary carcinomas, nine (69%) had wild-type K-ras genes. This included the medullary carcinoma with an admixed adenosquamous carcinoma; both components in this single tumor had wild-type K-ras genes. In contrast, all five of the microdissected carcinomas without medullary histology had K-ras mutations (P = 0.0294). A group of 77 previously xenografted pancreatic carcinomas, many previously studied by Goggins et al 4 and by others in our laboratory, were also used as a control. Five (6%) of these 77 had the medullary phenotype, and 72 (94%) were conventional ductal adenocarcinomas. Three (60%) of the five medullary carcinomas had wild-type K-ras genes, whereas only four (5.6%) of the 72 conventional ductal adenocarcinomas had wild-type K-ras genes. 35 Thus, medullary pancreatic carcinomas are more often wild type at the K-ras gene than are conventional ductal adenocarcinomas (P = 0.0044). Table 2 ▶ summarizes these data.
Table 2.
K-ras Gene Status and Microsatellite Instability of Medullary and Nonmedullary Adenocarcinomas of the Pancreas
| No. with wild-type K-ras gene/no. studied | No. with MSI/no. studied | |
|---|---|---|
| Microdissected medullary carcinomas | 9/13 (69%) | 1/13 (7.7%) |
| Xenografted medullary carcinomas | 3/5 (60%) | 3/5 (60%) |
| Microdissected nonmedullary carcinomas | 0/5 (0%) | 0/5 (0%) |
| Xenografted nonmedullary carcinomas | 4/72 (5.6%) | 0/72 (0%) |
| Total | 16/95 (17%) | 4/95 (4.2%) |
All of the K-ras gene mutations detected in the microdissected medullary carcinomas were at codon 12. Two of the four were GGT (gly) to GAT (asp) transition mutations, and the third was a GGT to CGT (arg) transversion. The fourth medullary carcinoma had an unusual dinucleotide substitution, resulting in a TTT (phe) at codon 12. This mutation had not previously been seen in a human tumor. This latter mutation was therefore confirmed by repeated direct sequencing of an independent PCR product and by cloning and sequencing of the PCR products. All five of the microdissected, nonmedullary carcinomas had K-ras gene mutations at codon 12; four (80%) were GAT mutations, and the remaining tumor had a CGT mutation.
Sixty-eight (94%) of the 72 xenografted conventional ductal adenocarcinoma controls harbored K-ras gene mutations. Sixty-six of these were at codon 12, two were at codon 13, and one was at codon 61. One xenografted conventional adenocarcinoma harbored both a GTT (val) mutation at codon 12 and a TGC (cys) mutation at codon 13. This dicodon mutation also had not previously been seen in a human tumor. It was confirmed by repeat direct sequencing of an independent PCR product. Cloning and sequencing of the PCR products established that these two mutations were on the same allele. Two of the five medullary carcinomas in the previous series collected by Goggins et al 4 had K-ras gene mutations, and these were GAT (asp) and GCT (ala) changes at codon 12, respectively.
Table 3 ▶ summarizes the types of mutations occurring in medullary and nonmedullary carcinomas of the pancreas. Figure 3 ▶ presents examples of a wild-type and a mutant K-ras codon 12 in a medullary carcinoma and a conventional ductal adenocarcinoma, respectively.
Table 3.
Distribution of K-ras Gene Mutations in Medullary and Nonmedullary Adenocarcinomas of the Pancreas
| GAT (codon 12) | GTT (codon 12) | CGT (codon 12) | GCT (codon 12) | AGT (codon 12) | TTT (codon 12) | GTTTGC (codons 12 and 13) | CTC (codon 13) | CAT (codon 61) | Wild type | |
|---|---|---|---|---|---|---|---|---|---|---|
| Microdissected medullary carcinomas (n = 13) | 2 | 0 | 1 | 0 | 0 | 1* | 0 | 0 | 0 | 9 |
| Xenografted medullary carcinomas (n = 5) | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 3 |
| Microdissected non-medullary carcinomas (n = 5) | 4 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Xenografted non-medullary carcinomas (n = 72) | 33 | 22 | 9 | 0 | 1 | 0 | 1† | 1 | 1 | 4 |
| Total (n = 95) | 40 | 22 | 11 | 1 | 1 | 1 | 1 | 1 | 1 | 16 |
*One microdissected medullary carcinoma contained a dinucleotide mutation (GGT to TTT) at codon 12.
†One xenografted nonmedullary carcinoma contained a di-codon mutation of one allele, comprising a GTT mutation in codon 12 and a TGC mutation in codon 13.
Figure 3.

K-ras sequencing of codon 12 of a medullary carcinoma (A) and a poorly differentiated conventional adenocarcinoma (B). The medullary carcinoma has a wild-type K-ras gene, but the conventional adenocarcinoma has a GGT (gly) to GAT (asp) mutation.
Prevalence of MSI
Each of the 18 microdissected pancreatic carcinomas also produced amplifiable BAT 25, BAT 26, and TGFB2 receptor gene products. One (7%) of the 13 microdissected medullary carcinomas had MSI. This carcinoma had shifts with both BAT 25 and BAT 26 markers and had only nine (versus the normal 10) thymidines in the polythymidine tract of its TGFBR2 receptor gene. None of the other 17 microdissected carcinomas had shifts with any of the three markers. Also, three of the five previously xenografted medullary carcinomas originally reported by Goggins et al 4 were MSI. If we combine these two series of patients, a total of 4 (22%) of 18 xenografted and microdissected medullary carcinomas had MSI.
This contrasts with a combined total of 77 carcinomas with nonmedullary histologies, wherein none had MSI. The difference in MSI frequency between medullary carcinomas and nonmedullary carcinomas was significant (P = 0.001). Table 2 ▶ summarizes these microsatellite data. Figure 4 ▶ illustrates the BAT 25 and BAT 26 PCR products of several tumors, including the microsatellite unstable tumor described in this study.
Figure 4.

BAT 25 and BAT 26 PCR products for two medullary carcinomas, a conventional adenocarcinoma, and normal pancreas. For both the BAT 25 and BAT 26 PCR products: lane 1, pancreatic medullary carcinoma with a microsatellite shift; lane 2, normal pancreas from the same patient; lane 3, a different pancreatic medullary carcinoma without a shift; and lane 4, a conventional ductal adenocarcinoma without a shift. The shifting medullary carcinoma and the normal pancreas are both from the patient with synchronous pancreatic and colonic carcinomas.
Immunohistochemistry for Mlh1 and Msh2
Immunohistochemistry for MLH1 and MSH2 gene products was performed on each of the 18 cases from this study and on the three MSI cases from the previous study of Goggins et al. 4 All four of the MSI cases from these two studies had absent Mlh1 expression and intact Msh2 expression (Figure 5) ▶ . Expression of both Mlh1 and Msh2 was intact in all of the MSS cases. Therefore, the sensitivity and specificity of immunohistochemistry for DNA repair gene enzymes, in the identification of the genetically proven MSI tumors, were both 100%.
Figure 5.
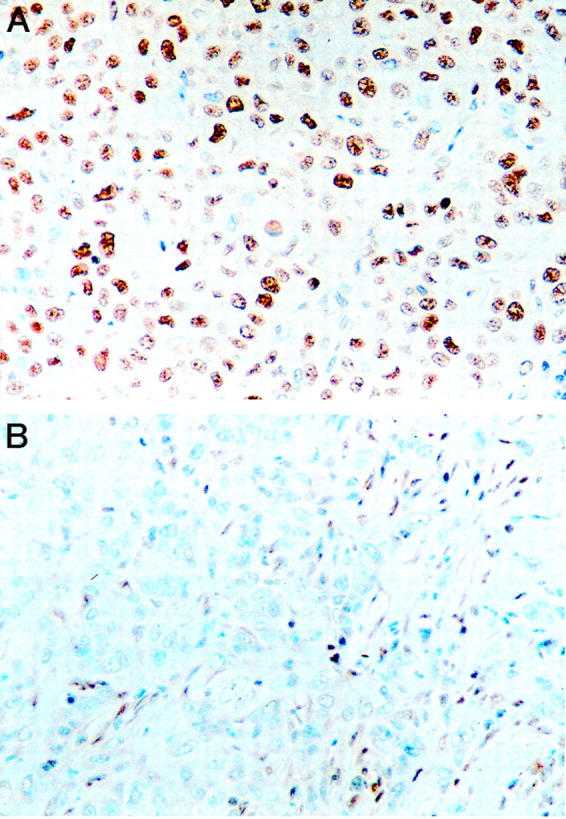
Immunohistochemistry for DNA repair-gene products in medullary carcinomas of the pancreas. A: This medullary carcinoma has intact nuclear labeling for the MSH2 gene product. B: Another medullary carcinoma has no expression of Mlh1. Note that the nuclei of stromal cells do express Mlh1.
Features of the Newly Identified MSI Case
The microdissected MSI carcinoma with no Mlh1 expression in the current series originated in a 34-year-old patient with synchronous carcinomas of the pancreas and colon. Three lines of evidence suggest that these two carcinomas represented two independent primaries. First, the pancreatic carcinoma had a medullary pattern with focal microglandular features, whereas the colonic tumor was, at least partially, frankly gland-forming. Second, the gross appearance of both tumors was typical of a primary carcinoma. Third, whereas the pancreatic primary was associated with multiple lymph node metastases to peripancreatic nodes, the colonic carcinoma had no lymph node metastases to surrounding mesenteric nodes. In addition, at the time of the operation to remove both primaries, no liver metastases were clinically present. Figure 6 ▶ compares the histology of both carcinomas.
Figure 6.
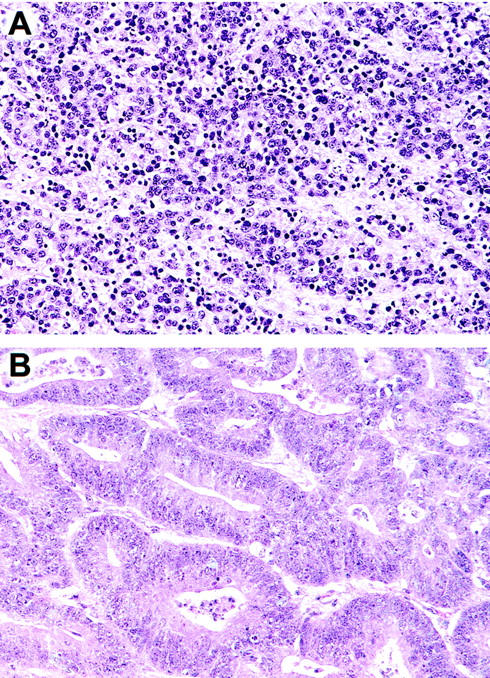
Histology of the Mlh1-deficient pancreatic (A) and colonic (B) carcinomas from the patient presumed to have HNPCC. Both carcinomas had MSI, and both had wild-type K-ras genes. Note that the histological patterns of the two cases are different: the pancreas has the medullary phenotype, whereas the colon cancer is gland-forming.
This 34-year-old patient with simultaneous carcinomas of the pancreas and colon met the Bethesda criteria for hereditary nonpolyposis colorectal carcinoma (HNPCC) syndrome. 36,37 He therefore underwent clinical genetic counseling, and informed consent was obtained for microsatellite testing of his colonic carcinoma by a commercial laboratory (Thomas Jefferson University, Philadelphia, PA). MSI was identified. Our laboratory subsequently re-evaluated and compared the MSI status of both the pancreatic and the colonic carcinomas; nonidentical shifts of the BAT 25, BAT 26, and the TGFBR2 gene markers were found. Both carcinomas also had wild-type K-ras genes.
Because both carcinomas were MSI, informed consent was also obtained for germline analysis of MLH1. Again, a commercial laboratory (University of Pennsylvania, Philadelphia, PA) performed this assay. Conformation-specific gel electrophoresis did not reveal an alteration in the germline copies of this patient’s MLH1 gene. 25-28 The patient and his family have undergone genetic counseling at The Johns Hopkins Oncology Center.
Analysis of Medullary Phenotype and Clinicopathological Variables
Multiple clinical, pathological, and genetic variables were analyzed for associations with the medullary phenotype. These analyses were performed using the combined total of 18 medullary carcinomas identified in this study and in the previous study of Goggins et al. 4 The medullary carcinomas were compared to the 77 well-characterized nonmedullary carcinomas (Table 2) ▶ .
The only variables significantly associated with the medullary phenotype were a family history of any cancer (excluding basal cell and squamous cell of skin) in a first-degree relative (P = 0.0004), MSI (P = 0.0010), and a wild-type K-ras gene (P < 0.0001). Table 4 ▶ summarizes these data.
Table 4.
Associations between Medullary Phenotype and Pathological, Genetic, and Clinical Parameters
| Factor | No. of medullary carcinomas | No. of nonmedullary carcinomas | P value |
|---|---|---|---|
| Family history of cancer | 0.0004 | ||
| Yes | 13 (93%) | 29 (42%) | |
| No | 1 (7%) | 40 (58%) | |
| K-ras gene | <0.0001 | ||
| Mutant | 6 (33%) | 73 (95%) | |
| Wild type | 12 (67%) | 4 (5%) | |
| MMR status | 0.0010 | ||
| MSI | 4 (22%) | 0 (0%) | |
| MSS | 14 (78%) | 77 (100%) | |
| Race | >0.1 | ||
| White | 17 (94%) | 70 (91%) | |
| Other* | 1 (6%) | 7 (9%) | |
| Gender | >0.1 | ||
| Male | 12 (67%) | 40 (52%) | |
| Female | 6 (33%) | 37 (48%) | |
| Smoking history | >0.1 | ||
| Yes | 9 (50%) | 32 (42%) | |
| No | 9 (50%) | 45 (58%) | |
| Alcohol history | >0.1 | ||
| Yes | 4 (22%) | 18 (24%) | |
| No | 14 (78%) | 57 (76%) | |
| Nodal metastases | >0.1 | ||
| Yes | 15 (83%) | 59 (77%) | |
| No | 3 (17%) | 18 (23%) | |
| Margin | >0.1 | ||
| Positive | 7 (39%) | 22 (29%) | |
| Negative | 11 (61%) | 55 (71%) | |
| Age | >0.1 | ||
| Over 60 | 13 (72%) | 55 (71%) | |
| Under 60 | 5 (28%) | 22 (29%) | |
| Tumor size | >0.1 | ||
| Over 3.5 cm | 10 (56%) | 40 (52%) | |
| Under 3.5 cm | 8 (44%) | 37 (48%) |
Statistics in this table were analyzed using Fisher exact tests.
*Other includes African-Americans and patients of Hispanic, Asian, and Arabic descent.
Abbreviations: MMR, mismatch repair; MSI, microsatellite unstable; MSS, microsatellite stable.
A variety of cancer types was seen in first-degree relatives of patients with pancreatic medullary carcinomas. The most common cancer in first-degree relatives was cancer primary to the breast (n = 5). Other cancers seen in family members included lung (n = 4), colon (n = 3), melanoma (n = 2), prostate (n = 2), pancreas (n = 1), liver (n = 1), and kidney cancers (n = 1). In two families the primary site of cancer in a first-degree relative was unknown.
We also examined the National Familial Pancreas Tumor Registry at The Johns Hopkins Hospital to determine whether any members of families with hereditary pancreas cancer syndromes had medullary cancers. These syndromes included the breast/pancreas cancer syndrome (associated with mutations in BRCA2), 38,39 familial atypical mole-malignant melanoma syndrome (associated with mutations in p16), 40 and Peutz-Jeghers syndrome (associated with mutations in STK11/LKB1). 41 No cases of medullary carcinoma were found among patients with these hereditary syndromes. 42
The presence of the medullary phenotype, a mutated K-ras gene, or MSI did not impact survival significantly. Overall 2- and 5-year survival for the 18 patients with medullary carcinoma was 29% and 13%, respectively.
Discussion
Five significant conclusions about medullary carcinomas can be drawn from these studies. First, pancreatic medullary carcinomas have a genetic profile different from that of conventional pancreatic ductal adenocarcinomas. Medullary carcinomas usually have K-ras wild-type genes and sometimes are microsatellite unstable. When combined with the previous study of Goggins et al, 4 12 (67%) of 18 medullary carcinomas reported to date have wild-type K-ras genes, and four (22%) of 18 have MSI. In contrast, conventional pancreatic ductal adenocarcinomas without the medullary phenotype almost always harbor K-ras gene mutations, and they virtually never have MSI. 4,9-13
The difference in MSI prevalence between the original study by Goggins et al 4 and the current study might be explained by the difference in study designs. The original study was a retrospective study in which a separate set of xenografted carcinomas having MSI were identified and subsequently studied histologically. In contrast, the present study represents a prospective analysis of the genetic changes within 13 cases specifically selected for their medullary histological phenotypes.
Second, this present study demonstrates that immunohistochemistry for DNA repair enzymes can identify patients whose medullary pancreatic carcinomas have MSI. Therefore, it may be reasonable to use immunohistochemistry for Mlh1 and Msh2 as a first step to screen medullary pancreatic carcinomas for MSI. Those that have a loss of labeling could then be tested for MSI using microsatellite markers.
Third, medullary carcinomas represent the first instance in which pancreatic tumor histology may be used to identify an inherited susceptibility to cancer. This inherited susceptibility may occur in two ways. For example, we found that the medullary phenotype correlates significantly with family history of any cancer in a first-degree relative. Therefore, where appropriate, the identification of a medullary carcinoma of the pancreas should spur investigation of the cancer incidence among a patient’s relatives. Also, one of the patients included in this study had synchronous MSI pancreatic and colonic cancers, suggesting that he has HNPCC. Indeed, Lynch et al 43 reported pancreatic cancer in some HNPCC kindreds. Although the patient in the present study met the clinical criteria for testing for HNPCC, conformation-specific gel electrophoresis studies have yet failed to identify a MLH1 germline mutation. Conformation-specific gel electrophoresis, however, is not 100% sensitive, and this patient may have an unidentified germline MLH1 mutation 25-28,44,45 . Sequencing of the entire MLH1 gene and Southern blot analysis would presumably answer this question, but the patient has not requested such studies.
Fourth, medullary carcinomas may represent a subset of ductal adenocarcinomas, rather than a distinct tumor class. This classification is entertained because PanINs are found in nonneoplastic pancreas surrounding almost half of the medullary carcinomas. PanINs are believed to be the precursors to pancreatic ductal adenocarcinoma, 14-16 and their presence in tissue surrounding medullary carcinomas suggests that medullary carcinomas may be derived from these precursor lesions. However, no high-grade (and presumably more advanced) PanINs were found near any of the medullary carcinomas. Nor does the mere presence of such precursor lesions necessarily denote that the medullary carcinomas arose from these PanINs.
Fifth, we report here the first case of a pancreatic carcinoma with latent EBV, as demonstrated by in situ hybridization. This medullary carcinoma had lymphoepithelioma-like features, with infiltration of the neoplasm by large numbers of lymphocytes. This finding is not surprising, in that latent EBV infection is common in lymphomas, nasopharyngeal lymphoepitheliomas, and lymphoepithelioma-like neoplasms in other sites, such as the stomach. 46-48 Therefore, medullary carcinomas may comprise two distinct subsets: those associated with and those not associated with EBV infection. A larger number of nonmedullary carcinomas will need to be studied to determine whether EBV infection also is associated with a minority of nonmedullary carcinomas as well.
In summary, we conclude that pathologists should distinguish medullary carcinomas from conventional ductal adenocarcinoma for two reasons. First, carcinomas with the medullary phenotype may have a distinct pathogenesis. Only by separating medullary carcinomas from conventional ductal adenocarcinomas will we be able to further define both the genetic changes and the role of EBV in these cancers. Second, medullary carcinomas can be a key clinical clue to the presence of an inherited cancer syndrome, including HNPCC. Further study of this familial association is justified. In the appropriate clinical setting, immunohistochemical studies, microsatellite analysis, germline testing, and genetic counseling may be warranted. In this scenario, immunohistochemical labeling for Mlh1 and Msh2 may help identify MSI in medullary carcinomas and characterize the specific underlying gene defect. More study will also be needed to see if the MSI subset of medullary carcinomas predicts a better survival, as to this date the number of cases is too small to draw conclusions regarding prognosis.
Acknowledgments
The authors would like to thank Jennifer Galford for her hard work and dedication and Drs. Zondervan, Drillenburg, and Nyhuis for providing specimens used in this study.
Footnotes
Address reprint requests to Scott E. Kern, M.D., The Johns Hopkins University School of Medicine, 1650 Orleans Street, 451 Cancer Research Building, Baltimore, MD 21231. E-mail: sk@jhmi.edu.
Supported by the National Institutes of Health Specialized Program in Research Excellence (SPORE) in gastrointestinal cancer (CA62924), the Public Health Service (CA67751–03), the Helen S. Heller and Daniel Kim memorial funds for pancreas cancer research, and The Netherlands Organization for Scientific Research (NWO) (950–10-625).
References
- 1.Hruban RH, Wilentz RE: The pancreas. Modern Surgical Pathology. Edited by N Weidner, RJ Cote, S Suster, LM Weiss. Philadelphia, Saunders, 2000
- 2.Solcia E, Capella C, Klöppel G: Tumors of the pancreas. Atlas of Tumor Pathology, Third Series, Fasicle 20. 1997, Armed Forces Institute of Pathology, Washington, DC,
- 3.Cubilla A, Fitzgerald PJ: Morphological patterns of primary nonendocrine human pancreas carcinoma. Cancer Res 1975, 35:2234-2248 [PubMed] [Google Scholar]
- 4.Goggins M, Offerhaus GJ, Hilgers W, Griffin CA, Shekher M, Tang D, Sohn TA, Yeo CJ, Kern SE, Hruban RH: Pancreatic adenocarcinomas with DNA replication errors (RER+) are associated with wild-type K-ras and characteristic histopathology. Poor differentiation, a syncytial growth pattern, and pushing borders suggest RER+. Am J Pathol 1998, 152:1501-1507 [PMC free article] [PubMed] [Google Scholar]
- 5.Lynch HT, Smyrk TC, Lynch JF: Overview of natural history, pathology, molecular genetics, and management of HNPCC (Lynch syndrome). Int J Cancer 1996, 69:38-43 [DOI] [PubMed] [Google Scholar]
- 6.Lothe RA, Peltomaki P, Meling GI, Aaltonen LA, Nystrom-Lahti M, Pylkkanen L, Heimdai K, Andersen TI, Moller P, Rognum TO, Fossa SD, Haldorsen TI, Langmark F, Brogger A, de la Chapelle A, Borresen A-L: Genomic instability in colorectal cancer: relationship of clinicopathological variables and family history. Cancer Res 1993, 53:5849-5852 [PubMed] [Google Scholar]
- 7.Jass JR, Smyrk TC, Stewart SM, Lane MR, Lanspa SJ, Lynch HT: Pathology of hereditary non-polyposis colon cancer. Anticancer Res 1994, 14:1631-1634 [PubMed] [Google Scholar]
- 8.Graham DM, Appelman HD: Crohn’s-like lymphoid reaction and colorectal carcinoma: a potential histologic prognosticator. Mod Pathol 1990, 3:332-335 [PubMed] [Google Scholar]
- 9.Hruban RH, van Mansfeld ADM, Offerhaus GJ, van Weering DHJ, Allison DC, Goodman SN, Kensler TW, Bose KK, Cameron JL, Bos JL: K-ras oncogene activation in adenocarcinoma of the human pancreas: a study of 82 carcinomas using a combination of mutant-enriched polymerase chain reaction analysis and allele-specific oligonucleotide hybridization. Am J Pathol 1993, 143:545-554 [PMC free article] [PubMed] [Google Scholar]
- 10.Almoguera C, Shibata D, Forrester K, Martin J, Arnheim N, Perucho M: Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell 1998, 53:549-554 [DOI] [PubMed] [Google Scholar]
- 11.Smit VT, Boot AJM, Smits AMM, Fleuren GJ, Cornelisse CJ, Bos JL: K-ras codon 12 mutations occur very frequently in pancreatic adenocarcinomas. Nucleic Acids Res 1988, 16:7773-7782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barbacid M: Ras genes. Annu Rev Biochem 1987, 56:779-827 [DOI] [PubMed] [Google Scholar]
- 13.Grünewald K, Lyons J, Frohlich A, Feichtinger H, Weger RA, Schwab G, Janssen JW, Bartram CR: High frequency of Ki-ras codon 12 mutations in pancreatic adenocarcinomas. Int J Cancer 1989, 43:1037-1041 [DOI] [PubMed] [Google Scholar]
- 14.Hruban RH, Wilentz RE, Goggins M, Offerhaus GJA, Yeo CJ, Kern SE: Pathology of incipient pancreatic cancer. Ann Oncol 1999, 10(Suppl):9-11 [PubMed] [Google Scholar]
- 15.Wilentz RE, Geradts J, Maynard R, Offerhaus GJA, Kang M, Goggins M, Yeo CJ, Kern SE, Hruban RH: Inactivation of the p16 (INK4A) tumor-suppressor gene in pancreatic duct lesions: loss of intranuclear expression. Cancer Res 1998, 58:4740-4744 [PubMed] [Google Scholar]
- 16.Wilentz RE, Iacobuzio-Donahue CA, Argani P, McCarthy DM, Parsons JL, Yeo CJ, Kern SE, Hruban RH: Loss of expression of Dpc4 in pancreatic intraepithelial neoplasia (PanIN): evidence that DPC4 inactivation occurs late in neoplastic progression. Cancer Res (in press) 2000 [PubMed]
- 17.Slebos RJC, Boerrigter L, Evers SG, Wisman P, Mooi WJ, Rodenhuis S: A rapid and simple procedure for the detection of ras mutations in formalin-fixed, paraffin-embedded tissue. Diagn Mol Pathol 1992, 1:136-141 [PubMed] [Google Scholar]
- 18.Wilentz RE, Kern SE: No H-ras gene mutations in pancreatic adenocarcinomas. NOGO 1999, 3:22 [Google Scholar]
- 19.Wilentz RE, Kern SE: No N-ras gene mutations in pancreatic adenocarcinomas. NOGO 1999, 3:23 [Google Scholar]
- 20.Hahn SA, Seymour AB, Hoque ATMS, Schutte M, da Costa LT, Redston MS, Caldas C, Weinstein CL, Fischer A, Yeo CJ, Hruban RH, Kern SE: Allelotype of pancreatic carcinoma using xenograft enrichment. Cancer Res 1995, 55:4670-4675 [PubMed] [Google Scholar]
- 21.Goggins M, Shekher M, Turnacioglu K, Yeo CJ, Hruban RH, Kern SE: Genetic alterations of the transforming growth factor beta receptor genes in pancreatic and biliary adenocarcinomas. Cancer Res 1998, 58:5329-5332 [PubMed] [Google Scholar]
- 22.Boland CR, Thibodeau SN, Hamilton SR, Sidransky D, Eshleman JR, Burt RW, Meltzer SJ, Rodriguez-Bigas MA, Fodde R, Ranzani GN, Srivastava S: A National Cancer Institute workshop on microsatellite instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res 1998, 58:5248-5257 [PubMed] [Google Scholar]
- 23.Perucho M: Correspondence. Cancer Res 1999, 59:249-255 [PubMed] [Google Scholar]
- 24.Marcus VA, Madlensky L, Gryfe R, et al: Immunohistochemistry for hMLH1 and hMLH2: a practical test for DNA mismatch repair deficient tumors. Am J Surg Pathol 1999, 23:1248-1255 [DOI] [PubMed] [Google Scholar]
- 25.Ganguly A, Williams C: Detection of mutations in multi-exon genes: comparison of conformation sensitive gel electrophoresis and sequencing strategies with respect to cost and time for finding mutations. Hum Mutat 1997, 9:339-343 [DOI] [PubMed] [Google Scholar]
- 26.Ganguly A, Prockop DJ: Detection of mismatched bases in double stranded DNA by gel electrophoresis. Electrophoresis 1995, 16:1830-1835 [DOI] [PubMed] [Google Scholar]
- 27.Ganguly A, Rock MJ, Prockop DJ: Conformation-sensitive gel electrophoresis for rapid detection of single-base differences in double-stranded PCR products and DNA fragments: evidence for solvent-induced bends in DNA heteroduplexes. Proc Natl Acad Sci USA 1993, 90:10325-10329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Korkko J, Annunen S, Pihlajamaa T, Prockop DJ, Ala-Kokko L: Conformation sensitive gel electrophoresis for simple and accurate detection of mutations: comparison with denaturing gradient gel electrophoresis and nucleotide sequencing. Proc Natl Acad Sci USA 1998, 95:1681-1685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kaplan EL, Meier P: Nonparametric estimation from incomplete observations. Am Stat Assoc J 1958, 53:457-480 [Google Scholar]
- 30.Mantel N, Haenszel W: Statistical aspects of the analysis of data from retrospective studies of disease. J Natl Cancer Inst 1959, 22:719-748 [PubMed] [Google Scholar]
- 31.Cox DR: Regression models and life tables (with discussion). J Roy Statist Soc (B) 1972, 34:187-220 [Google Scholar]
- 32.: SAS Institute I: SAS user’s guide: statistics, version 5 edition. 1985. SAS Institute, Inc., Cary,
- 33.Kawabata Y, Tomita N, Monden T, Ohue M, Ohnishi T, Sasaki M, Sekimoto M, Sakita I, Tamaki Y, Takahashi J, Yagyu T, Mishima H, Kikkawa N, Monden M: Molecular characteristics of poorly differentiated adenocarcinoma and signet-ring-cell carcinoma of colorectum. Int J Cancer 1999, 84A:33-38 [DOI] [PubMed] [Google Scholar]
- 34.: Statistics and Epidemiology Research Corporation: EGRET users’ manual. 1988. Statistics and Epidemiology Research Corporation, Seattle:
- 35.Goelz SE, Hamilton SR, Vogelstein B: Purification of DNA from formaldehyde-fixed and paraffin-embedded human tissue. Biochem Biophys Res Commun 1985, 130:118-126 [DOI] [PubMed] [Google Scholar]
- 36.Rodriguez-Bigas MA, Boland CR, Hamilton SR: A National Cancer Institute workshop on hereditary nonpolyposis colorectal cancer syndrome: meeting highlights and Bethesda guidelines. J Natl Cancer Inst 1997, 89:1758-1762 [DOI] [PubMed] [Google Scholar]
- 37.Syngal S, Fox EA, Li C, Dovidio M, Eng C, Kolodner RD, Garber JE: Interpretation of genetic test results for hereditary nonpolyposis colorectal cancer: implications for clinical predisposition testing. JAMA 1999, 282:247-253 [DOI] [PubMed] [Google Scholar]
- 38.Goggins M, Schutte M, Lu J, Moskaluk CA, Weinstein CL, Petersen GM, Yeo CJ, Jackson CE, Lynch HT, Hruban RH, Kern SE: Germline BRCA2 gene mutations in patients with apparently sporadic pancreatic carcinomas. Cancer Res 1996, 56:5360-5364 [PubMed] [Google Scholar]
- 39.Goggins M, Hruban RH, Kern SE: The late temporal pattern of BRCA2 inactivation in pancreatic intraepithelial neoplasia. Am J Pathol 2000, 156:1767-1771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bergman W, Watson P, de Jong J, Lynch HT, Fusaro RM: Systemic cancer and the FAMMM syndrome. Br J Cancer 1990, 61:932-936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Su GH, Hruban RH, Bova GS, Goggins M, Bansal RK, Tang DT, Shekher MC, Westerman A-M, Entius MM, Yeo CJ, Kern SE: Germline and somatic mutations of the STK11/LKB1 Peutz-Jeghers gene in pancreatic and biliary cancers. Am J Pathol 1999, 154:1835-1840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hruban RH, Petersen GM, Ha PK, Kern SE: Genetics of pancreatic cancer: from genes to families. Surg Oncol Clin North Am 1998, 7:1-23 [PubMed] [Google Scholar]
- 43.Lynch HT, Voorhees GJ, Lanspa SJ, McGreevy PS, Lynch JF: Pancreatic carcinoma and hereditary non-polyposis colorectal carcinoma: a family study. Br J Cancer 1985, 52:271-273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Thibodeau SN, French AJ, Roche PC, Cunningham JM, Tester DJ, Lindor NM, Moslein G, Baler SM, Liskay RM, Burgart LJ, Honchel R, Halling KC: Altered expression of hMSH2 and hMLH1 in tumors with microsatellite instability and genetic alterations in mismatch repair genes. Cancer Res 1996, 56:4836-4840 [PubMed] [Google Scholar]
- 45.Curia MC, Palmirotta R, Aceto G, Messerini L, Veri MC, Crognale S, Valanzano R, Ficari F, Fracasso P, Stigliano V, Tonelli F, Casale V, Guadagni F, Battista P, Mariani-Costantini R, Cama A: Unbalanced germ-line expression of hMLH1 and hMSH2 alleles in hereditary nonpolyposis colorectal cancer. Cancer Res 1999, 59:3570-3575 [PubMed] [Google Scholar]
- 46.Ambinder RF, Mann RB: Epstein-Barr-encoded RNA in situ hybridization: diagnostic applications. Hum Pathol 1994, 25:602-605 [DOI] [PubMed] [Google Scholar]
- 47.Ambinder RF, Mann RB: Detection and characterization of Epstein-Barr virus in clinical specimens. Am J Pathol 1994, 145:239-252 [PMC free article] [PubMed] [Google Scholar]
- 48.Herrera-Goepfert R, Reyes E, Hernandez-Avila M, Mohar A, Shinkura R, Fujiyama C, Akiba S, Eizuru Y, Harada Y, Tokunaga M: Epstein-Barr virus-associated gastric carcinoma in Mexico: analysis of 135 consecutive gastrectomies in two hospitals. Mod Pathol 1999, 12:873-878 [PubMed] [Google Scholar]