

Abstract
Miscarriage and pre-eclampsia are the most common disorders of human pregnancy. Both are placental-related and exceptional in other mammalian species. Ultrasound imaging has enabled events during early pregnancy to be visualized in vivo for the first time. As a result, a new understanding of the early materno–fetal relationship has emerged and, with it, new insight into the pathogenesis of these disorders. Unifying the two is the concept of placental oxidative stress, with associated necrosis and apoptosis of the trophoblastic epithelium of the placental villous tree. In normal pregnancies, the earliest stages of development take place in a low oxygen (O2) environment. This physiological hypoxia of the early gestational sac protects the developing fetus against the deleterious and teratogenic effects of O2 free radicals (OFRs). In miscarriage, development of the placento–decidual interface is severely impaired leading to early and widespread onset of maternal blood flow and major oxidative degeneration. This mechanism is common to all miscarriages, with the time at which it occurs in the first trimester depending on the aetiology. In contrast, in pre-eclampsia the trophoblastic invasion is sufficient to allow early pregnancy phases of placentation but too shallow for complete transformation of the arterial utero–placental circulation, predisposing to a repetitive ischaemia–reperfusion (I/R) phenomenon. We suggest that pre-eclampsia is a three-stage disorder with the primary pathology being an excessive or atypical maternal immune response. This would impair the placentation process leading to chronic oxidative stress in the placenta and finally to diffuse maternal endothelial cell dysfunction.
Keywords: fetus, miscarriage, oxygen, placenta, pre-eclampsia
Introduction
Placental-related disorders of pregnancy are almost unique to the human species. These disorders, which affect around a third of human pregnancies, primarily include miscarriage and pre-eclampsia. In other mammalian species, the incidence of both disorders is extremely low. Although epidemiological data on animals living in the wild, such as monkeys, are limited, laboratory rodents are known to have post-implantation pregnancy loss rates of less than 10% (Wilmut et al., 1986), and spontaneous pre-eclampsia has been exceptionally described only in ‘patas’ monkeys (Palmer et al., 1979) and guinea pigs (Seidl et al., 1979). In humans, both disorders have been known to medical science since ancient times with clear descriptions recorded in ancient Egypt and Greece (Kuller and Katz, 1994; Pukerson and Vekerdy, 1999). Recent changes in human lifestyle, such as delayed childbirth and hypercaloric diets, may have increased the global incidence of placental-related disorders over the last decades (Dietl, 2005). However, the fact that populations of hunter-gatherers still in existence today are affected by these pregnancy complications (Roy, 2003) suggests that they are not a direct consequence of modern human lifestyle but are more likely because of an evolutionary step in human reproduction and development.
There is mounting evidence that oxidative stress or an imbalance in the oxidant/antioxidant activity in utero–placental tissues plays a pivotal role in the development of placental-related diseases. We have reviewed the experimental and clinical data and the mechanisms involved in the alterations of oxygen (O2) homeostasis and evaluated the consequences for human pregnancy.
Environmental O2 and mammalian evolution
Evolution for mammals living on dry land has been closely linked to adaptation to changes in O2 concentration in the environment (Burton et al., 2003). Life on earth evolved under anaerobic conditions, with metabolic pathways centred on nitrogen, sulphur and carbon. O2 was initially released as a by-product of photosynthesis following the emergence of blue-green algae, and as atmospheric concentrations increased, it became possible to support more complex, multicellular life forms, including placental mammals (Falkowski et al., 2005). O2 enables more efficient energy transformation from dietary proteins, carbohydrates and fats to ATP. ATP molecules provide the chemical energy required to conduct the biochemical reactions essential to cellular life including protein biosynthesis, active transport of molecules across cellular membranes and muscular contraction (Burton et al., 2003). Most of the O2 used during the oxidation of dietary organic molecules is converted into water via the combined action of the enzymes of the respiratory chain. Around 1–2% of the O2 consumed escapes this process and is diverted into highly reactive O2 free radicals (OFRs) and other reactive O2 species (ROS) at a rate dependent on the prevailing O2 tension (Halliwell and Gutteridge, 1999). When the production of OFRs exceeds the natural cellular protection, indiscriminate damage can occur to proteins, lipids and DNA.
The placenta in mammals is the essential interface between the maternal circulation carrying O2-rich blood and nutrients and the fetal circulation. In the past, it has been assumed that the principal function of the organ is to supply the fetus with as much O2 as possible, and to a large extent that is the case in the second half of pregnancy when fetal weight gain is greatest. Our recent combined in vivo and in vitro investigations have resulted in a new understanding of the materno–fetal relationship during the first trimester of pregnancy and led to the hypothesis that the placenta limits, rather than facilitates, O2 supply to the fetus during the period of organogenesis (Jauniaux et al., 2003a). The earliest stages of development therefore take place in a low O2 environment, reflecting to some extent the evolutionary path (Figure 1). In most species, organogenesis is complete and development is very advanced before placental attachment. However, in the human placenta, development is precocious, and the conceptus is fully embedded in the uterine wall before the primitive streak has formed. Consequently, other strategies need to be employed to restrict exposure of the fetus to O2, and as a result, the human placenta is exposed to major changes in O2 concentration from conception to delivery (Jauniaux et al., 2000, 2001; Burton et al., 2003). In normal pregnancies, this is a very well controlled phenomenon that has to provide a delicate balance between the metabolic needs of the fetus and its placenta and the potential danger of OFRs. Embryonic and placental cells are sensitive to oxidative stress because of their extensive cell divisions and the concomitant exposure of their DNA (Burton et al., 2003). The placental syncytiotrophoblast is particularly sensitive, partly because it is the outermost tissue of the conceptus and so exposed to the highest concentrations of O2 coming from the mother, and partly because it contains surprisingly low concentrations of the principal antioxidant enzymes, particularly in early pregnancy (Watson et al., 1998). Thus, it is well established that maternal metabolic disorders such as diabetes, which are associated with an increased generation of OFRs, are known to be associated with a higher incidence of miscarriages, vasculopathy and fetal structural defects (Burton et al., 2003; Jawerbaum and Gonzales, 2005), indicating that the mammalian conceptus can be irreversibly damaged by oxidative stress.
Figure 1.

Diagram showing the oxygen (O2) delivery system in the adult body (left) to ensure that cells are not exposed to the full atmospheric concentration and excessive oxidative stress; plugging of the maternal spiral arteries and the presence of the large exocoelomic cavity (right), devoid of an O2 carrier, could serve as a similar protective mechanism for embryonic tissues during the first trimester.
Human early placentation
Human implantation is almost unique amongst mammals in that it is highly invasive and the conceptus embeds itself completely within the maternal uterine endometrium and superficial myometrium. The chorionic villi, the basic structures of the early placenta, form during the 4th and 5th weeks post-menstruation (Boyd and Hamilton, 1970) and surround the entire gestational sac until the 8th to 9th week of gestation. Between the 3rd and 4th month, the villi at the implantation site become elaborately branched and form the definitive placenta, whereas the villi on the opposite pole degenerate to form the placental membranes (Figure 2). Towards the end of pregnancy, the villi present a surface area of 12–14 m2, providing an extensive and intimate interface for materno–fetal exchange (Boyd and Hamilton, 1970).
Figure 2.

Diagram of a gestational sac at the end of the 2nd month showing the myometrium (M), the decidua (D), the placenta (P), the exo-coelomic cavity (ECC), the amniotic cavity (AC) and the secondary yolk sac (SYS).
The trophoblast lineage gives rise to three main cell types in the human placenta: (i) the syncytiotrophoblast which forms the epithelial covering of the villous tree and is the main endocrine component of the placenta, (ii) the villous cytotrophoblast cells which represent a germinative population that proliferate throughout pregnancy and fuse to generate the syncytiotrophoblast (iii) and the extravillous trophoblast cells which are non-proliferative and invade the maternal endometrium (Kaufmann and Burton, 1994). These extravillous trophoblasts can be first found both within and around the spiral arteries in the central area of the placenta. They gradually extend laterally, reaching the periphery of the placenta around mid-gestation (Figure 2). Depth-wise changes normally extend as far as the inner third of the uterine myometrium within the central region of the placental bed, but the extent of invasion is progressively shallower towards the periphery (Pijnenborg et al., 1980). Human placentation is also characterized by a remodelling of the spiral arteries during which the vessels lose their elastic lamina and smooth muscle, and consequently their responsiveness to circulating vasoactive compounds. In normal pregnancies, the transformation of spiral arteries into utero–placental arteries is completed around mid-gestation. The main aim of these vascular changes is to optimize the distribution of maternal blood into a low-resistance uterine vascular network and ultimately inside the placental intervillous chamber. At term, the utero–placental circulation carries approximately 600 ml of maternal blood per minute (Ramsey and Donner, 1980).
Recent anatomic and in vivo studies have shown that human placentation is in fact not truly haemochorial in early pregnancy. From the time of implantation, the extravillous trophoblast not only invades the uterine tissues but also forms a continuous shell at the level of the decidua (Figure 2). The cells of this shell anchor the placenta to the maternal tissue but also form plugs in the tips of the utero–placental arteries (Hustin and Schaaps, 1987; Burton et al., 1999). The shell and the plugs act like a labyrinthine interface that filters maternal blood, permitting a slow seepage of plasma, but no true blood flow, into the intervillous space. This is supplemented by secretions from the uterine glands, which are discharged into the intervillous space until at least 10 weeks (Burton et al., 2002). During that period, the placental villi display only a few capillaries and fetal erythrocytes are nucleated (Boyd and Hamilton, 1970), suggesting that the fetal blood is extremely viscous, and consequently the feto–placental blood flow is limited. Furthermore, during the first trimester, the villous membrane is twice the thickness it will be in the second, and the early placenta and fetus are separated by the exocoelomic cavity, which occupies most of the space inside the gestational sac (Figure 2) (Jauniaux and Gulbis, 2000).
At the end of the first trimester, the trophoblastic plugs are progressively dislocated, allowing maternal blood to flow progressively more freely and continuously within the intervillous space. During the transitional phase of 10–14 weeks gestation, two-thirds of the primitive placenta disappears, the exocoelomic cavity is obliterated by the growth of the amniotic sac and maternal blood flows progressively throughout the entire placenta (Jauniaux et al., 2003a). These events bring the maternal blood closer to the fetal tissues, facilitating nutrient and gaseous exchange between the maternal and fetal circulations.
Human placentation and oxidative stress
The comparison of morphological features with physiological data reveals that the architecture of the human first trimester gestational sac is designed to limit fetal exposure to O2 to that which is strictly necessary for its development (Jauniaux et al., 2003a). In vivo data have demonstrated that values for the intraplacental partial pressure of O2 (PO2) are two to three times lower at 8–10 weeks than after 12 weeks (Jauniaux et al., 2000, 2001). As pregnancy advances between 7 and 16 weeks, there is a progressive, but independent, increase in decidual PO2, which most probably reflects the increase in maternal blood flow volume within the uterine circulation during the first half of pregnancy. At 13–16 weeks, the PO2 in the fetal blood is still only 24 mmHg, whereas during the second half of pregnancy, umbilical venous values ranges between 35 and 55 mmHg. These are relatively low compared with the maternal circulation, suggesting a significant maternal to fetal O2 gradient throughout pregnancy. The gradual rise in intraplacental PO2 change observed between 8 and 14 weeks of gestation is accompanied by almost parallel increases in mRNA concentrations and the activity of the major antioxidant enzymes in the villous tissue (Jauniaux et al., 2000). The first trimester uterine O2 gradient exerts a regulatory effect on placental tissue development and function. In particular it influences cytotrophoblast proliferation and differentiation along the invasive pathway (Genbacev et al., 1997) and villous vasculogenesis (Charnock-Jones and Burton, 2000).
The physiological hypoxia of the first trimester gestational sac may protect the developing fetus against the deleterious and teratogenic effects of OFRs. Recent evidence also indicates that the hypoxia is necessary to maintain stem cells in a fully pluripotent state (Ezashi et al., 2005), for at physiological levels free radicals regulate a wide variety of cell functions, in particular transcription factors (Burton et al., 2003). Excessive production of OFRs results in oxidative stress, and there are two examples when this occurs physiologically during human pregnancy.
Firstly, at the end of the first trimester, a burst of oxidative stress is evidenced in the periphery of the early placenta (Jauniaux et al., 2000). The underlying utero–placental circulation in this area is never plugged by the trophoblastic shell allowing limited maternal blood flow to enter the placenta from 8 to 9 weeks of gestation (Figures 2 and 3). This leads to higher local O2 concentrations at a stage of pregnancy when the trophoblast possesses low concentrations and activities of the main antioxidant enzymes superoxide dismutase, catalase and glutathione peroxidase. Focal trophoblastic oxidative damage and progressive villous degeneration trigger the formation of the fetal membranes (Jauniaux et al., 2003b), which is an essential developmental step enabling vaginal delivery. The oxidative stress and rise in oxygenation may also stimulate the synthesis of various trophoblastic proteins, such as hCG and estrogens. Maternal serum concentrations of hCG peak towards the end of the first trimester, and oxidising conditions promote assembly of the subunits in vitro (Xing et al., 2001). Concentrations of hCG are further increased in cases of trisomy 21, where there is evidence of trophoblastic oxidative stress through an imbalance in expression of the antioxidant enzymes (Pidoux et al., 2004). Recently, it has been demonstrated that the enzyme P-450 cytochrome aromatase (CYP19) which is involved in the synthesis of estrogens is transcriptionally regulated by O2 (Mendelson et al., 2005), and this may account for the significant increase in production at the start of the second trimester.
Figure 3.
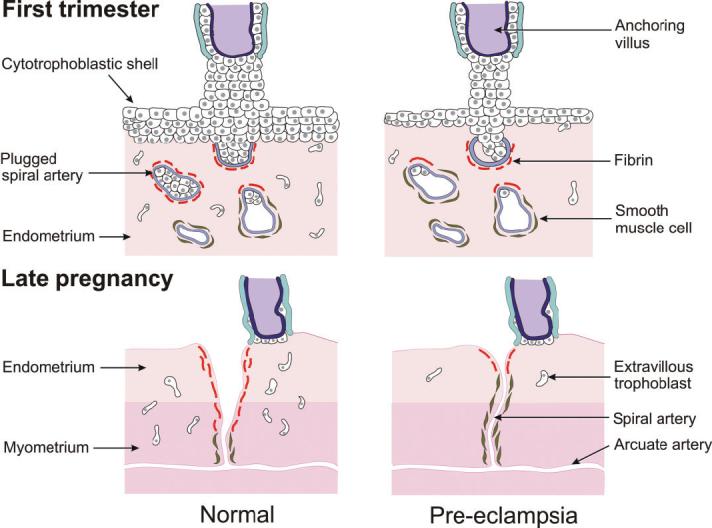
Diagram of the utero–placental interface in the first trimester and later in pregnancy showing the reduced cytotrophoblastic plugging and incomplete transformation of the spiral arteries in pregnancies complicated by pre-eclampsia.
The second example involves an ischaemia–reperfusion (I/R) phenomenon. Angiographic studies of the uterine vasculature of the rhesus monkey have demonstrated that during normal pregnancy, flow from spiral arteries into the intervillous space is often intermittent, arising from spontaneous vasoconstriction (Martin et al., 1964). Placental inflow may also be compromised by external compression of the arteries during uterine contractions in both the rhesus and human and even by postural changes (Ramsey and Donner, 1980). Some degree of I/R stimulus may therefore be a feature of normal human pregnancy, especially towards term when the fetus and placenta are extracting large quantities of O2 from the intervillous space (Hung et al., 2001). This chronic stimulus could lead to up-regulation of the anti-OFR defence in the placenta, thus reducing oxidant stress. As in early pregnancy, this well-controlled oxidative stress may play a role in continuous placental remodelling and essential placental functions such as transport and hormonal synthesis. Within this context, miscarriages and pre-eclampsia could be a temporary maladaptation to a changing O2 environment.
Miscarriages and oxidative stress
Early anatomical and histopathological studies have almost exclusively focused on abnormal villous development in early pregnancy failure (Boue et al., 1976). There is now clear evidence that miscarriages are a placentation disorder and that the villous changes described previously are the consequence, rather than the cause (Jauniaux and Burton, 2005). In about two-thirds of early pregnancy failures, there is anatomical evidence of defective placentation which is mainly characterized by a thinner and fragmented trophoblast shell, reduced cytotrophoblast invasion of the endometrium and incomplete plugging of the lumen at the tips of the spiral arteries (Hustin et al., 1990; Jauniaux et al., 1994). This is associated with the absence of physiological changes in most spiral arteries and leads to a premature onset of the maternal circulation throughout the entire placenta.
Independent of the cause of the miscarriage, the excessive entry of maternal blood into the intervillous space has two effects: (i) a direct mechanical effect on the villous tissue which becomes progressively enmeshed inside large intervillous blood thrombi (ii) and a widespread and indirect O2-mediated trophoblastic damage and increased apoptosis (Kokawa et al., 1998; Hempstock et al., 2003; Jauniaux et al., 2003b). Concentrations of lipid peroxides have also been shown to increase in the villous and decidual tissues of women undergoing early pregnancy loss (Nicol et al., 2000; Sugino et al., 2000). Overall, the consequences are placental degeneration with complete loss of syncytiotrophoblast function and detachment of the placenta from the uterine wall. This mechanism is common to all miscarriages, with the time at which it occurs in the first trimester depending on the aetiology.
Any factor causing abnormally high or rapidly fluctuating concentrations of O2 will have a harmful and rapid effect on the early villous tissue. We have proposed separation of the aetiologies of early pregnancy failure into primary and secondary causes of O2 stress (Hempstock et al., 2003). Primary causes can be clearly defined and include mainly chromosomal abnormalities which are found in at least 50% of miscarriages and are often associated with abnormal trophoblastic invasion of the uterine decidua (Hustin et al., 1990; Jauniaux and Burton, 2005). There is also increasing evidence showing an association between miscarriages and an anomaly of one of the enzymes involved in the metabolism of OFRs (Nicol et al., 2000; Sugino et al., 2000; Tempfer et al., 2001; Sata et al., 2003). These data support the concept that an early pregnancy failure could arise from a primary defect of placentation subsequent to a genetic anomaly of the enzymes or co-factors involved in the metabolism of O2.
Secondary causes are more complex and often multifactorial. For example, the role of maternal leukocytes and other immune factors such as cytokines in the trophoblast–decidual interaction remains unclear. There is evidence that the circulating cytokine levels and the cytokine profile in the decidua are different in women who experience recurrent miscarriages (Jenkins et al., 2000; Makhseed et al., 2000; von Wolff et al., 2000; Baxter et al., 2001), but the exact interaction of each of these cytokines with the invading trophoblast is not well defined. Some diseases such as maternal diabetes can generate OFRs in larger quantities than the early placental limited antioxidant defences can cope with (Burton et al., 2003), leading to DNA damage and oxidation of proteins and lipids, thus resulting in secondary trophoblast dysfunction. Although is has been shown that women with naturally higher levels of antioxidant enzymes are less likely to miscarry (Baxter et al., 2001), the impact of peri-conceptional antioxidant supplementation on early pregnancy failure rates in the general population remains to be investigated. Theoretically, major fetal abnormalities leading to fetal demise within the first 2 months of pregnancy could also lead to a secondary placental dysfunction as the placental development becomes increasingly dependent on fetal synthesis towards the end of the first trimester (Shurtz-Swirski et al., 1991). This may be modulated by tumour necrosis factor (TNF)-α, a multifunctional cytokine which has been identified in the maternal reproductive tract, the placenta and fetal tissues (Toder et al., 2003).
Pre-eclampsia and oxidative stress
Pre-eclampsia stems from a similar, though lesser, defect in early trophoblast invasion, for although invasion is sufficient to anchor the conceptus, it is insufficient to fully convert the spiral arteries into low-resistance channels (Robertson et al., 1985; Khong et al., 1986; Meekins et al., 1994). Incomplete conversion of the spiral arteries results in retention of smooth muscle cells within their walls (Figure 3), and so some vasoreactivity persists in 30–50% of the placental vascular bed. This may lead not only to diminished perfusion of the intervillous space but more importantly to intermittent perfusion. Because the placenta and fetus continually extract O2, it is expected that transient hypoxia will result (Hung et al., 2001) and that consequently the placenta suffers a chronic low-grade I/R type injury.
Increased O2 stress in the placenta of women with pre-eclampsia is well documented (Hubel et al., 1989; Wang et al., 1991; Uotila et al., 1993; Lorentzen et al., 1994; Walsh, 1994; Walsh and Wang, 1995; Myatt et al., 1996; Poranen et al., 1996; Wang and Walsh, 1998; Staff et al., 1999; Many et al., 2000; Wang and Walsh, 2001; Zusterzeel et al., 2001; Vanderlie et al., 2005). The disturbance in the oxidant–antioxidant balance renders the tissue more vulnerable to OFR injury. OFRs lead to the formation of lipid peroxides which alter cell membranes by increasing incorporation of cholesterol and oxidized free fatty acids (FFAs) and low-density lipoproteins (LDLs) (Hubel et al., 1989). Lipid peroxides are directly involved in mediating maternal endothelial dysfunction by increasing the production of thromboxane A2 and the expression of cell adhesion molecules in the utero–placental vasculature and also in the maternal peripheral vasculature (Uotila et al., 1993; Walsh and Wang, 1995; Myatt et al., 1996; Poranen et al., 1996; Wang and Walsh, 2001). Lipid peroxides are increased in the maternal circulation above non-pregnant levels even in normal pregnancy, indicating a certain degree of physiological oxidative stress (Walsh, 1994). FFAs are particularly susceptible to oxidation and are increased in pre-eclampsia, 15–20 weeks before the onset of the clinical disease (Lorentzen et al., 1994), and may contribute to the susceptibility to maternal oxidant stress.
In pre-eclampsia, antioxidant nutrients are presumed to be utilized to a greater extent to counteract OFRs, and this may provide an explanation for the repeated demonstration of reduced concentrations of ascorbic acid in the maternal circulation (Mikhail et al., 1994). This rapid decrease in antioxidant capacity would propagate lipid peroxidation leading to the biological cascade of leukocyte activation, platelet adhesion and aggregation and the consequent release of vasoconstriction agents. Although it is clear that maternal and placental antioxidant defences are reduced in pre-eclampsia, it is not known whether this is a primary or secondary event to excessive depletion through the increased generation of OFRs. Changes in some circulating markers of O2 stress precede the development of the clinical symptoms (Wang et al., 1991; Lorentzen et al., 1994; Mikhail et al., 1994; Chappell et al., 2002a; Scholl et al., 2005), suggesting a primary phenomenon of chronic oxidative stress. By contrast to miscarriage where there is rapid and generalized placental tissue degeneration, in pre-eclampsia the placental damage is progressive and can be compensated for sometime depending on the severity of initial placental defect and the intrinsic placental antioxidant capacity. This is supported by a clinical trial in women at risk of pre-eclampsia which has shown that antioxidant vitamin C and E supplementation during the second trimester of pregnancy improves biochemical incidences of oxidative stress and decreases the occurrence of the clinical disease in high-risk women (Chappell et al., 1999, 2002b).
It has been known for more than 100 years that during pregnancy trophoblastic cells are deported into the maternal venous circulation and that these may be detected peripherally; indeed they have been detected in the lung (Schmorl, 1883). Although this is not exclusively a human phenomenon, this so-called physiological desquamation of the villous syncytiotrophoblast is found in all human pregnancies. Normal placentation is associated with a certain degree of trophoblastic necrosis and apoptosis with shedding of placental debris in the maternal circulation. Apoptotic or necrotic cellular debris could constitute the basis of the inflammatory stimulus in all pregnancies and is found to increase with advancing gestation (Redman and Sargent, 2000). This model could explain some features of pre-eclampsia including its development in multiple pregnancies or in small oxidatively stressed placentae (Redman et al., 1999).
There is little doubt that the pathophysiology of pre-eclampsia in humans involves an imbalance in the maternal immune response to the placenta, and one prominent classical hypothesis is that pre-eclampsia is caused by immune maladaptation. First-time mothers are at increased risk of pre-eclampsia, and the risk decreases with the length of time she is exposed to the semen of the father of her baby (Einarsson et al., 2003). In black populations, the high incidence of pre-eclampsia could be because of subtle mutations in human leukocyte antigen (HLA)-G, one of the major histocompatibility complex (MHC) genes (van der Ken and Ober, 1994). This could lead to an increased cytolytic activation of decidual and blood leukocytes, which are generally inhibited by MHC molecules expressed on the cell surface (Shorter et al., 1993). Extravillous trophoblast cells express HLA-G, unlike syncytiotrophoblast, and would therefore be more vulnerable to mutations. Thus, defective HLA-G function could contribute to the low trophoblast invasion and vascular abnormalities observed in pre-eclamptic placentas (Le Bouteiller et al., 2003). HLA-G genotypes and expression seem to have a significant influence on the development of pre-eclampsia (Hylenius et al., 2004), and it has recently been shown that HLA-G levels in plasma from women who subsequently develop pre-eclampsia are lower than in control patients, as early as the first trimester (Yie et al., 2005). Similarly, combinations of killer immunoglobulin receptors on maternal natural killer cells and fetal HLA-C genes influence the risk of pre-eclampsia (Hiby et al., 2004).
There is also mounting evidence indicating that chronic maternal infection including cytomegalovirus-related villitis and periodontal disease is associated with a higher risk of pre-eclampsia (Loez et al., 2002; Von Dadelszen and Magee, 2002; Boggess et al., 2003; Hasegawa et al., 2003; Moore et al., 2004). Some authors have also found an association between these chronic infections and late miscarriage, fetal growth restriction and pre-term labour which could all be linked to chronic leukocyte activation and cytokine production (Hasegawa et al., 2003). The placental bed of women with pre-eclampsia is infiltrated with activated macrophages which release molecules that are capable of reducing trophoblastic invasiveness and even initiating apoptosis (King et al., 1991; Trundley and Moffett, 2004). Activated decidual leukocytes may also be a major source of OFRs, and they may produce cytokines which increase the inflammatory reaction (Stark, 1993).
The above findings suggest that pre-eclampsia is a three-stage rather than a two-stage disorder (Roberts and Hubel, 1999), with the primary pathology being an excessive or atypical maternal immune response. This would impair the placentation process leading to chromic oxidative stress in the placenta and finally to diffuse maternal endothelial cell dysfunction (Hung et al., 2004). The nature of the inflammatory response and oxidative stress seen in pre-eclampsia are not intrinsically different from those seen in normal pregnancies. The difference is one of degree, which strongly suggests that pre-eclampsia is at the extreme end of a continuum of changes seen in every pregnancy.
Human evolution and placental-related disorders
Whatever the factors involved in controlling the materno–placental interaction, the aim is to enable implantation, placentation and the subsequent progressive transformation of the maternal vasoreactive spiral arteries into flaccid, distended utero–placental arteries needed to supply the developing fetus and its placenta with an increasing amount of maternal blood in later pregnancy.
The form of placentation varies widely across mammalian species, but as yet no satisfactory explanation of the evolutionary driving forces has been provided. In most species, there is no invasion of the maternal endometrium, and the conceptus remains within the uterine lumen throughout gestation. The extraembryonic membranes are simply apposed to the uterine epithelium, establishing a relationship classified as epitheliochorial. In others, particularly the carnivores, there is limited erosion of the maternal tissues so that the trophoblast is apposed to the endothelium of the maternal vessels, forming an endotheliochorial placenta. In the past, it has been assumed that the increasing depth of invasion was evidence of evolutionary progression, with the haemochorial situation representing the most advanced state. By providing trophoblast with direct access to the maternal blood, it was thought that the haemochorial situation permitted more efficient transfer of nutrients, including O2, which allowed for greater development of the brain (Martin, 2003). However, dolphins which have epitheliochorial placentae produce large-brained fetuses second only in size to human fetuses (Martin, 2003). More recently, cladistic analyses have suggested that in fact the endotheliochorial relationship is the most primitive form and that the epitheliochorial and haemochorial relationships are parallel advancements (Vogel, 2005).
Despite the potential advantages for materno–fetal transfer it provides, the haemochorial relationship undoubtedly has a number of potential drawbacks. Firstly, there is a risk that the fetal capillaries within the placental villi may be compressed by the higher pressure inherent in the maternal circulation (Karimu and Burton, 1994). It is therefore essential that the pressure in the intervillous space is kept lower than fetal venous pressure, requiring a pressure drop from mean maternal arterial pressure to approximately 10 mmHg. Trophoblast invasion and conversion of the uterine arteries in haemochorial placentation is more likely to be important in this respect and in ensuring constancy of flow, rather than simply increasing uterine artery blood flow. The latter occurs in all species irrespective of trophoblast invasion and is an endocrine-mediated effect. As we have seen, however, the phenomenon of trophoblast invasion brings with it another set of immunological problems, and these have to be balanced evolutionarily against the benefits of the haemochorial relationship for nutrient transfer. The precise details of the latter remain obscure at present.
Secondly, in the haemochorial state, once maternal blood has been released into the placenta, there is little control over the direction or rate of flow, save for the uterine aterio–venous pressure differential. In contrast, retaining the maternal blood within a capillary network in the epitheliochorial and endotheliochorial situations allows maternal and fetal perfusion of the placenta to be matched more closely, and hence major fluctuations in oxygenation are likely to be less common. Finally, the haemochorial situation allows the trophoblast uniquely intimate and extensive access to the maternal circulation. Inflammatory cytokines, soluble receptors, fetal DNA and trophoblastic debris can all be released with ease directly into the maternal blood and hence disseminated widely throughout the mother's body. This is in stark contrast to the situation in, for example, an epitheliochorial placenta where trophoblast microparticles would have to cross the uterine epithelium, its basement membrane, any intervening stromal tissue and finally the capillary endothelium in order to gain similar access.
Miscarriages are the most common human pregnancy disorder with 50% of all conceptions lost before or around implantation and another 20% lost between implantation and completion of the first trimester (Edmonds et al., 1982; Norwitz et al., 2001). The difference in miscarriage rates between humans and other mammals including primates is linked to a difference in chromosomal abnormalities which occur at a rate of more than 50% in humans compared with less than 5% in rats, rabbits and hamsters (Hassold, 1986). The high rate of chromosomal abnormalities in humans could be because of the longer reproductive spans of humans which would lead to an accumulation of chromosomal errors not observed in short-lived mammalian species (Hassold, 1986). Indeed, the incidence of chromosomal abnormalities in the human conceptus increases with maternal and paternal age. Other known causes of miscarriages such as infections have been described in both humans and other mammals but account for only a small percentage of early pregnancy losses. Modern lifestyle and in particular delayed childbirth, maternal smoking and hypercaloric diets leading to diabetes have most certainly increased the incidence of miscarriage in the general population worldwide. The high incidence of miscarriages in the human could also reflect the separation of coitus from ovulation, with the risk of post-ovulatory aged oocytes and post-ejaculatory aged sperm throwing up genetic abnormalities. Overall, all causes of miscarriages lead to major trophoblastic dysfunction with major disturbances of the early phases of the placentation process.
Threatened miscarriages are to be considered separately from other forms of miscarriages as they result from a focal bleed in the periphery of the developing placenta. This bleed occurs at the time of the formation of the membranes and can lead to a complete miscarriage if the hematoma extends into the definitive placenta (Johns et al., 2003). Very early (in the first 8 weeks) vaginal bleeding is not associated with subsequent pregnancy complications, whereas the formation of an intrauterine hematoma at 13–14 weeks increases the risk of pre-eclampsia (Harville et al., 2003). The presence of a haematoma may also be associated with a chronic inflammatory reaction in the decidua resulting in persistent myometrial activity and expulsion of the pregnancy or progressive cellular dysfunction and/or damage to cellular layers of the membranes leading to pre-term rupture and delivery (Silver et al., 2005). Whatever the evolutionary benefits of haemochorial placentation, it appears to be a key factor in the high rate of this very common complication of human gestation, affecting between 10 and 15% of ongoing pregnancies.
Human physiological adjustments to pregnancy resemble those of our primate relatives but differ in several key aspects; most notably, humans have earlier, deeper and more extensive placentation. The human placenta is also larger relative to the uterine size (Martin, 2003). It has been suggested that the human placenta is designed as a means for overcoming the mechanical constraints and consequent reduction in cardiac output because of bipedal posture (Rockwell et al., 2003). The genetic conflict hypothesis predicts that maternal blood pressure is determined by the balance between fetal and placental paternally-inherited factors increasing blood pressure and maternal factors decreasing blood pressure (Haig, 1993). The paternal contribution tends to favour invasiveness of the placenta and thus increases feto–placental capture of maternal resources, whereas the maternal contribution tends to favour a lesser degree of placental response and to promote fetal brain development (Keverne et al., 1996). The placental response invasion may be a secondary strategy to increase non-placental resistance when the utero–placental blood supply is inadequate, but if so, it would appear to be a highly risky one.
Acknowledgements
This work is supported by a grant from The Wellcome Trust, London, UK.
The authors thank Ms Angela Scott (UCL illustration centre) for her assistance in preparing Figure 2.
References
- Baxter N, Sumiya M, Cheng S, Erlich H, Regan L, Simons A, Summerfield JA. Recurrent miscarriage and variant alleles of mannose binding lectin, tumour necrosis factor and lymphotoxin α genes. Clin Exp Immunol. 2001;126:529–534. doi: 10.1046/j.1365-2249.2001.01663.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boggess KA, Lieff S, Murtha AP, Moss K, Beck J, Offenbacher S. Maternal periodontal disease is associated with an increased risk for preeclampsia. Obstet Gynecol. 2003;101:227–231. doi: 10.1016/s0029-7844(02)02314-1. [DOI] [PubMed] [Google Scholar]
- Boue J, Philippe E, Giroud A, Boue A. Phenotypic expression of lethal anomalies in human abortuses. Teratology. 1976;14:3–20. doi: 10.1002/tera.1420140103. [DOI] [PubMed] [Google Scholar]
- Boyd JD, Hamilton WJ. The Human Placenta. Heffer and Sons; Cambridge, UK; 1970. [Google Scholar]
- Burton GJ, Jauniaux E, Watson AL. Maternal arterial connections to the placental intervillous space during the first trimester of human pregnancy: the Boyd Collection revisited. Am J Obstet Gynecol. 1999;181:718–724. doi: 10.1016/s0002-9378(99)70518-1. [DOI] [PubMed] [Google Scholar]
- Burton GJ, Watson AL, Hempstock J, Skepper JN, Jauniaux E. Uterine glands provide histiotrophic nutrition for the human fetus during the first trimester of pregnancy. J Clin Endocrinol Metab. 2002;87:2954–2959. doi: 10.1210/jcem.87.6.8563. [DOI] [PubMed] [Google Scholar]
- Burton GJ, Hempstock J, Jauniaux E. Oxygen, early embryonic metabolism and free radical-mediated embryopathies. Reprod Biomed Online. 2003;6:84–96. doi: 10.1016/s1472-6483(10)62060-3. [DOI] [PubMed] [Google Scholar]
- Chappell LC, Seed PT, Briley AL, Kelly FJ, Lee R, Hunt BJ, Parmar K, Bewley SJ, Shennan AH, Steer PJ, et al. Effects of antioxidants on the occurrence of pre-eclampsia in women at increased risk: a randomised trial. Lancet. 1999;354:810–816. doi: 10.1016/S0140-6736(99)80010-5. [DOI] [PubMed] [Google Scholar]
- Chappell LC, Seed PT, Briley A, Kelly FJ, Hunt BJ, Charnock-Jones DS, Mallet A, Poston L. A longitudinal study of biochemical variables in women at risk of preeclampsia. Am J Obstet Gynecol. 2002a;187:127–136. doi: 10.1067/mob.2002.122969. [DOI] [PubMed] [Google Scholar]
- Chappell LC, Seed PT, Kelly FJ, Briley A, Hunt BJ, Charnock-Jones DS, Mallet A, Poston L. Vitamin C and E supplementation in women at risk of preeclampsia is associated with changes in indices of oxidative stress and placental function. Am J Obstet Gynecol. 2002b;187:777–784. doi: 10.1067/mob.2002.125735. [DOI] [PubMed] [Google Scholar]
- Charnock-Jones S, Burton GJ. Placental vascular morphogenesis. Baillieres Best Pract Res Clin Obstet Gynaecol. 2000;14:953–968. doi: 10.1053/beog.2000.0137. [DOI] [PubMed] [Google Scholar]
- Dietl J. Maternal obesity and complications during pregnancy. J Perinat Med. 2005;33:100–105. doi: 10.1515/JPM.2005.018. [DOI] [PubMed] [Google Scholar]
- Edmonds DK, Lindsay KS, Miller JF, Williamson E, Wood P. Early embryonic mortality in woman. Fertil Steril. 1982;38:447–453. [PubMed] [Google Scholar]
- Einarsson JL, Sangi-Haghpeykar H, Gardner MO. Sperm exposure and development of preeclampsia. Am J Obstet Gynecol. 2003;188:1241–1243. doi: 10.1067/mob.2003.401. [DOI] [PubMed] [Google Scholar]
- Ezashi T, Das P, Roberts RM. Low O2 tensions and the prevention of hES cells. Proc Natl Acad Sci USA. 2005;102:4783–4788. doi: 10.1073/pnas.0501283102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falkowski PG, Katz ME, Milligan AJ, Fennel K, Cramer BS, Aubry MP, Berner RA, Novacek MJ, Zapol WM. The rise of oxygen over the past 205 million years and the evolution of large placental mammals. Science. 2005;2005:2202–2204. doi: 10.1126/science.1116047. [DOI] [PubMed] [Google Scholar]
- Genbacev O, Zhou Y, Ludlow JW, Fisher SJ. Regulation of human placental development by oxygen tension. Science. 1997;277:1669–1672. doi: 10.1126/science.277.5332.1669. [DOI] [PubMed] [Google Scholar]
- Haig D. Genetic conflicts in human pregnancy. Q Rev Biol. 1993;68:495–532. doi: 10.1086/418300. [DOI] [PubMed] [Google Scholar]
- Halliwell B, Gutteridge JMC. Free Radicals in Biology and Medicine. 3rd edn. Oxford, UK: Oxford Science Publications; 1999. [Google Scholar]
- Harville EW, Wilkox AJ, Baird DD, Weinberg CR. Vaginal bleeding in very early pregnancy. Hum Reprod. 2003;18:1944–1947. doi: 10.1093/humrep/deg379. [DOI] [PubMed] [Google Scholar]
- Hasegawa K, Furuichi Y, Shimotsu A, Nakamura M, Yoshinaga M, Kamitomo M, Hatae M, Maruyama I, Izumi Y. Associations between systemic status, periodontal status, serum cytokines levels, and delivery outcome in pregnant women with a diagnosis of threatened premature labour. J Periodontol. 2003;74:1764–1770. doi: 10.1902/jop.2003.74.12.1764. [DOI] [PubMed] [Google Scholar]
- Hassold T. Chromosome abnormalities in human reproductive wastage. Trends Genet. 1986;2:105–110. [Google Scholar]
- Hempstock J, Jauniaux E, Greenwold N, Burton GJ. The contribution of placental oxidative stress to early pregnancy failure. Hum Pathol. 2003;34:1265–1275. doi: 10.1016/j.humpath.2003.08.006. [DOI] [PubMed] [Google Scholar]
- Hiby SE, Walker JJ, O'shaughnessy KM, Redman CW, Carrington M, Trowsdale J, Moffett A. Combinations of maternal KIR and fetal HLA-C genes influences the risk of preeclampsia and reproductive success. J Exp Med. 2004;200:957–965. doi: 10.1084/jem.20041214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubel CA, Roberts JM, Taylor RN, Musci TJ, Rogers GM, McLaughlin MK. Lipid peroxidation in pregnancy: New perspectives on preeclampsia. Am J Obstet Gynecol. 1989;161:1025–1034. doi: 10.1016/0002-9378(89)90778-3. [DOI] [PubMed] [Google Scholar]
- Hung TH, Skepper JN, Burton G. In vitro ischemia-reperfusion injury in term human placenta as a model for oxidative stress in pathological pregnancies. Am J Pathol. 2001;159:1031–1043. doi: 10.1016/S0002-9440(10)61778-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung TH, Charnock-Jones DS, Skepper JN, Burton GJ. Secretion of tumor necrosis factor-alpha from human placental tissues induced by hypoxia-reoxygenation causes endothelial activation in vitro: a potential mediator of the inflammatory response in preeclampsia. Am J Pathol. 2004;164:1049–1061. doi: 10.1016/s0002-9440(10)63192-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hustin J, Schaaps JP. Echographic and anatomic studies of the maternotrophoblastic border during the first trimester of pregnancy. Am J Obstet Gynecol. 1987;157:162–168. doi: 10.1016/s0002-9378(87)80371-x. [DOI] [PubMed] [Google Scholar]
- Hustin J, Jauniaux E, Schaaps JP. Histological study of the materno–embryonic interface in spontaneous abortion. Placenta. 1990;11:477–486. doi: 10.1016/s0143-4004(05)80193-6. [DOI] [PubMed] [Google Scholar]
- Hylenius S, Andersen AM, Melbye M, Hviid TV. Association between HLA-G genotype and risk of pre-eclampsia: a case-control study using family triads. Mol Hum Reprod. 2004;10:237–246. doi: 10.1093/molehr/gah035. [DOI] [PubMed] [Google Scholar]
- Jauniaux E, Burton GJ. Pathophysiology of histological changes in early pregnancy loss. Placenta. 2005;26:114–123. doi: 10.1016/j.placenta.2004.05.011. [DOI] [PubMed] [Google Scholar]
- Jauniaux E, Gulbis B. Fluid compartments of the embryonic environment. Hum Reprod Update. 2000;6:268–278. doi: 10.1093/humupd/6.3.268. [DOI] [PubMed] [Google Scholar]
- Jauniaux E, Zaidi J, Jurkovic D, Campbell S, Hustin J. Comparison of colour Doppler features and pathologic findings in complicated early pregnancy. Hum Reprod. 1994;9:243–247. doi: 10.1093/oxfordjournals.humrep.a138465. [DOI] [PubMed] [Google Scholar]
- Jauniaux E, Watson AL, Hempstock J, Bao Y-P, Skepper JN, Burton GJ. Onset of maternal arterial blood flow and placental oxidative stress: a possible factor in human early pregnancy failure. Am J Pathol. 2000;157:2111–2122. doi: 10.1016/S0002-9440(10)64849-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jauniaux E, Watson AL, Burton GJ. Evaluation of respiratory gases and acid-base gradients in fetal fluids and uteroplacental tissue between 7 and 16 weeks. Am J Obstet Gynecol. 2001;184:998–1003. doi: 10.1067/mob.2001.111935. [DOI] [PubMed] [Google Scholar]
- Jauniaux E, Gulbis B, Burton GJ. The human first trimester gestational sac limits rather than facilities oxygen transfer to the foetus: a review. Placenta-Trophoblast Res. 2003a;24:S86–S93. doi: 10.1053/plac.2002.0932. [DOI] [PubMed] [Google Scholar]
- Jauniaux E, Hempstock J, Greenwold N, Burton GJ. Trophoblastic oxidative stress in relation to temporal and regional differences in maternal placental blood flow in normal and abnormal early pregnancies. Am J Pathol. 2003b;162:115–125. doi: 10.1016/S0002-9440(10)63803-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jawerbaum A, Gonzales E. The role of alterations in arachidonic acid metabolism and nitric oxide homeostasis in rat models of diabetes during early pregnancy. Curr Pharm Des. 2005;11:1327–1342. doi: 10.2174/1381612053507503. [DOI] [PubMed] [Google Scholar]
- Jenkins C, Wilson R, Roberts J, Miller H, McKillop JH, Walker JJ. Antioxidants: their role in pregnancy and miscarriage. Antioxid Redox Signal. 2000;2:623–628. doi: 10.1089/15230860050192369. [DOI] [PubMed] [Google Scholar]
- Johns J, Hyett J, Jauniaux E. Obstetric outcome after threatened miscarriage with and without a hematoma on ultrasound. Obstet Gynecol. 2003;102:483–487. doi: 10.1016/s0029-7844(03)00580-5. [DOI] [PubMed] [Google Scholar]
- Karimu AL, Burton GJ. Significance of changes in fetal perfusion to factors controlling angiogenesis in the human term placenta. J Reprod Fertil. 1994;102:447–450. doi: 10.1530/jrf.0.1020447. [DOI] [PubMed] [Google Scholar]
- Kaufmann P, Burton GJ. Anatomy and genesis of the placenta. In: Knobil E, Neill JD, editors. The Physiology of Reproduction. 2nd edn. New York, USA: Raven Press; 1994. pp. 441–484. [Google Scholar]
- Keverne EB, Fundele R, Narasimha M, Barton SC, Surani MA. Genomic imprinting and the differential roles of parental genomes in brain development. Dev Brain Res. 1996;92:91–100. doi: 10.1016/0165-3806(95)00209-x. [DOI] [PubMed] [Google Scholar]
- Khong TY, De Wolf F, Robertson WB, Brosens I. Inadequate maternal vascular response to placentation in pregnancies complicated by preeclampsia and small-for-gestational age infants. Br J Obstet Gynaecol. 1986;93:1049–1059. doi: 10.1111/j.1471-0528.1986.tb07830.x. [DOI] [PubMed] [Google Scholar]
- King A, Balendran N, Wooding P, Carter NP, Loke YW. CD3 leucocytes present in the human uterus during early placentation: phenotypic and morphologic characterization of the CD56++ population. Dev Immunol. 1991;1:169–190. doi: 10.1155/1991/83493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokawa K, Shikone T, Nakano R. Apoptosis in human chorionic villi and decidua during normal embryonic development and spontaneous abortion in the first trimester. Placenta. 1998;19:21–26. doi: 10.1016/s0143-4004(98)90094-7. [DOI] [PubMed] [Google Scholar]
- Kuller JA, Katz VL. Miscarriage: a historical perspective. Birth. 1994;24:227–228. doi: 10.1111/j.1523-536x.1994.tb00535.x. [DOI] [PubMed] [Google Scholar]
- Le Bouteiller P, Pizzato N, Barakonyi A, Solier CI. HLA-G, preeclampsia, immunity and vascular events. J Reprod Immunol. 2003;59:219–234. doi: 10.1016/s0165-0378(03)00049-4. [DOI] [PubMed] [Google Scholar]
- Loez NJ, Smith PC, Gutierrez J. Periodontal therapy may reduce the risk of preterm low birth weight in women with periodontal disease: a randomized controlled trial. J Periodontol. 2002;73:911–924. doi: 10.1902/jop.2002.73.8.911. [DOI] [PubMed] [Google Scholar]
- Lorentzen B, Endersen MJ, Clausen T, Henriksen T. Fasting serum free fatty acids and triglycerides are increased before 20 weeks of gestation in women who later develop preeclampsia. Hypertens Pregnancy. 1994;13:103–109. [Google Scholar]
- Makhseed M, Raghupathy R, Azizieh F, Farhat R, Hassan N, Bandar A. Circulating cytokines and CD30 in normal human pregnancy and recurrent spontaneous abortions. Hum Reprod. 2000;15:2011–2017. doi: 10.1093/humrep/15.9.2011. [DOI] [PubMed] [Google Scholar]
- Many A, Hubel CA, Fisher SJ, Roberts JM, Zhou Y. Invasive cytotrophoblasts manifest evidence of oxidative stress in preeclampsia. Am J Pathol. 2000;156:321–331. doi: 10.1016/S0002-9440(10)64733-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin RD. Human reproduction: a comparative background for medical hypothesis. J Reprod Immunol. 2003;59:111–135. doi: 10.1016/s0165-0378(03)00042-1. [DOI] [PubMed] [Google Scholar]
- Martin CB, McGaughey HS, Kaiser IH, Donner MW, Ramsey EM. Intermittent functioning of the uteroplacental arteries. Am J Obstet Gynecol. 1964;90:819–823. doi: 10.1016/0002-9378(64)90948-2. [DOI] [PubMed] [Google Scholar]
- Meekins JW, Pijnenborg R, Hanssens M, McFadyen IR, Van Assche FA. A study of placental bed spiral arteries and trophoblast invasion in normal and severe pre-eclamptic pregnancies. Br J Obstet Gynaecol. 1994;101:669–674. doi: 10.1111/j.1471-0528.1994.tb13182.x. [DOI] [PubMed] [Google Scholar]
- Mendelson CR, Jiang B, Shelton JM, Richardson JA, Hinshelwood MM. Transcriptional regulation of aromatase in placenta and ovary. J Steroid Biochem Mol Biol. 2005;95:25–33. doi: 10.1016/j.jsbmb.2005.04.016. [DOI] [PubMed] [Google Scholar]
- Mikhail MS, Anyaegbunam A, Garfinkiel D, Palan PR, Basu J, Romney SL. Preeclampsia and antioxidant nutrients: decreased plasma levels of reduced ascorbic acid, alpha-tocopherol, and beta-carotene in women with preeclampsia. Am J Obstet Gynecol. 1994;171:150–157. doi: 10.1016/0002-9378(94)90462-6. [DOI] [PubMed] [Google Scholar]
- Moore S, Ide M, Coward PY, Randhawa M, Borkowska E, Baylis R, Wilson RF. A prospective study to investigate the relationship between periodontal disease and adverse pregnancy outcome. Br Dent J. 2004;197:251–258. doi: 10.1038/sj.bdj.4811620. [DOI] [PubMed] [Google Scholar]
- Myatt L, Rosenfield RB, Eis ALW, Brockman DE, Greer I, Lyall F. Nitrotyrosine residues in placenta. Evidence of peroxynitrite formation and action. Hypertension. 1996;28:488–493. doi: 10.1161/01.hyp.28.3.488. [DOI] [PubMed] [Google Scholar]
- Nicol CJ, Zielenski J, Tsui LC, Wells PG. An embryoprotective role for glucose-6-phosphate dehydrogenase in developmental oxidative stress and clinical teratogenesis. FASEB J. 2000;14:111–127. doi: 10.1096/fasebj.14.1.111. [DOI] [PubMed] [Google Scholar]
- Norwitz ER, Schust DJ, Fisher SJ. Implantation and the survival of early pregnancy. N Engl J Med. 2001;345:1400–1408. doi: 10.1056/NEJMra000763. [DOI] [PubMed] [Google Scholar]
- Palmer AE, London WT, Sly DL, Rice JM. Spontaneous preeclamptic toxaemia of pregnancy in the patas monkey (Erythrocebus patas) Lab Anim Sci. 1979;29:102–106. [PubMed] [Google Scholar]
- Pidoux G, Guibourdenche J, Frendo JL, Gerbaud P, Conti M, Luton D, Muller F, Evain-Brion D. Impact of trisomy 21 on human trophoblast behaviour and hormonal function. Placenta. 2004;25:S79–S84. doi: 10.1016/j.placenta.2004.01.007. [DOI] [PubMed] [Google Scholar]
- Pijnenborg R, Dixon G, Robertson WB, Brosens I. Trophoblastic invasion of human decidua from 8 to 18 weeks of pregnancy. Placenta. 1980;1:3–19. doi: 10.1016/s0143-4004(80)80012-9. [DOI] [PubMed] [Google Scholar]
- Poranen AK, Ekblad U, Uotila P, Ahotuba M. Lipid peroxidation and antioxidants in normal and pre-eclamptic pregnancies. Placenta. 1996;17:401–405. doi: 10.1016/s0143-4004(96)90021-1. [DOI] [PubMed] [Google Scholar]
- Pukerson ML, Vekerdy L. A history of eclampsia, toxaemia and the kidney in pregnancy. Am J Nephrol. 1999;19:313–319. doi: 10.1159/000013467. [DOI] [PubMed] [Google Scholar]
- Ramsey EM, Donner NW. Placental Vasculature and Circulation. Stuttgart, Germany: Georg Thieme; 1980. [Google Scholar]
- Redman CWG, Sargent IL. Placental debris, oxidative stress and pre-eclampsia. Placenta. 2000;21:597–602. doi: 10.1053/plac.2000.0560. [DOI] [PubMed] [Google Scholar]
- Redman CWG, Sacks GP, Sargent IL. Preeclampsia: An excessive maternal inflammatory response to pregnancy. Am J Obstet Gynecol. 1999;180:499–506. doi: 10.1016/s0002-9378(99)70239-5. [DOI] [PubMed] [Google Scholar]
- Roberts JM, Hubel CA. Is oxidative stress the link in the two-stage model of pre-eclampsia? Lancet. 1999;354:788–789. doi: 10.1016/S0140-6736(99)80002-6. [DOI] [PubMed] [Google Scholar]
- Robertson WB, Brosens I, Landells WN. Abnormal placentation. Obstet Gynecol Annu. 1985;14:411–426. [PubMed] [Google Scholar]
- Rockwell LC, Vargas E, Moore L. Human physiological adaptation to pregnancy: inter- and intraspecific perspectives. Am J Hum Biol. 2003;15:330–341. doi: 10.1002/ajhb.10151. [DOI] [PubMed] [Google Scholar]
- Roy RP. A Darwinian view of obstructed labor. Obstet Gynecol. 2003;101:397–401. doi: 10.1016/s0029-7844(02)02367-0. [DOI] [PubMed] [Google Scholar]
- Sata F, Yamada H, Kondo T, Gong Y, Tozaki S, Kobashi G, Kato EH, Fujimoto S, Kishi R. Glutathione S-transferase M1 and T1 polymorphism and the risk of recurrent pregnancy loss. Mol Hum Reprod. 2003;9:165–169. doi: 10.1093/molehr/gag021. [DOI] [PubMed] [Google Scholar]
- Schmorl G. Pathologishe-anatomische untersuchungen uber puerperaleclampsie. Leipzig, Germany: Vogel; 1883. [Google Scholar]
- Scholl TO, Leskiw M, Chen X, Sims M, Stein TP. Oxidative stress, diet, and the etiology of preeclampsia. Am J Clin Nutr. 2005;81:1390–1396. doi: 10.1093/ajcn/81.6.1390. [DOI] [PubMed] [Google Scholar]
- Seidl DC, Hughes HC, Bertolet R, Lang CM. True pregnancy toxaemia (preeclampsia) in the guinea pig (Cavia pocellus) Lab Anim Sci. 1979;29:472–478. [PubMed] [Google Scholar]
- Shorter SC, Starkey PM, Ferry BL, Clover LM, Sargent IL, Redman CWG. Antigenic heterogeneity of human cytotrophoblast and evidence for the transient expression of MHC class I antigens distinct from HLA-G. Placenta. 1993;14:571–582. doi: 10.1016/s0143-4004(05)80210-3. [DOI] [PubMed] [Google Scholar]
- Shurtz-Swirski R, Simon RJ, Cohen Y, Barnea ER. Human embryo modulates placental function in the first trimester, effects of neural tissues upon chorionic gonadotropin and progesterone secretion. Placenta. 1991;12:521–531. doi: 10.1016/0143-4004(91)90028-e. [DOI] [PubMed] [Google Scholar]
- Silver RK, Wilson D, Philip J, Thom EA, Zachary JM, Mohide P, NICHD EATA Trial Group Late first-trimester placental disruption and subsequent gestational hypertension/preeclampsia. Obstet Gynecol. 2005;105:587–592. doi: 10.1097/01.AOG.0000152343.08096.c3. [DOI] [PubMed] [Google Scholar]
- Staff AC, Ranheim T, Khoury J, Henriksen T. Increased contents of phospholipids, cholesterol, and lipid peroxides in decidua basalis in women with preeclampsia. Am J Obstet Gynecol. 1999;180:587–592. doi: 10.1016/s0002-9378(99)70259-0. [DOI] [PubMed] [Google Scholar]
- Stark JM. Pre-eclampsia and cytokine induced oxidative stress. Br J Obstet Gynaecol. 1993;100:105–109. doi: 10.1111/j.1471-0528.1993.tb15203.x. [DOI] [PubMed] [Google Scholar]
- Sugino N, Kakata M, Kashida S, Karube A, Takigushi S, Kato H. Decreased superoxide dismutase expression and increased concentrations of lipid peroxide and prostaglandin F (2alpha) in the decidua of failed pregnancy. Mol Hum Reprod. 2000;6:642–647. doi: 10.1093/molehr/6.7.642. [DOI] [PubMed] [Google Scholar]
- Tempfer C, Unfried G, Zeillinger R, Hefler L, Nagele F, Huber JC. Endothelial nitric oxide synthase gene polymorphism in women with idiopathic recurrent miscarriage. Hum Reprod. 2001;16:1644–1647. doi: 10.1093/humrep/16.8.1644. [DOI] [PubMed] [Google Scholar]
- Toder V, Fein A, Carp H, Torchinsky A. TNF-α in pregnancy loss and embryo maldevelopment: a mediator of detrimental stimuli or a protector of the fetoplacental unit? J Assist Reprod Genet. 2003;20:73–81. doi: 10.1023/A:1021740108284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trundley A, Moffett A. Human uterine leucocytes and pregnancy. Tissue Antigens. 2004;63:1–12. doi: 10.1111/j.1399-0039.2004.00170.x. [DOI] [PubMed] [Google Scholar]
- Uotila JT, Tuimala RJ, Aarnio TM. Findings on lipid peroxidation and antioxidant function in hypertensive complications of pregnancy. Br J Obstet Gynaecol. 1993;100:270–276. doi: 10.1111/j.1471-0528.1993.tb15242.x. [DOI] [PubMed] [Google Scholar]
- Vanderlie J, Venardos K, Clifton VL, Gude NM, Clarke FM, Perkins AV. Increased biological oxidation and reduced anti-oxidant enzyme activity in pre-eclamptic placentae. Placenta. 2005;26:53–58. doi: 10.1016/j.placenta.2004.04.002. [DOI] [PubMed] [Google Scholar]
- van der Ken K, Ober C. HLA-G polymorphism in African Americans. J Immunol. 1994;153:5628–5633. [PubMed] [Google Scholar]
- Vogel P. The current molecular phylogeny of Eutherian mammals challenges previous interpretations of placental evolution. Placenta. 2005;26:591–596. doi: 10.1016/j.placenta.2004.11.005. [DOI] [PubMed] [Google Scholar]
- Von Dadelszen P, Magee LA. Could an infectious trigger explain the differential maternal response to the shared placental pathology of preeclampsia and normotensive intrauterine growth restriction? Acta Obstet Gynecol Scand. 2002;81:642–648. [PubMed] [Google Scholar]
- von Wolff M, Thaler CJ, Strowitzki T, Broome J, Stolz W, Tabibzadeh S. Regulated expression of cytokines in human endometrium throughout the menstrual cycle: dysregulation in habitual abortion. Mol Hum Reprod. 2000;6:627–634. doi: 10.1093/molehr/6.7.627. [DOI] [PubMed] [Google Scholar]
- Walsh SW. Lipid peroxidation in pregnancy. Hypertens Pregnancy. 1994;13:1–31. [Google Scholar]
- Walsh SW, Wang Y. Trophoblast and placental villous core production of lipid peroxides, thromboxane, and prostacyclin in preeclampsia. J Clin Endocrinol Metab. 1995;80:1888–1893. doi: 10.1210/jcem.80.6.7775637. [DOI] [PubMed] [Google Scholar]
- Wang Y, Walsh SW. Placental mitochondria as a source of oxidative stress in pre-eclampsia. Placenta. 1998;19:581–586. doi: 10.1016/s0143-4004(98)90018-2. [DOI] [PubMed] [Google Scholar]
- Wang Y, Walsh SW. Increased superoxide generation is associated with decreased superoxide dismutase activity and mRNA expression in placental trophoblast cells in preeclampsia. Placenta. 2001;22:206–212. doi: 10.1053/plac.2000.0608. [DOI] [PubMed] [Google Scholar]
- Wang Y, Walsh SW, Guo J, Zhang J. The imbalance between thromboxane and prostacyclin in preeclampsia is associated with an imbalance between lipid peroxides and vitamin E in maternal blood. Am J Obstet Gynecol. 1991;165:1695–1700. doi: 10.1016/0002-9378(91)90017-l. [DOI] [PubMed] [Google Scholar]
- Watson AL, Palmer ME, Skepper JN, Jauniaux E, Burton GJ. Susceptibility of human placental syncytiotrophoblast mitochondria to oxygen-mediated damage in relation to gestational age. J Clin Endocrinol Metabol. 1998;83:1697–1705. doi: 10.1210/jcem.83.5.4830. [DOI] [PubMed] [Google Scholar]
- Wilmut I, Sales DI, Ashworth CJ. Maternal and embryonic factors associated with prenatal loss in mammals. J Reprod Fert. 1986;76:851–864. doi: 10.1530/jrf.0.0760851. [DOI] [PubMed] [Google Scholar]
- Xing Y, Williams C, Campbell RK, Cook S, Knoppers M, Addona T, Altarocca V, Moyle WR. Threading of a glycosylated protein loop through a protein hole: implications for combination of human chorionic gonadotropin subunits. Protein Sci. 2001;10:226–235. doi: 10.1110/ps.25901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yie SM, Taylor RN, Librach C. Low plasma HLA-G protein concentrations in early gestation indicate the development of preeclampsia later in pregnancy. Am J Obstet Gynecol. 2005;193:204–208. doi: 10.1016/j.ajog.2004.11.062. [DOI] [PubMed] [Google Scholar]
- Zusterzeel PLM, Rutten H, Roelofs HMJ, Peters WHM, Steegers EAP. Protein carbonyls in decidua and placenta of pre-eclamptic women as markers for oxidative stress. Placenta. 2001;22:213–219. doi: 10.1053/plac.2000.0606. [DOI] [PubMed] [Google Scholar]