

Abstract
Objective:
To determine if 24-hour blood concentrations of macrophage migration inhibitory factor (MIF), soluble CD14, and CD163 receptors could predict complications associated with acute pancreatitis (AP).
Summary Background Data:
Soluble receptor proteins derived from the macrophage-monocyte lineage potentiate the inflammatory cytokine response early in AP. Understanding the temporal expression of these molecules could afford better measures for therapeutic intervention.
Methods:
Patients with AP (amylase >5 times normal) were recruited within 24-hour of onset of pain. Peripheral blood was analyzed for MIF, sCD163, and sCD14 levels and levels correlated with CRP, APACHE-II score, and clinical disease severity (Atlanta criteria); subclassified as multiorgan dysfunction (MOF), pancreatic necrosis (PN >30% on contrast CT), and death.
Results:
In total, 64 patients with AP (severe, 19: 8 had MOF alone, 7 both PN and MOF, 2 PN without MOF, and 2 single-organ failures with local septic complications) were recruited. Both sCD14 and MIF concentrations were elevated in patients with severe attacks (P = 0.004 and P < 0.001 respectively), and patients who developed MOF (P = 0.004 and P < 0.001). However, only serum MIF was significantly raised in patients who subsequently developed PN (median, 92.5 ng/mL; IQR, 26–181 vs. 31.1 ng/mL; IQR, 5–82, P < 0.001), independently of MOF (P = 0.01). Multivariate analysis demonstrated serum MIF as an independent predictor of PN (P = 0.01; OR = 2.73; 95% CI, 2.72–2.74).
Conclusion:
The prognostic utility of 24-hour plasma MIF concentration in predicting PN has major clinical and healthcare resource implications. Its mechanistic pathway may afford novel therapeutic interventions in clinical disease by using blocking agents to ameliorate the systemic manifestations of AP.
Serum MIF concentration within 24 hours of onset of abdominal pain associated with acute pancreatitis was elevated in patients with severe attacks, those who had an associated multiorgan failure, and could predict patients likely to develop pancreatic necrosis.
Acute pancreatitis (AP) is a life-threatening illness with an annual incidence of 30 to 50 attacks per 100,000 inhabitants.1,2 The clinical presentation ranges from a mild edematous, self-limiting disease with good prognosis to severe necrotizing inflammation, fatal in about 15% to 20% of instances. In patients with severe AP, multiorgan failure (MOF) is responsible for early disease mortality while sepsis supervenes later and remains the major cause of death.2–4 Secondary infection occurs in 30% to 70% of patients with pancreatic necrosis,5–8 and is associated with significantly increased mortality.5 Both the risk of MOF and infective complications appear related to the degree of pancreatic necrosis.9
The current British Society of Gastroenterology Working Party Guidelines has made a number of recommendations on the management of AP,10 which have major implications on healthcare resources for local and specialist centers. These guidelines recommend patients with severe attacks should be managed on a high dependency unit/intensive care with full monitoring facilities recommendation grade B, and specialist center referral should be considered for patients with extensive necrotizing pancreatitis or complications requiring interventional radiology, endoscopic, or surgical procedures recommendation grade B. Among the greatest difficulties in implementing these guidelines is the early identification of patients with severe acute pancreatitis and those likely to benefit from early transfer to specialist units. An idea first coined in 1974 by the late John H. C. Ranson introduced the concept of a “prognostic” scoring system for early identification of patients with severe pancreatitis.11 Multifactorial scoring systems have since evolved, attempting to identify an “at risk” group that may potentially benefit from aggressive medical or surgical management.12 Such stratification is initially a prediction only and becomes a fact (by definition) when systems failure or local complications supervene. A number of laboratory markers predicting disease severity have been reported, either based on the degree of the inflammatory reaction,13,14 tests that relate to the activation of trypsinogen and other pancreatic proenzymes,15–18 tests that measure leakage of certain pancreatic enzymes,19,20 or scoring systems (Ranson's, APACHE-II).11,21 However, none can accurately predict disease severity within 24-hour of onset, and the prediction of pancreatic necrosis at this stage has certainly not been reported.
We have previously shown an association between monocyte expression of the CD14 receptor, a glycosyl-phosphatidylinositol anchored cell surface molecule, and severity of an attack of AP.22 Furthermore, it appeared that plasma concentrations of the soluble receptor, sCD14, correlated with the magnitude of the systemic inflammatory response as early as 24-hour after the onset of disease, and with the subsequent development of MOF.
Given the importance of the monocyte-macrophage lineage in the pathogenesis of the local and systemic complications of AP, we sought to investigate if peripheral venous blood concentrations of sCD163, a glycoprotein belonging to group B of the scavenger-receptor cysteine-rich superfamily,23 and macrophage migratory inhibitory factor (MIF), a key effector molecule of the innate and acquired immune system released by macrophages, are associated with severity of an attack of AP.
CD163 is a highly specific marker for cells of the macrophage-monocyte lineage, and is regulated by both pro- and anti-inflammatory mediators.23,24 It has been shown to be present as a soluble receptor in plasma (sCD163) and elevated levels have been found in patients with sepsis complicating hematologic malignancy.25 Its role, however, in an acute inflammatory disorder is as yet undetermined.
Macrophage MIF was originally discovered in 1966 as a cytokine derived from activated T lymphocytes, that prevents the random migration of macrophages at the site of inflammation.26 MIF is secreted by T cells and macrophages in response to gram-negative and gram-positive bacteria, pro-inflammatory cytokines (TNF-α, IL-2 and IFN-γ), and through stimulation by glucocorticoids.27 MIF release produces a pro-inflammatory cytokine response and serum levels in patients with severe AP have been demonstrated to be significantly higher than those with a mild attack.28 A MIF gene Knock-Out (MIF −/−) rodent model has shown significant amelioration of disease severity.28
The current study aims to determine the relationship between sCD163 and MIF, in addition to sCD14 to the early inflammatory response associated with AP, and there prognostic utility to assess the local (pancreatic necrosis) and systemic complications (SIRS, MOF) associated with a severe attack.
METHODS
Patients were recruited over an 18-month period from the Leeds Teaching Hospitals NHS Trust, Leeds, U.K. For the purposes of the study, acute pancreatitis was defined as an attack of acute upper abdominal pain accompanied by hyperamylasemia exceeding 5 times the upper normal range, diagnostic verification by computer tomography (CT) and a consistent clinical course. Patients were recruited only if the onset of abdominal pain was reported to be within 24-hour of presentation. Attacks were stratified by:
a. Etiology: Alcohol-related (if consumption exceeded 50 g/day); gallstones (detected by ultrasonography, MRCP, EUS, or ERCP); other identified causes or idiopathic (cause unknown despite investigation).
b. Severity of the attack: attacks were classed as “severe” if there was failure of one or more organ, or if there was pancreatic necrosis (> 30% on intravenous contrast-enhanced CT performed after 72-hour of attack), or sepsis. This classification was consistent with the Atlanta Criteria.
Consent: Inclusion and Exclusions
Written, informed consent was obtained according to a protocol approved by both institutional ethical committees. Patients with malignant disease, chronic inflammatory disease, including established or suspected chronic pancreatitis, preexisting chronic organ failure (renal failure; creatinine >200 mg/dL or requiring dialysis, heart failure, NYHA class >3, recent myocardial infarction [<6 months]), unstable coronary syndromes, liver failure (Childs grade B+), chronic obstructive airways disease, and immunosuppressive disorders (drugs, hematologic malignancies, HIV) were excluded due to potential variations in the systemic inflammatory responses and their potential influence on treatment decisions.
Determination of Severity
A 5-mL sample of heparin treated venous blood was obtained from all patients every 24 hours for at least 4 days from the onset of pain, from which C-reactive protein levels were measured using an enzyme-linked immunosorbent assay (DAKO, High Wycombe, UK). The peak (0–72 hours) CRP concentration (Peak CRP) was used as a biochemical severity marker.
The Acute Physiology and Chronic Health Evaluation score (APACHE-II), and its subcomponent, the Acute Physiology Score (APS) were determined at 24- and 48-hour from the onset of pain, to estimate the magnitude of the systemic inflammatory response.
The 24-hour plasma sCD14, sCD163, and serum MIF concentration was determined from a peripheral blood samples taken at approximately 24-hour after the reported onset of abdominal pain.
Plasma sCD14 Assay
Plasma sCD14 concentration was measured from a heparin treated peripheral venous blood by a CD14 capture sandwich enzyme linked immunosorbent assay (IBL, Hamburg, Germany). The lower limit of detection was <1 ng/mL.
Plasma sCD163 Assay
Determination of sCD163 concentration in serum sCD163 was measured in enzyme-linked immunosorbent assay. In brief, rabbit anti-CD163, 4 mg/L was coated onto microtitre wells. After wash, 100 μL of sample (diluted 1:50 in PBS with albumin, pH 7.2) was added and incubated for 1 hour. The wells were washed and 100 μL of monoclonal anti-CD163 (GHI/61, diluted 1:500) was added and incubated for 1 hour. After wash, 100 μL of peroxidase-labeled antibody (goat anti-mouse immunoglobulins, DAKO P447, diluted 1:4000) was added and incubated for 1 hour. The wells were washed, and 100 μL of a H2O2/1,2-phenylenediamine dihydrochloride substrate solution was added. After 15 minutes, 50 μL of 1 mol/L H2SO4 was added, and the plates were read at 492/620 nm. Control samples and standards of purified CD163 were coanalyzed in each run.
Serum Macrophage MIF Assay
Serum MIF concentration was measured by a quantitative sandwich enzyme linked immunosorbent assay (Quantikine, R&D systems, Abingdon, U.K.). The lower limit of detection was <0.05 ng/mL and overall intra-assay variation was <5%.
Statistical Analysis
Results obtained for genotypes were analyzed with reference to etiology and severity using Pearson's χ2 contingency tables and Fisher exact test. Comparison of continuous variables with genotype was analyzed using the Mann-Whitney U test. A receiver operating characteristic (ROC) curve was used to explore the relationship between the sensitivity and specificity of the prognostic indices in determining disease severity. An index of the goodness of the test is the area under the curve; a perfect test has area 1.0, while a nondiscriminating test has area 0.5. All statistical analyses were performed using SPSS v8.00 statistical analysis software (SPSS Inc., Cary, NC).
RESULTS
In total, 64 patients with AP (19 severe) were studied. Age and gender were similar in all groups (Table 1) and, as expected, the APACHE-II scores at 48 hours were significantly higher in patients with a severe attack (median, 10.5; range, 4–26) compared with those with a mild attack of AP (median, 4.5; range, 1–10, P < 0.001). The cause of AP was gallstones in 40 patients (63%), and ethanol in 19 patients (29%). In the remaining 5 patients (8%), AP was due to hyperlipidemia, pancreas divisum, or unknown (designated idiopathic). Among patients with a severe attack, 8 had MOF alone, 7 had pancreatic necrosis and MOF, 2 patients had pancreatic necrosis without MOF, and 2 had single-organ failure with local septic complications. Secondary infection of pancreatic necrosis (confirmed by both CT-guided fine needle aspiration and surgical debridement) occurred in 2 patients. Overall mortality was 9.4% (6 patients).
TABLE 1. Demographic Details of the Study Group

Soluble Receptors and Acute Pancreatitis
Plasma sCD14 levels were significantly higher at 24 hours in patients with a severe attack (median, 71.3 ng/mL; range, 25–215 ng/mL) compared with mild attack (median, 51.2 ng/mL; range, 24–103 ng/mL, P = 0.004; Fig. 1). Plasma concentrations in healthy volunteers (median, 50.5 ng/mL; range, 32–76 ng/mL) were similar to mild attacks. Furthermore, plasma sCD14 levels correlated significantly with APS (r = 0.59, P < 0.001), 24-hour (r = 0.42, P = 0.002), and 48-hour APACHE-II (r = 0.43, P < 0.001). Although there was no correlation with 24-hour CRP, sCD14 did correlate significantly albeit weakly with Peak CRP (r = 0.28, P = 0.03).

FIGURE 1. a-c, Box-whisker plots (95% CI) showing 24-hour serum MIF concentrations in patients with AP and severity of attack, MOF, and pancreatic necrosis.
Serum MIF levels were also raised in patients with a severe attack (median, 58.8 ng/mL; range, 13–181 ng/mL) compared with mild attack (median, 20.3 ng/mL; range, 5–80 ng/mL, P < 0.001; Table 2). The latter was similar to levels in healthy controls (median, 18.2 ng/mL; range, 12–57 ng/mL). Serum MIF levels correlated significantly with serum 24-hour CRP (r = 0.36, P = 0.02), Peak CRP (r = 0.36, P = 0.003), and with 48-hour APACHE-II (r = 0.29, P = 0.03). There was no significant correlation with 24-hour APS or APACHE-II score. There was no significant correlations between plasma sCD163 and either clinical or biochemical markers of disease severity (Table 2).
TABLE 2. Correlation of 24-Hour Disease Markers With Complications of Acute Pancreatitis

Monocyte Receptors and MOF
Patients who either had or developed MOF had significant elevations in plasma sCD14 (median, 71.3 ng/mL; range, 25–216 ng/mL) compared with those who did not (median, 51.6 ng/mL; range, 24–132 ng/mL, P = 0.004).
Serum MIF levels were also increased in patients with MOF (median, 58.8 ng/mL; range, 13–118 ng/mL) compared with those without MOF (median, 27.3 ng/mL; range, 5–181 ng/mL, P < 0.001, Figure 1b). Although 24-hour APACHE-II score and CRP was also raised (Table 2), levels of sCD163 failed to significantly differentiate between the 2 groups.
Monocyte Receptors and Pancreatic Necrosis
In total, 9 of 64 patients developed pancreatic necrosis (14%). From the monocyte-macrophage markers (Table 2), only 24-hour serum MIF concentration was able to significantly differentiate between patients who developed pancreatic necrosis (median, 92.5 ng/mL; range, 26–181 ng/mL) and those who did not (median, 31.1 ng/mL; range, 5–82 ng/mL, P < 0.001, Figure 1c). Serum MIF concentrations were significantly higher among patients with PN independent of the presence of MOF (P = 0.01). No such relationship was evident for sCD14 or sCD163. Binary logistic regression analysis reported serum MIF concentration (P = 0.01, OR, 2.73; 95% CI, 2.72–2.74) and MOF (P = 0.017, OR, 1.07; 95% CI, 1.01–1.86) as independent predictors of pancreatic necrosis.
Monocyte Receptors and Survival
Five of 38 patients died (13%), 4 had both MOF and pancreatic necrosis and 1 with MOF alone. Plasma MIF was significantly lower in survivors compared with patients who died (Fig. 2). Soluble CD14 and CD163 concentrations were similar in both groups.
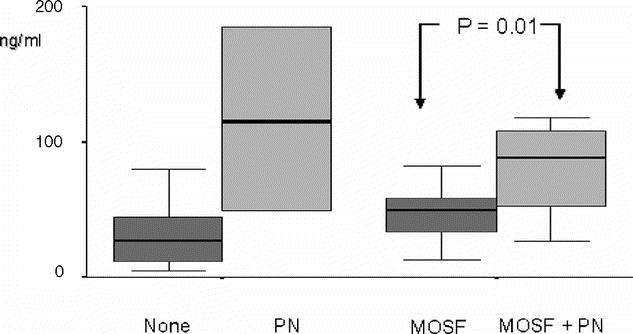
FIGURE 2. Box-whisker plots (95% CI) showing 24-hour serum MIF concentrations in patients with AP stratified to MOF and the presence of pancreatic necrosis.
ROC Curve Analysis for Severity, MOF, and Pancreatic Necrosis
Table 3 shows the area under the ROC curves for each 24-hour predictive markers with respect to the local and systemic complications of AP. Although the APACHE-II score and sCD14 levels were comparable in terms of disease severity and MOF, serum macrophage MIF concentrations were far superior in predicting complications of AP, particularly pancreatic necrosis (area under ROC, 0.86; P < 0.001).
TABLE 3. Area Under Receiver Operated Curve (ROC) Analysis for All Markers Based on 24-Hour Values Based on Disease Severity, Presence of MOF, and Pancreatic Necrosis

DISCUSSION
The identification of molecular prognostic markers early in the course of AP has several benefits: risk stratification for the allocation of costly and limited healthcare resources, patient selection for clinical trials, and for potential development of therapies targeted at such mediators to ameliorate disease severity.
Fenton-Lee and Imrie29 estimated the mean quality of life year (QALY) gained per patient with severe AP to be 8.6 years, at an average cost per QALY of £2156. The overall cost of treating these patients ranges from £9000 to £ 34,000, and these costs are likely to be an even greater burden on health resources given that Trinto et al30 reported that the age-standardized hospital admission rates between 1990 and 2000 had increased by 43%. The increased incidence of gallstone disease, obesity, and alcohol consumption will also inevitably add to the human and financial costs. Most of patients have mild self-limiting disease, which settles within 3 to 5 days; however, patients with severe attacks often require aggressive fluid resuscitation, enteral nutrition, and should be managed on a high dependency unit with full monitoring facilities.31 Early mortality is often due to MOF, and sepsis supervenes at a later stage and contributes to over 80% of late deaths; therefore, early identification of “at risk” patients allows more efficient allocation of resources.31
In the current study, we examined the potential utility of soluble receptors and the secretory protein MIF derived from the monocyte-macrophage lineage, in predicting local and systemic complications associated with an attack of AP.
We confirm our previous reports of a strong correlation between sCD14 concentration, severity of attack of AP, and the physiologic component of the APACHE-II score, the acute physiologic score.22 Plasma soluble CD14 receptor concentration at 24 hours was also predictive of MOF but not PN, suggesting that LPS interaction with sCD14 may be an important mediator of SIRS and multiorgan failure associated with AP. Soluble sCD163 levels failed to correlate with the severity of AP, and levels in healthy controls were similar to those in AP. This would suggest that sCD163 is not associated with this disease.
MIF was originally described as a product of T-lymphocytes in 1966, and later demonstrated to be secreted from a variety of epithelial, endocrine, and endothelial cells.26,32–34 Unlike other cytokines, for example, TNF and IL-1 that require de novo synthesis,35,36 MIF is preformed and stored as intracellular pools within secretory vesicles.32 Its secretion from monocytes and macrophages is part of the innate immune response and occurs in response to lipopolysaccharides, gram-positive exotoxins, and pro-inflammatory cytokines (TNF-α, IFN-γ). It has both autocrine and paracrine effects; pro-inflammatory effects38 include induction of cytokines (TNF-α, IL-1b, IL-2, IL-6, IL-8, and IFN-γ),38–40 NO,41 MMP-2 expression,42 and on the COX-2 pathway.43 It counteracts glucocorticoid induced inhibition of inflammatory cytokine secretion in macrophages and T cells, and as such is thought to be a critical regulator of the host inflammatory response.44,45
Elevations in serum and ascitic MIF levels have been demonstrated in experimental pancreatitis, with increased survival observed in rodents treated with prophylactic administration of anti-MIF antibody.45,46 A number of clinical studies have also examined the prognostic role of MIF in septic patients. Beishuizen et al47 measured MIF, cortisol, ACTH, TNF-α, and IL-6 levels in 40 critically ill patients. Serum MIF concentrations were significantly elevated in patients with septic shock and correlated with mortality. Interestingly, however, MIF levels in multitrauma nonseptic patients were similar to healthy controls, and Joshi et al later confirmed that MIF levels are elevated in trauma patients only in the presence of positive bacterial cultures.48
We found serum macrophage MIF levels were elevated in patients with a severe attack of AP, particularly among those who were subsequently diagnosed as having pancreatic necrosis (>30%) on contrast enhanced CT (P = 0.002). Stratification of attacks to MOF with and without pancreatic necrosis demonstrated clearly that elevations in serum MIF concentration were related to the presence of PN and not MOF (OR, 2.72; P = 0.01). This is the first report of an early prognostic marker for the development of pancreatic necrosis.
A significant correlation exists between the development of pancreatic necrosis, the frequency of bacterial contamination of necrosis and the evolution of systemic complications.9 Secondary infection is thought to originate from enteric bacterial translocation secondary to disruption of the gut mucosal barrier during acute pancreatitis,49 and this has been shown to occur within 24 hours of hospitalization. Clinical interventions that may limit this process are early enteral nutrition and the controversial use of prophylactic antibiotics. A recent Cochrane review reported there was strong evidence that intravenous antibiotic prophylactic therapy up to 14 days decreased the risk of superinfection of necrotic tissue and mortality in patients with severe acute pancreatitis with proven pancreatic necrosis at CT.50 Furthermore, it is becoming increasingly recognized that misuse of antibiotics may lead to devastating superinfections caused by Candida.51 Limiting prophylactic antibiotic use in severe pancreatitis will minimize the development of resistance and superinfections in vulnerable hosts, and also avoids unnecessary costs.
Maintenance of the intestinal structure and function is supported by enteral feeding rich in glutamine and short chain fatty acids to stimulate the proliferation of mucosal cells and enhance gut integrity. Enteral nutrition may benefit patients with a severe attack and theoretically reduce secondary infection of pancreatic necrosis. Given that gut mucosal barrier dysfunction is observed as early as 24 hours after the onset of abdominal pain, enteral nutrition may have greatest benefit if commenced in the “at risk” group as early as possible following an attack.
Considerable interest has grown in the development of reliable biochemical markers that reflect the severity of acute pancreatitis as improved outcome in the severe form of the disease is based on early identification of disease severity and subsequent focused management of these high-risk patients. A meta-analysis of the prognostic utility of some of the more reliable markers in predicting severe AP is shown in Table 4. 21 Of the current markers, serum CRP concentration appears to be the only one widely used in clinical practice. Although a late severity marker, it is, however, fairly useless in predicting disease outcome when measured on the first day of admission.52 Interleukin-6 (IL-6), however, the principal mediator of the synthesis of C-reactive protein and other acute phase reactants, has been reported to discriminate severe from mild disease as early as 24 hours,13,14,53 as have trypsin-alpha 1-protease inhibitor complexes16–20 and carboxypeptidase activation peptide15 (Table 4). The current study suggests that although macrophage MIF compares less favorably in predicting disease severity (sensitivity, 79%; specificity, 74%; cutoff, 420 mg/L), it is the only reported early prognostic index of pancreatic necrosis. However, the current study is limited by the relative small population cohort and the true potential of MIF in predicting the complications of severe AP needs to be validated by larger studies.
TABLE 4. Meta-analysis of Prognostic Inflammatory Mediators

Furthermore, macrophage MIF unlike TAP and carboxypeptidase activation peptide has other advantages, its widespread availability, it is relatively inexpensive, and has the major advantage in that it is a key mediator of the inflammatory process and therefore may be a potential therapeutic target at the molecular level. MIF exacerbates LPS-induced cytotoxicity, and anti-MIF neutralizing antibodies have been shown to rescue mice from lethal endotoxemia.45,46 Both MIF gene knock-out and antisense oligonucleotide experimental models demonstrate reduced endogenous MIF expression and reduction in the pro-inflammatory cytokine production following LPS stimulation.54 The mechanisms, although as yet unclear, involve sustained activation of ERK MAP kinase and thus cytoplasmic phospholipase A2, a target anti-inflammatory for glucocorticoids.55 MIF may also act to activate NFkappaB, a regulator cytokine gene expression.56,57 The antagonistic effect of glucocorticoids is to inhibit the binding of the p65 subunit of NFkappaB to the transcriptional machinery of the target gene, and to induce IkappaB synthesis, thereby inhibiting NFkappaB translocation to the nucleus.58,59 Furthermore, MIF inhibits tumor suppressor p53, leading to defective apoptosis of activated macrophages.60 This prolonged pro-inflammatory response may contribute to the development of pancreatic necrosis and systemic organ failure.
CONCLUSION
The observed correlations between MIF and sCD14 blood concentrations and the local and systemic complications of severe AP further implicate endotoxemia as a central mechanism in this disease. Serum MIF concentration may serve as important prognostic marker that could identify patients likely to benefit from aggressive medical treatment and early transfer to specialist pancreatic units as suggested by the current BSG working party guidelines. At the molecular level, targeted disruption of these proteins with monoclonal antibodies may afford some therapeutic benefit in preventing the development of MOF, pancreatic necrosis, and septic complications.
Footnotes
Reprints: Sakhawat H. Rahman, MD, MRCS, Division of Surgery, University of Leeds School of Medicine, Clinical Sciences Building, St. James's University Hospital, Leeds LS7 9TF, U.K. E-mail: zak-rahman@lineone.net.
REFERENCES
- 1.McKay CJ, Imrie CW. The continuing challenge of early mortality in acute pancreatitis. Br J Surg. 2004;91:1243–1244. [DOI] [PubMed] [Google Scholar]
- 2.Mann DV, Hershman MJ, Hittinger R, et al. Multicentre audit of death in acute pancreatitis. Br J Surg. 1994;81:890–893. [DOI] [PubMed] [Google Scholar]
- 3.Renner IG, Savage WT III, et al. Deaths due to acute pancreatitis: a prospective analysis of 405 autopsy cases. Dig Dis Sci. 1985;30:1005. [DOI] [PubMed] [Google Scholar]
- 4.Buggy BP, Nostrant TT. Lethal pancreatitis. Am J Gastroenterol. 1983;78:810–814. [PubMed] [Google Scholar]
- 5.Beger HG, Bittner R, Block S, et al. Bacterial contamination of pancreatic necrosis: a prospective clinical study. Gastroenterology. 1986;91:433–438. [DOI] [PubMed] [Google Scholar]
- 6.Gerzof SA, Banks PA, Robbins AH, et al. Early diagnosis of pancreatic infection by computed tomography-guided aspiration. Gastroenterology. 1987;93:1315–1320. [DOI] [PubMed] [Google Scholar]
- 7.Bassi C, Falconi M, Girelli R, et al. Microbiological findings in severe pancreatitis. Surg Res Commun. 1989;5:1–4. [Google Scholar]
- 8.Bradley EL III. A clinically based classification system for acute pancreatitis. Arch Surg. 1993;128:586–590. [DOI] [PubMed] [Google Scholar]
- 9.Isenmann R, Rau B, Beger HG. Bacterial infection and extent of necrosis in severe acute pancreatitis are determinants of organ failure in patients with acute necrotizing pancreatitis. Br J Surg. 1999;86:1020–1024. [DOI] [PubMed] [Google Scholar]
- 10.UK Working Party on Acute Pancreatitis. UK guidelines for the management of acute pancreatitis. Gut 2005;54:iii1–iii9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ranson JHC, Rifkind KM, Roses DF, et al. Prognostic signs and the role of operative management in acute pancreatitis. Surg Gynecol Obstet. 1974;139:69–81. [PubMed] [Google Scholar]
- 12.Karimgani I, Porter K, Langevin R, et al. Prognostic factors in sterile pancreatic necrosis. Gastroenterology. 1992;103:1636–1640. [DOI] [PubMed] [Google Scholar]
- 13.Wilson C, Heads A, Shenkin A, et al. C-reactive protein, antiproteases and complement factors as objective markers of severity in acute pancreatitis. Br J Surg. 1989;76:177–181. [DOI] [PubMed] [Google Scholar]
- 14.Mayer JM, Rau B, Siech M, et al. Local and systemic zymogen activation in human acute pancreatitis. Digestion. 2000;62:164–170. [DOI] [PubMed] [Google Scholar]
- 15.Appelros S, Petersson U, Toh S, et al. Activation peptide of carboxypeptidase B and anionic trypsinogen as early predictors of the severity of acute pancreatitis. Br J Surg. 2001;88:216–221. [DOI] [PubMed] [Google Scholar]
- 16.Borgstrom A, Lasson A. Trypsin-alpha 1-protease inhibitor complexes in serum and clinical course of acute pancreatitis. Scand J Gastroenterol. 1984;19:1119–1122. [PubMed] [Google Scholar]
- 17.Gudgeon AM, Heath D, Hurley P, et al. Trypsinogen activation peptide assay in the early prediction of severity of acute pancreatitis. Lancet. 1990;335:4–8. [DOI] [PubMed] [Google Scholar]
- 18.Hedstrom J, Sainio V, Kemppainen E, et al. Serum complex of trypsin 2 and alpha 1 anti-trypsin as diagnostic and prognostic marker of acute pancreatitis: clinical study in consecutive patients. BMJ. 1996;313:333–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hedstrom J, Haglund C, Leinonen J, et al. Trypsinogen-1, -2 and tumour-associated trypsin-inhibitor in bile and biliary tract tissues from patients with biliary tract diseases and pancreatic carcinomas. Scand J Clin Lab Invest. 2001;61:111–118. [DOI] [PubMed] [Google Scholar]
- 20.Sainio V, Puolakkainen P, Kemppainen E, et al. Serum trypsinogen-2 in the prediction of outcome in acute necrotizing pancreatitis. Scand J Gastroenterol. 1996;31:818–824. [DOI] [PubMed] [Google Scholar]
- 21.Larvin M, McMahon MJ. Apache-II score for assessment and monitoring of acute pancreatitis. Lancet. 1989;22:201–205. [DOI] [PubMed] [Google Scholar]
- 22.Rahman SH, Salter G, Holmfield JHM, et al. Soluble CD14 receptor expression and monocyte heterogeneity but not the C-260T CD14 genotype is associated with severe acute pancreatitis. Crit Care Med. 2004;32:2457–2463. [DOI] [PubMed] [Google Scholar]
- 23.Kristiansen M, Graversen J, Jacobsen J, et al. Identification of the hemoglobin scavenger receptor. Nature. 2001;409:198–201. [DOI] [PubMed] [Google Scholar]
- 24.Zwadlo G, Voegeli R, Osthoff KS, et al. A mAb to a novel differentiation antigen on human macrophages associated with the down-regulatory phase of the inflammatory process. Exp Cell Biol. 1987;55:295–304. [DOI] [PubMed] [Google Scholar]
- 25.Møller HJ, Peterslund NA, Graversen JH, et al. Identification of the hemoglobin scavenger receptor/CD163 as a natural soluble protein in plasma. Blood. 2002;99:378–380. [DOI] [PubMed] [Google Scholar]
- 26.Bloom BR, Bennett B. Mechanism of a reaction in vitro associated with delayed-type hypersensitivity. Science. 1966;153:80–82. [DOI] [PubMed] [Google Scholar]
- 27.Calandra T, Bernhagen J, Metz CN, et al. MIF as a glucocorticoid-induced modulator of cytokine production. Nature. 1995;377:68–71. [DOI] [PubMed] [Google Scholar]
- 28.Sakai Y, Masamune A, Satoh A, et al. Macrophage migration inhibitory factor is a critical mediator of severe acute pancreatitis. Gastroenterology. 2003;124:725–736. [DOI] [PubMed] [Google Scholar]
- 29.Fenton-Lee D, Imrie CW. Pancreatic necrosis: assessment of outcome related to quality of life and cost of management. Br J Surg. 1993;80:1579–1582. [DOI] [PubMed] [Google Scholar]
- 30.Trinto A, Lloyd DA, Kang JY, et al. Acute and chronic pancreatitis—diseases on the rise: a study of hospital admissions in England 1989/90–1999/2000. Aliment Pharmacol Ther. 2002;16:2097–2105. [DOI] [PubMed] [Google Scholar]
- 31.McKay CJ, Buter A. Natural history of organ failure in acute pancreatitis. Pancreatology. 2003;3:111–114. [DOI] [PubMed] [Google Scholar]
- 32.Nishino T, Bernhagen J, Shiiki H, et al. Localization of macrophage-migration inhibitory factor (MIF) to secretory granules within the corticotropic and thyrotropic cells of the pituitary-gland. Mol Med. 1995;1:781–788. [PMC free article] [PubMed] [Google Scholar]
- 33.Bacher M, Meinhardt A, Lan HY, et al. Migration inhibitory factor expression in experimentally induced endotoxemia. Am J Pathol. 1997;150:235–246. [PMC free article] [PubMed] [Google Scholar]
- 34.Donnelly SC, Haslett C, Reid PT, et al. Regulatory role for macrophage migration inhibitory factor in acute respiratory distress syndrome. Nat Med. 1997;3:320–323. [DOI] [PubMed] [Google Scholar]
- 35.Chesney J, Metz C, Bacher M, et al. An essential role for macrophage migration inhibitory factor (MIF) in angiogenesis and the growth of a murine lymphoma. Mol Med. 1999;5:181–191. [PMC free article] [PubMed] [Google Scholar]
- 36.Shimizu T, Abe R, Nakamura H, et al. High expression of macrophage migration inhibitory factor in human melanoma cells and its role in tumor cell growth and angiogenesis. Biochem Biophys Res Commun. 1999;264:751–758. [DOI] [PubMed] [Google Scholar]
- 37.Calandra T, Spiegel LA, Metz CN, et al. Macrophage migration inhibitory factor is a critical mediator of the activation of immune cells by exotoxins of gram-positive bacteria. Proc Natl Acad Sci USA. 1998;95:11383–11388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Calandra T, Bernhagen J, Mitchell RA, et al. The macrophage is an important and previously unrecognized source of macrophage-migration inhibitory factor. J Exp Med. 1994;179:1895–1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bacher M, Metz CN, Calandra T, et al. An essential regulatory role for macrophage migration inhibitory factor in T-cell activation. Proc Natl Acad Sci USA. 1996;93:7849–7854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Benigni F, Atsumi T, Calandra T, et al. The proinflammatory mediator macrophage migration inhibitory factor induces glucose catabolism in muscle. J Clin Invest. 2000;106:1291–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bernhagen J, Calandra T, Mitchell RA, et al. Purification and characterization of the cytokine macrophage- migration inhibitory factor (MIF). FASEB J. 1994;8:A1417. [Google Scholar]
- 42.Onodera S, Kaneda K, Mizue Y, et al. Macrophage migration inhibitory factor upregulates expression of matrix metalloproteinases in synovial fibroblasts of rheumatoid arthritis. J Biol Chem. 2000;275:444–450. [DOI] [PubMed] [Google Scholar]
- 43.Mitchell RA, Metz CN, Peng T, et al. Sustained mitogen-activated protein kinase (MAPK) and cytoplasmic phospholipase A2 activation by macrophage migration inhibitory factor (MIF): regulatory role in cell proliferation and glucocorticoid action. J Biol Chem. 1999;274:18100–18106. [DOI] [PubMed] [Google Scholar]
- 44.Bucala R. MIF rediscovered: cytokine, pituitary hormone, and glucocorticoid-induced regulator of the immune response. FASEB J. 1996;10:1607–1613. [DOI] [PubMed] [Google Scholar]
- 45.Calandra T, Echtenacher B, Roy DL, et al. Protection from septic shock by neutralization of macrophage migration inhibitory factor. Nat Med. 2000;6:164–170. [DOI] [PubMed] [Google Scholar]
- 46.Echtenacher B, Falk W, Mannel DN, et al. Requirement of endogenous tumor necrosis factor/cachectin for recovery from experimental peritonitis. J Immunol. 1990;145:3762–3766. [PubMed] [Google Scholar]
- 47.Beishuizen A, Thijs LG, Haanen C, et al. Macrophage migration inhibitory factor and hypothalamo-pituitary-adrenal function during critical illness. J Clin Endocrinol Metab. 2001;86:2811–2816. [DOI] [PubMed] [Google Scholar]
- 48.Joshi PC, Poole GV, Sachdev V, et al. Trauma patients with positive cultures have higher levels of circulating macrophage migration inhibitory factor (MIF). Res Commun Mol Pathol Pharmacol. 2000;107:13–20. [PubMed] [Google Scholar]
- 49.Rahman SH, Ammori BJ, Holmfield JHM, et al. Intestinal hypoperfusion contributes to gut barrier failure in severe acute pancreatitis. J Gastrointest Surg. 2003;7:26–36. [DOI] [PubMed] [Google Scholar]
- 50.Bassi C, Larvin M, Villatoro E. Antibiotic therapy for prophylaxis against infection of pancreatic necrosis in acute pancreatitis. Cochrane Database Syst Rev. 2003;(4):CD002941. [DOI] [PubMed] [Google Scholar]
- 51.Isenmann R, Schwarz M, Rau B, et al. Characteristics of infection with candida species in patients with necrotizing pancreatitis. World J Surg. 2002;25:372–376. [DOI] [PubMed] [Google Scholar]
- 52.de Beaux AC, Goldie AS, Ross JA, et al. Serum concentrations of inflammatory mediators related to organ failure in patients with acute pancreatitis. Br J Surg. 1996;83:349–353. [DOI] [PubMed] [Google Scholar]
- 53.Pezzilli R, Billi P, Miniero R, et al. Serum interleukin-6, interleukin-8, and beta2-microglobulin in early assessment of severity of acute pancreatitis: comparison with C-reactive protein. Dig Dis Sci. 1995;40:2341–2348. [DOI] [PubMed] [Google Scholar]
- 54.Froidevaux C, Roger T, Martin C, et al. Macrophage migration inhibitory factor and innate immune responses to bacterial infections. Crit Care Med. 2001;297(suppl):13–15. [DOI] [PubMed] [Google Scholar]
- 55.Denhardt DT. Signal-transducing protein phosphorylation cascades mediated by Ras/Rho proteins in the mammalian cell: the potential for multiplex signalling. Biochem J. 1996;318:729–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Barnes PJ, Karin M. Nuclear factor-kappaB: a pivotal transcription factor in chronic inflammatory diseases. N Engl J Med. 1997;336:1066–1071. [DOI] [PubMed] [Google Scholar]
- 57.De Bosscher K, Vanden Berghe W, Vermeulen L, et al. Glucocorticoids repress NFkappaB- driven genes by disturbing the interaction of p65 with the basal transcription machinery, irrespective of coactivator levels in the cell. Proc Natl Acad Sci USA. 2000;97:3919–3924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Scheinman RI, Cogswell PC, Lofquist AK, et al. Role of transcriptional activation of I kappa B alpha in mediation of immunosuppression by glucocorticoids. Science. 1995;270:283–286. [DOI] [PubMed] [Google Scholar]
- 59.Auphan N, DiDonato JA, Rosette C, et al. Immunosuppression by glucocorticoids: inhibition of NF-kappa B activity through induction of I kappa B synthesis. Science. 1995;270:286–290. [DOI] [PubMed] [Google Scholar]
- 60.Hudson JD, Shoaibi MA, Maestro R, et al. A proinflammatory cytokine inhibits p53 tumor suppressor activity. J Exp Med. 1999;190:1375–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]