

Abstract
Objective:
It was the aim of this study to characterize the influence of isoflurane-induced heme oxygenase-1 (HO-1) expression on hepatocellular integrity after ischemia and reperfusion.
Summary Background Data:
Abundant experimental data characterize HO-1 as one of the most powerful inducible enzymes that contribute to the protection of the liver and other organs after harmful stimuli. Therapeutic strategies aimed at utilizing the protective effects of HO-1 are hampered by the fact that most pharmacological inducers of this enzyme perturb organ function by themselves and are not available for use in patients because of their toxicity and undesirable or unknown side effects.
Methods:
Rats were pretreated with isoflurane before induction of partial hepatic ischemia (1 hour) and reperfusion (1 hour). At the end of each experiment, blood and liver tissue were obtained for molecular biologic, histologic, and immunohistochemical analyses.
Results:
Isoflurane pretreatment increased hepatic HO-1 mRNA, HO-1 protein, HO enzyme activity, and decreased plasma levels of AST, ALT, and α-GST. Histologic analysis of livers obtained from isoflurane-pretreated rats showed a reduction of necrotic areas, particularly in the perivenular region, the predominant site of isoflurane-induced HO-1 expression. In addition, sinusoidal congestion that could otherwise be observed after ischemia/reperfusion was inhibited by the anesthetic. Furthermore, isoflurane augmented hepatic microvascular blood flow and lowered the malondialdehyde content within the liver compared with control animals. Administration of tin protoporphyrin IX inhibited HO activity and abolished the isoflurane-induced protective effects.
Conclusions:
This study provides first evidence that pretreatment with the nontoxic and clinically approved anesthetic isoflurane induces hepatic HO-1 expression, and thereby protects rat livers from ischemia/reperfusion injury.
Ischemia/reperfusion injury during liver surgery and transplantation is the main cause of postoperative liver failure and the subsequent rise in mortality in these patients. Our study provides first evidence that pretreatment with the nontoxic and clinically approved anesthetic isoflurane induces hepatic heme oxygenase-1 expression, and thereby protects livers from ischemia/reperfusion injury.
Ischemia/reperfusion (IR) injury during liver surgery and transplantation is the main cause of postoperative liver failure and the subsequent rise in mortality in these patients.1,2 Furthermore, a recently published report by van der Bilt et al3 shows that IR induced by vascular clamping, a frequently used technique to avoid blood loss during hepatic surgery, is a strong stimulus that promotes the outgrowth of micrometastases in the liver. Therefore, therapeutic strategies to prevent liver tissue damage after IR have become the focus of extensive research efforts. Accumulating evidence suggests that the heme oxygenase (HO) enzyme system plays a pivotal role in the maintenance of cellular function after a sublethal stress in nearly all organ systems, including the liver.4 To date 3 members of the HO-family have been identified. Heme oxygenase-1 (HO-1), also known as heat shock protein-32, represents the inducible isoform and is up-regulated in response to many different clinically relevant pathologic stimuli, including endotoxemia, hemorrhagic shock, or IR. HO-2 and HO-3 are constitutively expressed isoforms. The HO system metabolizes heme into 3 products: Fe2+, carbon monoxide (CO), and biliverdin, which is subsequently converted to bilirubin. The accumulation of free iron leads to the induction of ferritin, an iron-binding protein. Each of the 3 products has protective functions. Both the induction of HO-1 and administration of biliverdin, bilirubin, carbon monoxide, or iron-binding compounds confer protection in experimental models of IR injury, allograft and xenograft survival, intimal hyperplasia following balloon injury, and others.5
The development of therapeutic strategies that use the protective effect of HO-1 induction is hampered by the fact that most pharmacological inducers of this enzyme perturb organ function by themselves and that gene therapy for up-regulation of HO-1 has potential negative side effects, which currently preclude its clinical application under these conditions. Hence, the substances used for induction of HO-1 in many promising experiments published over the last years are not available for use in patients because of their toxicity and undesirable or unknown side effects. We6–8 have previously shown that the volatile anesthetic isoflurane (ISO) leads to an expression of HO-1 in the normal rat liver and thus augments hepatic macro- and microvascular blood flow under physiological conditions in vivo. ISO is used in daily clinical practice for induction and maintenance of general anesthesia. Since no effective pharmacological approach to prevent hepatic IR injury has been developed for practical use in clinical settings so far,9 there are compelling reasons to identify a suitable compound that specifically induces HO-1, has nontoxic properties, is clinically approved, and confers protection against organ injury. Therefore, this study was designed to determine the functional relevance of hepatic HO-1 induction by the nontoxic and clinically available substance ISO on liver integrity in a rat model of IR. Here, we show a profound protective effect of ISO after hepatic IR and demonstrate the dependence of this quality on the induction of the cytoprotective enzyme HO-1.
MATERIALS AND METHODS
Reagents
Isoflurane was obtained from Abbott (Wiesbaden, Germany) and pentobarbital sodium from Alvetra (Neumuenster, Germany). Pancuronium was purchased from Organon (BH Oss, the Netherlands) and tin protoporphyrin IX (SnPP) from Frontier Scientific Europe (Carnforth, UK). All other reagents used were purchased from Sigma Aldrich (Deisenhofen, Germany) if not specified otherwise.
Animals
Male Sprague Dawley rats (Charles River, Sulzfeld, Germany), weighing 345 ± 36 g, were used for all experiments. The experimental protocol was approved by the local animal care and use committee, and all animals received humane care according to the criteria outlined in the Guide for the Care and Use of Laboratory Animals.10
Experimental Protocol
After inhalational induction of anesthesia with ISO, a tail vein was cannulated and the animals were randomized into 4 groups (Fig. 1): group 1: PEN (pentobarbital sodium, 40 mg/kg/h i.v.) + vehicle (0.5 mL NaHCO3 8.4%, i.v.); group 2: ISO (2.4 MAC) + vehicle; group 3: PEN + SnPP (HO-inhibitor, 50 μmol/kg i.v.)11; group 4: ISO + SnPP. In addition, for each of the intervention groups, an additional set of experiments with sham-operated control animals was performed (n = 3 animals per group). A tracheostomy was performed and after relaxation (pancuronium 1 mg/kg i.v.) all animals were mechanically ventilated (Rodent Ventilator UB 7025-10; Harvard Apparatus, March-Hugstetten, Germany). Doses of 0.5 mg/kg pancuronium were repeated every 3 hours to maintain relaxation. Cannulation of the left carotid artery with polyethylene (PE-50) tubing was performed for arterial blood pressure monitoring and blood gas analysis. There were no significant differences in blood pressure measurements between the groups during the entire experiment (data not shown). The body temperature was maintained in a normothermic range at 37 ± 0.5°C during the entire experiment.
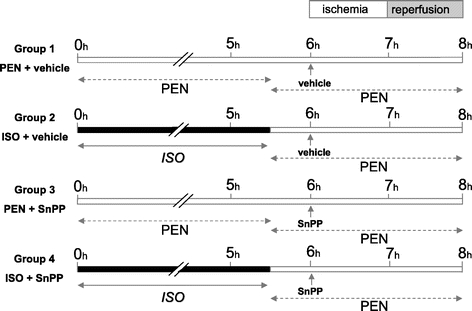
FIGURE 1. Experimental protocol. Animals were randomized into 4 groups (n = 8 animals per group). Group 1 and group 3 received pentobarbital (PEN) during the entire experiment. Group 2 and group 4 were pretreated with isoflurane (ISO) for 5 hours and 30 minutes followed by PEN anesthesia. Six hours after onset, partial hepatic ischemia was induced for 1 hour followed by 1 hour of reperfusion. Ten minutes before ischemia, baseline measurements were obtained and vehicle (groups 1 and 2) or tin protoporphyrin IX ((SnPP) groups 3 and 4) was applied. For each of the groups, an additional experimental series of sham-operated control animals was performed (n = 3 animals per group).
Animal Preparation for Induction of Partial Hepatic IR
Five hours and 30 minutes after onset of anesthesia, the ISO administration in group 2 and group 4 was stopped and all animals received PEN (40 mg/kg/h i.v.) to avoid any direct actions of ISO during the further experiment. A series of additional experiments showed that ISO was not harmful in regard to the IR injury when it was continued during laparotomy and IR (data not shown). For induction of hepatic partial IR, bile flow, and microvascular blood flow measurements, a midline laparotomy was performed and the common bile duct was cannulated with polyethylene tubing (PE-10). Two microvascular flow probes were placed on the same defined positions on the surface of the right and left liver lobe. For compensation of the higher evaporative losses during and after the laparotomy, the infusion rate of crystalloid solution was increased from 4 to 10 mL/kg/h until the end of the experiment. After a stabilization period, 5 hours and 50 minutes after onset, baseline measurements were obtained and either vehicle or SnPP was administered. Five minutes later, the portal vein and hepatic artery supplying the middle and left hepatic lobe were clamped with an atraumatic vascular clip (Aesculap, Tuttlingen, Germany) for induction of partial hepatic ischemia. After 1 hour of ischemia, a 60-minute reperfusion period was initiated on removal of the clip. At the end of reperfusion, animals were killed by an overdose of PEN, and blood and liver tissue were removed for subsequent analyses. Sham-operated control animals underwent identical procedures, with the exception that the vascular clip was not applied.
RNA Isolation and Northern Blot Analysis
Total RNA was extracted from approximately 100 mg of liver tissue according to the manufacturer’s recommendation (TRIZOL; Gibco, Grand Island, NY). Aliquots of total RNA (10 μg per lane) were size-fractionated on an agarose gel (1% agarose, 90 mL diethylpyrocarbonate-treated water, 1.8 mL formaldehyde, 10 mL MOPS/ethylenediaminetetraacetate), transferred to a nylon membrane (Hybond-N; Amersham Pharmacia, Freiburg, Germany) by capillary blotting in 20× sodium saline citrate (3 mol/L NaCl, 0.3 mol/L sodium citrate) and cross-linked to the membrane by ultraviolet irradiation. The membrane was preincubated for 30 minutes in hybridization solution (ExpressHyb; Clontech, Palo Alto, CA) and incubated overnight at 68°C with a 32P-labeled (Prime-It labeling kit; Stratagene, La Jolla, CA) HO-1 complementary DNA (cDNA) probe. All blots were stripped and reprobed with 18S ribosomal RNA cDNA to verify equal loading and transfer of the RNA. Autoradiographs were analyzed by laser scanning densitometry (Personal Densitometer; Molecular Dynamics, Krefeld, Germany). Results were expressed as relative densitometric units calculated as dividends of the background-corrected densitometric values of HO-1/18S.
Western Blot Analysis
Frozen liver tissue was homogenized on ice in TOTEX buffer, incubated for 1 hour at 4°C, and centrifuged at 13,000 rpm for 5 minutes. Total protein concentration was determined in the supernatant using the Bradford assay (Bio-Rad, Munich, Germany). For each lane, 100 μg of protein were dissolved in 10 μL of 1× sodium dodecyl sulfate loading dye and boiled for 5 minutes. After addition of a prestained protein marker (Bio-Rad Laboratories, Munich, Germany), samples were separated on a 10% sodium dodecyl sulfate-polyacrylamide gel and then transferred to a polyvinylidene fluoride membrane (Immobilon-P; Millipore, Bedford, MA) by electroblotting for 2.5 hours (Semidry Transblot; Bio-Rad, Hercules, CA). The membrane was blocked in 1× TBST, and 5% low fat dry milk powder overnight at 4°C and then incubated with a rabbit polyclonal anti-HO-1 antibody (1:1000 dilution; SPA 895, Stressgen Biotechnologies, Victoria, British Colombia, Canada) for 2 hours at room temperature. After 3 washing steps, a secondary anti-rabbit antibody (1:10,000 dilution; ECL detection kit, Amersham Pharmacia, Freiburg, Germany) was added and incubated for 1 hour. Detection was performed by the ECL detection kit (Amersham Pharmacia, Freiburg, Germany) according to the manufacturer’s instructions.
Determination of HO Enzyme Activity
Frozen liver tissue was homogenized (Ultraturrax; Janke and Kunkel, Staufen, Germany) on ice in 4 volumes of 5:1 K:Na 100 mM phosphate buffer with 2 mM MgCl2 (HO activity buffer). After sonication, the homogenate was centrifuged at 13,000 rpm for 15 minutes. One hundred microliters of the supernatant were used to quantitate HO enzyme activity in a reaction volume of 500 μL, containing 100 μL liver cytosol (source of biliverdin reductase), 0.8 mM nicotinamide dinucleotide phosphate, 20 μM hemin, 2 mM glucose-6-phosphate, and 0.002 U/μL glucose-6-phosphate dehydrogenase. The reaction was performed at 37°C for 1 hour in the dark and stopped by addition of 500 μL chloroform. For extraction of bilirubin, the tubes were mixed thoroughly followed by centrifugation at 13,000 rpm for 5 minutes. The chloroform layers were scanned on a spectrophotometer at 464 nm minus the background at 530 nm. Based on the protein content within the reaction volume, the results of the measurements were expressed as formation of bilirubin (pmol) per milligram of protein within 1 hour. Spleen tissue of control animals served as a positive control but had no influence in the data presentation.
Hematoxylin and Eosin Staining of Liver Sections
For histologic analysis, defined sections of the ischemic and nonischemic liver were fixed with 4% buffered formalin (pH 6.9) and embedded in paraffin. Livers were sliced (5 μm) and stained with hematoxylin and eosin. These slices were evaluated for the presence of liver injury by a pathologist who was blinded to the nature of treatment.
Immunohistochemistry
For assessment of cell-type specific expression pattern of HO-1, slides from the same paraffin-embedded livers as used for hematoxylin and eosin staining were used. Briefly, the sections underwent an antigen retrieval using microwave irradiation in a citrate buffer according to a standard protocol.12 Endogenous peroxidase activity was blocked by incubation in 3% H2O2/methanol. After subsequent treatment with normal goat serum, slides were incubated at 37°C for 1 hour with the same primary anti-HO-1 antibody (dilution 1:200) as used in Western blot analysis. As secondary antibody, a biotinylated goat anti-rabbit antibody was used for streptavidine-biotin-complex peroxidase staining. 3,3′-diaminobenzidine and 0.02% cobalt chloride were used as chromogens and slides were counterstained with hematoxylin.
Determination of Bile Flow
Bile was collected in preweighed graduated tubes in 10-minute intervals during the reperfusion period. The results were expressed as mg/10 minutes, and the volume of bile was estimated by weighing the previously tared collection vials.
Determination of Microvascular Blood Flow
Hepatic microvascular blood flow (flux) was determined by the laser Doppler technique (Moor DRT-4; Moor Instruments, Axminster, UK) as described previously.13 Data were acquired 10 minutes before ischemia (baseline) and then continuously throughout the reperfusion period. In each animal, 2 laser Doppler probes were placed at the same defined position on the surface of the right liver lobe (nonischemic) and the left liver lobe (ischemic). The mean flux values at the respective time points were calculated and expressed as a percentage of the baseline values.
Determination of Liver Enzymes
Blood samples were collected at the end of each experiment and immediately centrifuged at 4°C. Enzyme activities of serum ALT and AST were determined by standard clinical automated analyses (Roche, Basel, Switzerland), and the results were expressed in international units per liter. α-GST serum concentration was measured using an antirat α-GST enzyme immunoassay (Hepkit alpha GST; Biotrin, Dublin, Ireland). The procedures were performed according to the manufacturer’s instructions.
Determination of Hepatic Malondialdehyde
Hepatic content of malondialdehyde (MDA) was determined as an index of lipid peroxidation by a colorimetric assay kit according to the manufacturer’s instructions (LPO-586; OXIS International, Portland, Oregon). Briefly, frozen liver tissue was weighed and homogenized in ice-cold phosphate-buffered saline (pH 7.4) containing 5 mM butylated hydroxytoluene. After centrifugation, 200 μL of the supernatant were added to the reaction mixture containing 650 μL N-methyl-2-phenylindole and 150 μL hydrochloric acid (12 mol/L). The samples were incubated for 1 hour at 45°C and centrifuged at 11,700 rpm for 10 minutes. The absorbance of the supernatant was measured at 586 nm. Hepatic MDA concentrations were calculated and normalized by protein concentration and are expressed as nmol/mg protein.
Data Analysis
Statistical differences within each group were determined using a one-way ANOVA for repeated measurements and among the different groups by one-way ANOVA followed by the posthoc Student–Newman–Keuls test for pairwise comparisons. The data are presented as mean ± SEM with n = 8 animals per group. When criteria for parametric tests were not met, Kruskal–Wallis ANOVA on ranks followed by the Student–Newman–Keuls test was used. These data are presented as median (box: 25th and 75th percentiles; error bars: 5th and 95th percentiles) for n = 8 animals per group. One animal had to be excluded from the study (ISO + vehicle group) because the transaminase values differed by more than 12 (AST) and 15 (ALT) standard deviations from the mean of the group. Data were considered significant when P < 0.05. Statistical analysis was performed using the Sigma Stat and Sigma Plot software package (Jandel Scientific, San Rafael, CA).
RESULTS
Effect of ISO Pretreatment on Hepatic HO-1 mRNA Expression
Northern blot analyses of hepatic HO-1 mRNA isolated from ischemic (Fig. 2A) and nonischemic liver tissue (Fig. 2D) are shown in Figure 2. ISO pretreatment led to an accumulation of HO-1 transcripts in both ischemic and nonischemic liver lobes (Figs. 2A and D, lanes 2 and 4), resulting in HO-1 mRNA levels that were higher than those of animals anesthetized with PEN (lanes 1 and 3). Administration of SnPP 10 minutes before induction of partial hepatic ischemia had no influence on HO-1 mRNA levels compared with the vehicle groups (lane 3: PEN and SnPP; lane 4: ISO and SnPP). Equal loading was verified by 18S rRNA labeling and relative HO-1 mRNA levels were quantified by densitometric analyses.
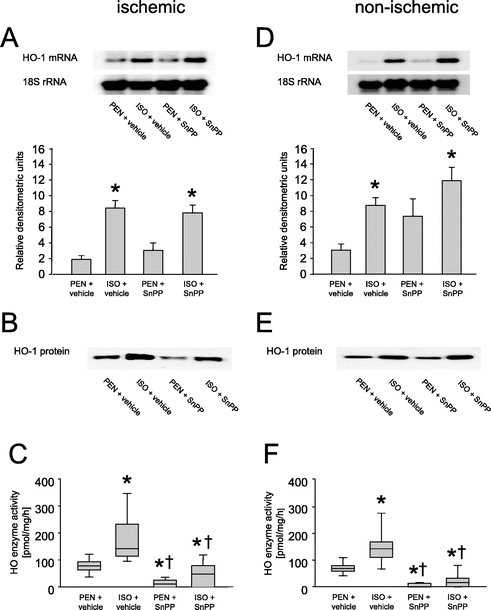
FIGURE 2. A and D, Northern blot analysis of hepatic HO-1 messenger RNA and 18S ribosomal RNA expression of representative animals subjected to pentobarbital anesthesia (PEN) or isoflurane pretreatment (ISO). Data are presented as mean ± SEM for n = 8 animals per group. B and E, Western blot analysis of hepatic HO-1 protein. C and F, Measurements of HO enzyme activity in rat liver. Data are presented as median (box: 25th and 75th percentiles; error bars: 5th and 95th percentiles) for n = 8 animals per group. *P < 0.05 versus PEN + vehicle; †P < 0.05 versus ISO + vehicle.
Effect of ISO Pretreatment on Hepatic HO-1 Protein Levels and HO Enzyme Activity
Western blot analyses showed an accumulation of HO-1 protein after ISO pretreatment (Figs. 2B and E, lanes 2 and 4) compared with PEN anesthetized animals (lanes 1 and 3). Administration of SnPP had no influence on HO-1 protein expression levels (lane 3: PEN and SnPP; lane 4: ISO and SnPP). ISO pretreatment led to a strong up-regulation of hepatic HO enzyme activity in both ischemic (Fig. 2C) and nonischemic (Fig. 2F) liver tissue compared with the PEN anesthetized control animals. Administration of SnPP almost completely inactivated the HO enzyme in both groups (Figs. 2C and F; P < 0.05).
Expression Pattern of Hepatic HO-1 Protein After ISO Pretreatment
HO-1 immunoreactive protein was restricted to spindle-shaped sinusoidal lining cells and isolated hepatocytes in PEN-anesthetized control animals (Fig. 3A). In contrast, HO-1 protein was markedly up-regulated in hepatocytes predominantly located in the perivenular area after ISO pretreatment (Fig. 3B). The administration of SnPP prior to hepatic IR did not alter this expression pattern of HO-1 immunoreactive protein (data not shown).
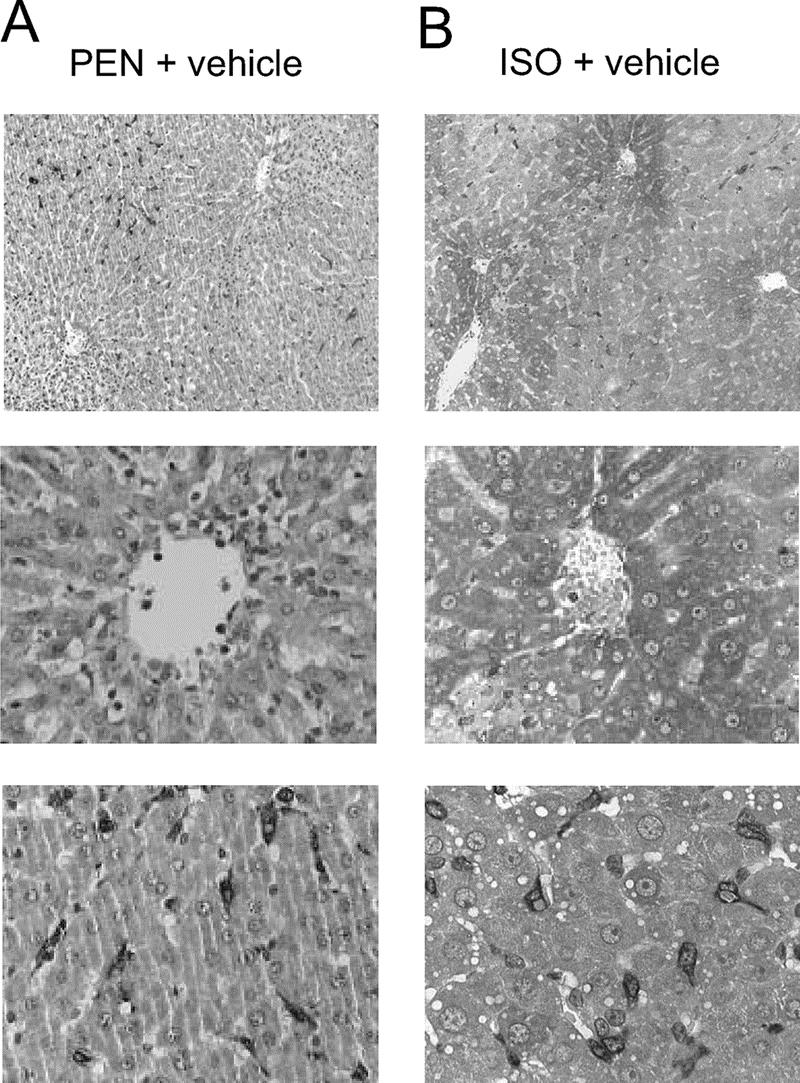
FIGURE 3. Cell-specific expression pattern of HO-1 protein after hepatic ischemia and reperfusion in pentobarbital-anesthetized (PEN) and isoflurane-pretreated (ISO) animals. A, HO-1 protein was restricted to spindle-shaped sinusoidal lining cells and isolated hepatocytes in PEN controls. B, ISO pretreatment led to a markedly up-regulated HO-1 protein expression in hepatocytes of the perivenular area.
Effect of ISO Pretreatment on Hepatic Excretory Function During Reperfusion
Hepatic excretory function was assessed by measurement of hepatic bile production. Data were obtained prior to administration of vehicle or SnPP, 10 minutes before induction of partial hepatic ischemia (baseline), and then during reperfusion in 10-minute intervals. The assessed values are shown in Figure 4. ISO pretreatment tended to improve hepatic excretory function in the early reperfusion period. However, this tendency did not reach statistical significance.
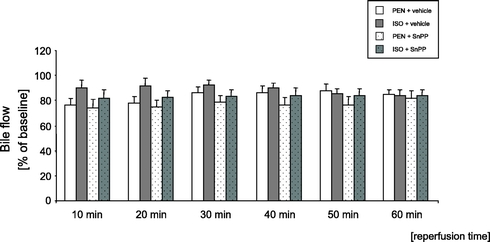
FIGURE 4. Effects of pentobarbital anesthesia (PEN) and isoflurane pretreatment (ISO) on hepatic bile flow measurements during reperfusion. Data are presented as mean ± SEM percent changes of baseline for n = 8 animals per group.
Effect of ISO Pretreatment on Plasma Levels of Liver Enzymes
The plasma levels of α-glutathione S-transferase (α-GST), aspartate aminotransferase (AST), and alanine aminotransferase (ALT) were measured at the end of each experiment. As shown in Figure 5A, ISO pretreatment led to a profound decrease in plasma enzyme levels compared with PEN anesthetized controls (P < 0.05). Blockade of the HO metabolism by administration of SnPP increased markers of hepatocellular injury to control levels (P < 0.05). In case of the α-GST plasma concentration, SnPP caused an even higher increase above the level of the PEN control group (P <0.05).

FIGURE 5. Effects of pentobarbital anesthesia (PEN) and isoflurane pretreatment (ISO) on liver injury after IR. A, Plasma levels of α-glutathione-s-transferase (α-GST), aspartate-aminotransferase (AST), and alanine-aminotransferase (ALT). Data are presented as median (box: 25th and 75th percentiles; error bars: 5th and 95th percentiles) for n = 8 animals per group. *P < 0.05 versus PEN + vehicle; †P < 0.05 versus ISO + vehicle. B, Representative photomicrographs of rat livers from each group. Histopathological examination revealed large necrotic areas predominantly in the perivenular zone and sinusoidal congestion after IR in the PEN group. ISO pretreatment strongly protects livers from these changes. Inhibition of the HO enzyme by administration of SnPP further deteriorated liver injury.
Effect of ISO Pretreatment on Liver Histology After IR
Histopathological examination revealed large necrotic areas predominantly in the perivenular zone and sinusoidal congestion after IR in the PEN group (Fig. 5B). ISO pretreatment strongly protected livers from these changes. Inhibition of the HO enzyme by administration of SnPP aggravated the morphologic features of liver injury (Fig. 5B, PEN and SnPP) and abolished the ISO-mediated protection (Fig. 5B, ISO and SnPP).
Effect of ISO Pretreatment on Hepatic Microvascular Blood Flow During Reperfusion
As shown in Figure 6A, ISO pretreatment led to an improvement of hepatic microvascular blood flow during early reperfusion in the ischemic lobe, as compared with all other groups (P < 0.05). Data were assessed prior to administration of vehicle or SnPP, 10 minutes before induction of partial hepatic ischemia (baseline), and during reperfusion in 10-minute intervals. In contrast, no significant differences in flux could be observed among the groups in the nonischemic liver lobe (Fig. 6B).
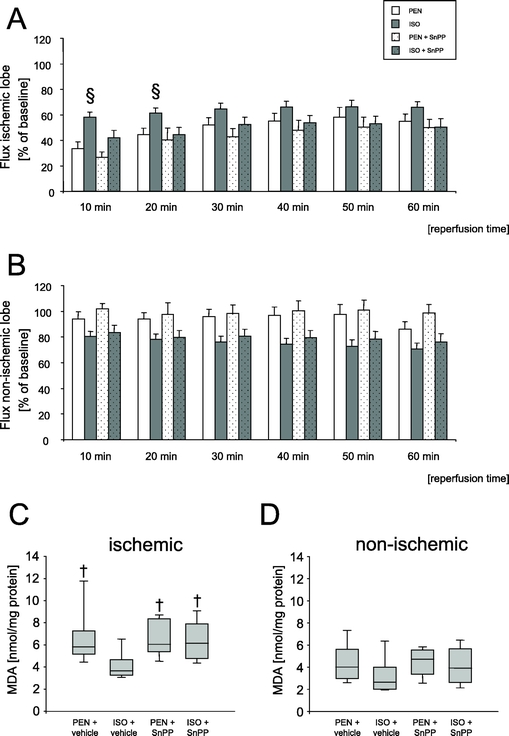
FIGURE 6. A and B, Effects of pentobarbital anesthesia (PEN) and isoflurane pretreatment (ISO) on changes of hepatic microvascular blood flow during reperfusion. Data are presented as mean ± SEM for n = 8 animals per group. §P < 0.05 versus all other groups at the respective time points. C and D, Hepatic malondialdehyde (MDA) content after IR. Data are presented as median (box: 25th and 75th percentiles; error bars: 5th and 95th percentiles) for n = 8 animals per group. †P < 0.05 versus ISO + vehicle.
Effect of ISO Pretreatment on Hepatic Malondialdehyde Content
Hepatic MDA levels were analyzed in tissue specimens of both the ischemic and nonischemic liver lobes. Results are depicted in Figures 6C and D. No significant differences in MDA content could be demonstrated among the groups within the nonischemic tissue (Fig. 6D). In contrast, ISO pretreatment resulted in a lower hepatic MDA level within the ischemic lobe as compared with PEN-anesthesized controls and SnPP-treated animals (Fig. 6C).
Sham-Operated Control Animals
All measured parameters of sham controls are depicted in Table 1. ISO treatment as well as the administration of SnPP led to an induction of hepatic HO-1 mRNA compared with PEN-anesthetized animals (P < 0.05). HO enzyme activity increased in the ISO + vehicle group and differed significantly from all other sham groups (P < 0.05). Administration of SnPP almost completely inhibited HO activity.
TABLE 1. Parameters of Sham-Operated Control Animals

DISCUSSION
Our laboratory has previously demonstrated that administration of volatile anesthetics differentially induces hepatic HO-1 mRNA and protein.6 More recently, we8 could show that treatment with ISO enhances hepatic macro- and microvascular blood flow by increasing HO enzyme activity in the normal liver. Based on these results obtained under physiological conditions, we performed the present study to evaluate the influence of ISO-induced HO-1 expression on hepatocellular integrity after warm IR. Our results provide first evidence that pretreatment with the nontoxic and clinically approved anesthetic ISO induces hepatic HO-1 mRNA and protein, increases HO enzyme activity, and thereby protects rat livers from IR injury. Hepatic organ protection is indicated by a decrease in plasma liver enzymes (α-GST, ALT, and AST) and reduced histopathological features of liver damage. Although we could not detect any significant net changes in bile secretion rates between the different groups, hepatic excretory function after ISO pretreatment tended to be higher than in PEN-anesthetized and SnPP-treated animals during early reperfusion. This overall protective effect is accompanied by an induction of HO-1 at all levels, including HO-1 mRNA, HO-1 protein, and HO enzymatic activity after ISO treatment. Blockade of the HO enzyme by administration of SnPP abolished protection in ISO-pretreated animals, implicating a direct involvement of HO.
In view of a potential clinical importance of the presented animal data, it has to be noted that these results were obtained with a rather long ISO pretreatment time of 5.5 hours and a high dose of 2.4 MAC. So far, no results are available in humans regarding whether ISO administration during standard procedures of liver resection also induces hepatic HO-1. However, such pilot studies could be performed. High inspiratory concentrations of ISO decrease blood pressure and, therefore, it could be necessary to use catecholamines simultaneously to stabilize the circulation. Regarding that point, Rensing et al14 provided evidence that the β-1 adrenoceptor agonist, dobutamine, could also act as an inducer of hepatic HO-1 in rats under in vivo conditions, which at least offers the possibility of a combined use also in humans. The simultaneous use of 2 HO-1–inducing substances may reduce the administered concentrations and/or times of pretreatment to obtain a beneficial effect. With regard to the time of pretreatment, a duration of 5.5 hours would not preclude its use in the treatment of organ donors before explanation, since that therapy could already begin in the intensive care unit. The same time of ISO administration in patients undergoing hepatic tumor surgery would likely be too long in most cases. Therefore, it has to be evaluated in further studies, whether shorter time intervals could be sufficient to get an HO-1–inducing and protective effect.
The pathophysiology of hepatic IR injury is characterized by an interplay of mainly 3 key mechanisms, including microcirculatory disturbances, the initiation of an inflammatory response, and the generation of reactive oxygen species (ROS).15 The maintenance of microvascular perfusion after hepatic ischemia plays a crucial role in the prevention of liver cell injury. As shown by Chun et al,16 early restoration of liver blood flow after ischemia is of particular importance to prevent hepatocellular death. We17 could previously show that the vasodilator CO, a metabolite of the HO pathway, is an important contributor to the maintenance of hepatic microvascular perfusion in stress-exposed livers. In agreement with recently published observations from our group, showing an improvement of hepatic perfusion after ISO pretreatment and the subsequent induction of HO-1 within the normal liver, we could demonstrate similar qualities of the anesthetic after IR. ISO administration led to a significant increase of hepatic flux in the early reperfusion period and could therefore contribute to the protective effect of the volatile anesthetic in our study. This improvement of microvascular perfusion is likely to be mediated by an increased production of CO that exerts its vasodilatory effect by activation of guanylate cyclase, which increases the synthesis of cyclic guanosine monophosphate (cGMP), leading to vasodilatation, a mechanism similar to that of nitric oxide. Other cGMP dependent actions, mediated by CO that could also contribute to the improvement of microcirculation after IR, are its inhibitory effect on platelet activation and aggregation, which suppresses thrombosis and the proinflammatory response stimulated by activated platelets, as shown by Brune et al.18 Furthermore, Fujita et al19 could recently demonstrate that CO down-regulates the expression of plasminogen activator inhibitor type 1 (PAI-1) and thus exerts a protective effect after IR of the lung by derepression of fibrinolysis. The histopathologic examination of liver slices revealed large areas of congested sinusoids in animals, which did not show an increased activity of the HO enzyme. In contrast, livers after ISO pretreatment and with increased HO activity depicted fewer areas of sinusoidal congestion, an observation that would be in line with the results of the aforementioned studies. The perivenular area of the hepatic lobule is most susceptible to ischemic injury due to the oxygen gradient from upstream toward downstream hepatocytes. Examination of ischemic liver sections in our study confirmed these observations, indicated by an improvement of histopathological features of liver injury especially in the perivenular area. In this regard, the expression pattern of the HO-1 protein that we could demonstrate by immunohistochemistry is of special interest. Previous reports revealed that constitutive HO-1 protein expression is restricted to spindle-shaped cells within the sinusoids, which could be identified as Kupffer cells.19a Despite this pattern, we could additionally show that ISO pretreatment led to an up-regulation of HO-1 protein expression in hepatocytes, predominantly around the central veins, which is the same area where we could detect necrosis in animals that were not pretreated and did not express HO-1. This promotes the hypotheses that hepatocytes which express HO-1 protein after ISO pretreatment are obviously protected against IR-related cell death. On the other hand, the changes in microvascular blood flow that we could detect in our study were only moderate and transient. Furthermore, the regional association between necrotic and protected areas and HO-1 expression is difficult to explain purely on a blood flow basis, based on the knowledge of the sites that determine sinusoidal blood flow, which are localized further upstream. Therefore, other “local” effects are more likely to be involved in the profound protection.
One of the main causes leading to the destructive effects of IR arises from the activation of Kupffer cells and the subsequent generation and release of ROS, lysosomal enzymes, and various cytokines like TNF-α or IL-1β.20 Teoh et al21 showed that the administration of low-dose TNF-α protects against hepatic IR injury in mice by preventing the Kupffer cell-mediated TNF-α burst in response to IR. Our group recently demonstrated that gadolinium chloride, an agent that inhibits Kupffer cell function, thereby reducing the expression of Kupffer cell related gene products, abrogates the ISO-induced HO-1 up-regulation in hepatocytes in vivo.7 Taking this into account, it is conceivable that ISO pretreatment may also lead to a modest activation of Kupffer cells, thereby reducing the IR-related cytokine burst, a mechanism similar to preconditioning. Furthermore, data published by Uchinami et al22 indicate that heat shock preconditioning could reduce nuclear binding of the proinflammatory transcription factor NFκB after hepatic IR, which may also contribute to an attenuated postischemic inflammatory response by a reduced formation of TNF-α or other cytokines. In line with these results, Fondevila et al23 demonstrated that biliverdin, a product of the HO pathway, could inhibit surface expression of adhesion molecules like P-selectin/E-selectin and ICAM-1 in an animal model of orthotopic liver transplantation. The expression of adhesion molecules is an important contributor to the development of IR injury. ROS, which are generated subsequent to reoxygenation, mediate direct tissue damage and initiate a cascade of deleterious cellular responses, leading to inflammation, cell death, and organ failure.15,24 ROS are capable of aggravating hepatic organ injury by initiating lipid peroxidation of polyunsaturated fatty acids, which leads to the destruction of cell membranes. Lipid peroxidation results in the formation of aldehyde by-products such as MDA, which has the potential to further amplify organ injury by inducing hepatocyte death, necrosis, and inflammation.25 Ample evidence supports the notion that the HO-generated bile pigments biliverdin and bilirubin are 2 of the most potent physiological antioxidant molecules and thus protect cells from oxidative injury.23,26,27 In addition, depletion of pro-oxidative heme by the action of HO further contributes to an overall antioxidative effect of an increased HO enzyme activity. We could demonstrate that ISO-induced HO-1 up-regulation led to a significant decrease of the MDA content within the ischemic liver tissue, which possibly contributes to the organ protective effect of the volatile anesthetic shown in this study. Consistent with our findings, it has been reported that induction of HO-1 and administration of metabolites of the HO pathway reduces lipid peroxidation products like MDA in various animal models.28,29 In addition, other consequences of the heme metabolism, like the induction of ferritin,30 and the depletion of heme itself,31 could contribute to the antioxidative effects of the HO enzyme and, therefore, might be responsible for the decrease of MDA after ISO pretreatment in our study.
Although we did not examine apoptotic cell death in the present study, it has been shown that HO-1 overexpression and the administration of CO or biliverdin exert major cytoprotective effects against hepatic IR injury via up-regulation of antiapoptotic (Bcl-x, Bcl-2, Bag-1) and suppression of pro-apoptotic (caspase-3) mediators, which could also contribute to the protective effect of ISO in our study.23,32,33 The p38 MAPK pathway has been identified as a key mechanism of CO-mediated protection against hepatic IR injury.34 In contrast, recent data published by Massip-Salcedo et al35 indicate that the inhibition of p38 MAPK decreased liver injury after partial hepatic IR. As already noted by these authors, the differentiation between MAPK isoforms might play an important role in that regard. Data obtained in the heart indicate that the p38α isoform activation has the potential to accelerate cell death, while the p38β pathway may be antiapoptotic.36 Similar data exist for the differential activation of JNK isoenzymes, which results in different functional roles.37
Many protective attempts against liver injury have been proposed, including surgical strategies, gene therapy, and the use of pharmacological agents. Nonetheless, in a recently published review article Selzner et al38 stated that gene therapy strategies are still far beyond introduction into clinical practice and that currently, only surgical strategies could be considered protective against IR injury routinely used in patients. Regarding ischemic preconditioning Clavien et al39 recently published the first prospective randomized study including 100 patients undergoing major liver resection with or without ischemic preconditioning. The authors could demonstrate a beneficial effect of ischemic preconditioning in young patients and in cases of a lower resected liver volume and concluded that other strategies are needed in older patients. Concerning pharmacological preconditioning, the induction of heat shock proteins and in particular of HO-1 (heat shock protein-32) has been extensively studied and conferred protection against hepatic IR injury under experimental conditions.40,41 Although helpful to define fundamental principles of HO-1 related mechanisms for organ protection, the pharmacological HO-1 inducers used in the majority of these studies will most likely never find a way into clinical practice because of their undesirable or unknown side effects. For example, Ito et al42 could recently demonstrate that administration of doxorubicin, another clinically available pharmacologic agent used for human cancer treatment, played a protective role in a rat model of warm IR via up-regulation of HO-1. Nevertheless, the toxicity of this substance limits its clinical usefulness. Therefore, the results of our study are especially relevant in view of a possible clinical impact. As a new inducer of the HO enzyme system, ISO offers advantages compared with other pharmacological HO-1 inducers that are currently described in the literature. ISO is an approved pharmacologic agent that is used in patients for induction and maintenance of general anesthesia and is the therapy for severe asthmatic events because of its bronchodilatory potency. The volatile properties of the substance would simplify its administration by inhalation in clinical settings. Furthermore, adverse side effects of ISO are rare and well characterized because of its frequent usage in daily clinical anesthesia practice for more than 30 years.
In summary, this study provides the first evidence that induction of the cytoprotective enzyme heme oxygenase-1 by the nontoxic and clinically approved substance isoflurane protects rat livers from ischemia/reperfusion injury in vivo. This protective effect seems to be mediated through an interaction of different properties of the heme oxygenase system, like improvement of microvascular blood flow and potent antioxidant effects. This in turn leads to a decrease of plasma liver enzymes and histopathological features of liver injury. Given the known safety of ISO as a clinically used anesthetic, our results reveal a potential new and safe way of induction of the cytoprotective enzyme HO-1 as a therapy against hepatic IR injury. Further studies are now required to confirm these effects in humans.
ACKNOWLEDGMENTS
We thank Prof. Dr. H. K. Koch, Institute of Pathology, Freiburg, Germany, for the assessment of histopathologic changes and Prof. Dr. S. Shibahara, Department of Applied Physiology and Molecular Biology, Tohoku University School of Medicine, Sendai, Japan, who kindly provided the HO-1 cDNA clone. We are grateful to Maria Eckenfels for her expert technical assistance.
Footnotes
This work was supported by grants from the Deutsche Forschungsgemeinschaft, Bonn, Germany to B.H.J. Pannen (DFG PA 533/3-2 and PA 533/4-1).
Reprints: Rene Schmidt, MD, Anaesthesiologische Universitätsklinik, Hugstetterstrasse 55, D-79106 Freiburg, Germany. E-mail: rene.schmidt@uniklinik-freiburg.de.
REFERENCES
- 1.Serracino-Inglott F, Habib NA, Mathie RT. Hepatic ischemia-reperfusion injury. Am J Surg. 2001;181:160–166. [DOI] [PubMed] [Google Scholar]
- 2.Lemasters JJ, Thurman RG. Reperfusion injury after liver preservation for transplantation. Annu Rev Pharmacol Toxicol. 1997;37:327–338. [DOI] [PubMed] [Google Scholar]
- 3.van der Bilt JD, Kranenburg O, Nijkamp MW, et al. Ischemia/reperfusion accelerates the outgrowth of hepatic micrometastases in a highly standardized murine model. Hepatology. 2005;42:165–175. [DOI] [PubMed] [Google Scholar]
- 4.Ryter SW, Alam J, Choi AM. Heme oxygenase-1/carbon monoxide: from basic science to therapeutic applications. Physiol Rev. 2006;86:583–650. [DOI] [PubMed] [Google Scholar]
- 5.Otterbein LE, Soares MP, Yamashita K, et al. Heme oxygenase-1: unleashing the protective properties of heme. Trends Immunol. 2003;24:449–455. [DOI] [PubMed] [Google Scholar]
- 6.Hoetzel A, Geiger S, Loop T, et al. Differential effects of volatile anesthetics on hepatic heme oxygenase-1 expression in the rat. Anesthesiology. 2002;97:1318–1321. [DOI] [PubMed] [Google Scholar]
- 7.Hoetzel A, Leitz D, Schmidt R, et al. Mechanism of hepatic heme oxygenase-1 induction by isoflurane. Anesthesiology. 2006;104:101–109. [DOI] [PubMed] [Google Scholar]
- 8.Schmidt R, Hoetzel A, Baechle T, et al. Isoflurane pretreatment lowers portal venous resistance by increasing hepatic heme oxygenase activity in the rat liver in vivo. J Hepatol. 2004;41:706–713. [DOI] [PubMed] [Google Scholar]
- 9.Carini R, Albano E. Recent insights on the mechanisms of liver preconditioning. Gastroenterology. 2003;125:1480–1491. [DOI] [PubMed] [Google Scholar]
- 10.American Association for Laboratory Animal Science. Guide for the Care and Use of Laboratory Animals. Bethesda, MD: National Institutes of Health; 1985. [Google Scholar]
- 11.Landaw SA, Sassa S, Drummond GS, et al. Proof that Sn-protoporphyrin inhibits the enzymatic catabolism of heme in vivo. Suppression of 14CO generation from radiolabeled endogenous and exogenous heme sources. J Exp Med. 1987;165:1195–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Evers P, Uylings HB. Microwave-stimulated antigen retrieval is pH and temperature dependent. J Histochem Cytochem. 1994;42:1555–1563. [DOI] [PubMed] [Google Scholar]
- 13.Schmidt R, Baechle T, Hoetzel A, et al. Dihydralazine treatment limits liver injury after hemorrhagic shock in rats. Crit Care Med. 2006;34:815–822. [DOI] [PubMed] [Google Scholar]
- 14.Rensing H, Bauer I, Kubulus D, et al. Heme oxygenase-1 gene expression in pericentral hepatocytes through beta1-adrenoceptor stimulation. Shock. 2004;21:376–387. [DOI] [PubMed] [Google Scholar]
- 15.Jaeschke H. Molecular mechanisms of hepatic ischemia-reperfusion injury and preconditioning. Am J Physiol Gastrointest Liver Physiol. 2003;284:G15–G26. [DOI] [PubMed] [Google Scholar]
- 16.Chun K, Zhang J, Biewer J, et al. Microcirculatory failure determines lethal hepatocyte injury in ischemic/reperfused rat livers. Shock. 1994;1:3–9. [DOI] [PubMed] [Google Scholar]
- 17.Pannen BH, Kohler N, Hole B, et al. Protective role of endogenous carbon monoxide in hepatic microcirculatory dysfunction after hemorrhagic shock in rats. J Clin Invest. 1998;102:1220–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brune B, Ullrich V. Inhibition of platelet aggregation by carbon monoxide is mediated by activation of guanylate cyclase. Mol Pharmacol. 1987;32:497–504. [PubMed] [Google Scholar]
- 19.Fujita T, Toda K, Karimova A, et al. Paradoxical rescue from ischemic lung injury by inhaled carbon monoxide driven by derepression of fibrinolysis. Nat Med. 2001;7:598–604. [DOI] [PubMed] [Google Scholar]
- 19a.Bauer I, Rensing H, Florax A, et al. Expression pattern and regulation of heme-oxygenase-1/heat shock protein 32 in human liver cells. Shock. 2003;20:116–122. [DOI] [PubMed] [Google Scholar]
- 20.Fan C, Zwacka RM, Engelhardt JF. Therapeutic approaches for ischemia/reperfusion injury in the liver. J Mol Med. 1999;77:577–592. [DOI] [PubMed] [Google Scholar]
- 21.Teoh N, Leclercq I, Pena AD, et al. Low-dose TNF-alpha protects against hepatic ischemia-reperfusion injury in mice: implications for preconditioning. Hepatology. 2003;37:118–128. [DOI] [PubMed] [Google Scholar]
- 22.Uchinami H, Yamamoto Y, Kume M, et al. Effect of heat shock preconditioning on NF-kappaB/I-kappaB pathway during I/R injury of the rat liver. Am J Physiol Gastrointest Liver Physiol. 2002;282:G962–G971. [DOI] [PubMed] [Google Scholar]
- 23.Fondevila C, Shen XD, Tsuchiyashi S, et al. Biliverdin therapy protects rat livers from ischemia and reperfusion injury. Hepatology. 2004;40:1333–1341. [DOI] [PubMed] [Google Scholar]
- 24.Jaeschke H. Preservation injury: mechanisms, prevention and consequences. J Hepatol. 1996;25:774–780. [DOI] [PubMed] [Google Scholar]
- 25.Browning JD, Horton JD. Molecular mediators of hepatic steatosis and liver injury. J Clin Invest. 2004;114:147–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stocker R, Yamamoto Y, McDonagh AF, et al. Bilirubin is an antioxidant of possible physiological importance. Science. 1987;235:1043–1046. [DOI] [PubMed] [Google Scholar]
- 27.Kato Y, Shimazu M, Kondo M, et al. Bilirubin rinse: a simple protectant against the rat liver graft injury mimicking heme oxygenase-1 preconditioning. Hepatology. 2003;38:364–373. [DOI] [PubMed] [Google Scholar]
- 28.Nakao A, Neto JS, Kanno S, et al. Protection against ischemia/reperfusion injury in cardiac and renal transplantation with carbon monoxide, biliverdin and both. Am J Transplant. 2005;5:282–291. [DOI] [PubMed] [Google Scholar]
- 29.Nakao A, Otterbein LE, Overhaus M, et al. Biliverdin protects the functional integrity of a transplanted syngeneic small bowel. Gastroenterology. 2004;127:595–606. [DOI] [PubMed] [Google Scholar]
- 30.Balla G, Jacob HS, Balla J, et al. Ferritin: a cytoprotective antioxidant strategem of endothelium. J Biol Chem. 1992;267:18148–18153. [PubMed] [Google Scholar]
- 31.Jeney V, Balla J, Yachie A, et al. Pro-oxidant and cytotoxic effects of circulating heme. Blood. 2002;100:879–887. [DOI] [PubMed] [Google Scholar]
- 32.Ke B, Buelow R, Shen XD, et al. Heme oxygenase 1 gene transfer prevents CD95/Fas ligand-mediated apoptosis and improves liver allograft survival via carbon monoxide signaling pathway. Hum Gene Ther. 2002;13:1189–1199. [DOI] [PubMed] [Google Scholar]
- 33.Sass G, Soares MC, Yamashita K, et al. Heme oxygenase-1 and its reaction product, carbon monoxide, prevent inflammation-related apoptotic liver damage in mice. Hepatology. 2003;38:909–918. [DOI] [PubMed] [Google Scholar]
- 34.Amersi F, Shen XD, Anselmo D, et al. Ex vivo exposure to carbon monoxide prevents hepatic ischemia/reperfusion injury through p38 MAP kinase pathway. Hepatology. 2002;35:815–823. [DOI] [PubMed] [Google Scholar]
- 35.Massip-Salcedo M, Casillas-Ramirez A, Franco-Gou R, et al. Heat shock proteins and mitogen-activated protein kinases in steatotic livers undergoing ischemia-reperfusion: some answers. Am J Pathol. 2006;168:1474–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ravingerova T, Barancik M, Strniskova M. Mitogen-activated protein kinases: a new therapeutic target in cardiac pathology. Mol Cell Biochem. 2003;247:127–138. [DOI] [PubMed] [Google Scholar]
- 37.Ping P, Zhang J, Huang S, et al. PKC-dependent activation of p46/p54 JNKs during ischemic preconditioning in conscious rabbits. Am J Physiol. 1999;277:H1771–H1785. [DOI] [PubMed] [Google Scholar]
- 38.Selzner N, Rudiger H, Graf R, et al. Protective strategies against ischemic injury of the liver. Gastroenterology. 2003;125:917–936. [DOI] [PubMed] [Google Scholar]
- 39.Clavien PA, Selzner M, Rudiger HA, et al. A prospective randomized study in 100 consecutive patients undergoing major liver resection with versus without ischemic preconditioning. Ann Surg. 2003;238:843–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Amersi F, Buelow R, Kato H, et al. Upregulation of heme oxygenase-1 protects genetically fat Zucker rat livers from ischemia/reperfusion injury. J Clin Invest. 1999;104:1631–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shen XD, Ke B, Zhai Y, et al. Toll-like receptor and heme oxygenase-1 signaling in hepatic ischemia/reperfusion injury. Am J Transplant. 2005;5:1793–1800. [DOI] [PubMed] [Google Scholar]
- 42.Ito K, Ozasa H, Sanada K, et al. Doxorubicin preconditioning: a protection against rat hepatic ischemia-reperfusion injury. Hepatology. 2000;31:416–419. [DOI] [PubMed] [Google Scholar]