

Abstract:
Apoptosis plays a critical role in intestinal mucosal homeostasis. We previously showed that the bile salt taurodeoxycholate has a beneficial effect on the intestinal mucosa through an increase in resistance to apoptosis mediated by nuclear factor (NF)-κB. The current study further characterizes the effect of bile salts on intestinal epithelial cell susceptibility to apoptosis and determines if the X-linked inhibitor of apoptosis protein (XIAP) regulates bile salt-induced resistance to apoptosis. Exposure of normal intestinal epithelial cells (IEC-6) to the conjugated bile salts taurodeoxycholate (TDCA) and taurochenodeoxycholate (TCDCA) resulted in an increase in resistance to tumor necrosis factor (TNF)-α and cycloheximide (CHX)-induced apoptosis, and NF-κB activation. Treatment with TDCA and TCDCA resulted in an increase in XIAP expression. Specific inhibition of NF-κB by infection with an adenoviral vector that expresses the IκBα super-repressor (IκBSR) prevented the induction of XIAP expression and the bile salt-mediated resistance to apoptosis. Treatment with the specific XIAP inhibitor Smac also overcame this increase in enterocyte resistance to apoptosis. Bile salts inhibited formation of the active caspase-3 from its precursor procaspase-3. Smac prevented the inhibitory effect of bile salts on caspase-3 activation. These results indicate that bile salts increase intestinal epithelial cell resistance to apoptosis through NF-κB-mediated XIAP expression. Bile salt-induced XIAP mediates resistance to TNF-α/CHX-induced apoptosis, at least partially, through inhibition of caspase-3 activity. These data support an important beneficial role of bile salts in regulation of mucosal integrity. Decreased enterocyte exposure to luminal bile salts, as occurs during starvation and parenteral nutrition, may have a detrimental effect on mucosal integrity.
Exposure of intestinal epithelial cells to conjugated bile salts leads to increased resistance to apoptosis. This appears to occur through activation of NF-κB and subsequent induction of the X-linked inhibitor of apoptosis protein (XIAP).
Maintenance of mucosal integrity is important for preservation of normal digestive and gut barrier functions. Normal mucosal epithelial integrity depends on a dynamic balance between cell proliferation, growth arrest, and apoptosis.1–5 Increasing evidence indicates that apoptosis, rather than exfoliation of enterocytes, accounts for the majority of cell loss at the luminal surface of the intestine.4,6,7 Apoptosis also occurs in the crypt, where cell division takes place, thereby contributing to the maintenance of homeostasis in this region of the mucosal epithelium as well.3,4,7 Tipping the balance between “cell death” (apoptosis) and “cell birth” (from cell division) in either direction has significant pathologic consequences. For example, mucosal hyperplasia may result from situations in which the rate of cell proliferation exceeds apoptosis or the rate of cell death falls below the rate of cell production. In contrast, mucosal atrophy may occur if the rate of cell renewal is reduced below the rate of apoptosis or if the rate of cell death is increased beyond the rate of cell proliferation. Therefore, apoptosis is an important regulator of intestinal mucosal integrity.
NF-κB is a ubiquitous transcription factor that regulates the activation of a number of genes involved in proinflammatory responses, differentiation, and growth.8–10 NF-κB is found in the cytoplasm bound to endogenous inhibitors, known as IκBs, and is activated after the phosphorylation of IκB. This results in IκB degradation, release of NF-κB, translocation of NF-κB into the nucleus, and induction of transcription.11–15 In intestinal epithelial cells, activated NF-κB induces the expression of numerous genes that affect mucosal inflammation and repair.
NF-κB has a proapoptotic or antiapoptotic function depending on the cell type and stimulus.16–21 Several proapoptotic genes, including p53, Fas ligand, and the IL-1β-converting enzyme, consistently have NF-κB binding sequences in their promoter.22–25 In contrast, NF-κB activity appears to be necessary for activation of genes that suppress some types of apoptosis. For example, in immature B cells exposed to anti-IgM and in the developing liver, inhibition of NF-κB activity markedly enhances apoptotic cell death.26,27 Antiapoptotic factors that are up-regulated by NF-κB include manganese superoxide dismutase, c-IAP, AKT kinase, and the zinc finger protein A20.28 Li et al21 demonstrated that polyamine depletion activates NF-κB, which, in turn, decreases the susceptibility of IEC-6 cells to TNF-α-induced apoptosis. Others16,20,27 have shown that NF-κB is a cell survival factor that protects cells from death stimuli. In general, NF-κB is thought to inhibit apoptosis through caspase-dependent pathways. Numerous reports have documented the antiapoptotic action of NF-κB as well as the proapoptotic effects of inactivating NF-κB in a variety of cell types.16,20,27,29
Previous studies in our laboratory revealed that bile salts have beneficial effects on the small intestinal mucosa. Following injury, bile salts augment intestinal epithelial cell migration. This effect is mediated at least partially by activating NF-κB, which increases the expression of TGF-β, a cytokine known to induce intestinal epithelial cell migration after injury.30–32 We also demonstrated that bile salts at physiologic concentrations stimulate the proliferation of the intestinal epithelium, which is regulated through an increase in c-Myc expression.33 We have shown that the bile salt taurodeoxycholate (TDCA) has a beneficial effect on intestinal mucosal integrity by increasing enterocyte resistance to apoptosis through NF-κB.34 Others have shown that a decrease in luminal bile salts exacerbates the mucosal atrophy seen with the absence of enteric nutrients.35–39 In concordance with these observations, animals with biliary diversion after small bowel resection demonstrate an impaired adaptive response.37–39 Thus, bile salts may also have potentially beneficial effects during mucosal renewal.
The inhibitor of apoptosis (IAP) family of proteins are potent natural factors that function by directly inhibiting the activity of caspases, the principal effectors of apoptosis. IAPs inhibit caspases through 2 distinct mechanisms: 1) by directly interacting with caspases and 2) by facilitating a RING-dependent ubiquination and proteasomal degradation of caspases.40 Members of the mammalian IAP family include: the X-linked inhibitor of apoptosis protein (XIAP), the cellular inhibitor of apoptosis protein-1 (cIAP-1), the cellular inhibitor of apoptosis protein-2 (cIAP-2), and others, which directly inhibit caspase-3, caspase-7, and caspase-9.40,41 The baculovirus IAP repeat (BIR) 3 domain is responsible for the inhibition of active caspase-9, while the linker between BIR 1 and BIR 2 inhibits active caspase-3. Further, XIAP has additional antiapoptotic activities because mutant XIAP proteins unable to inhibit caspase-3 and caspase-9 retain their ability to inhibit apoptosis.42,43
Recently, it has been shown that the expression of cIAP-2 and XIAP is regulated by NF-κB and that the NF-κB-mediated IAPs are involved in protecting endothelial cells from TNF-α/cycloheximide (CHX)-induced apoptosis.44 Moreover, NF-κB-mediated IAP expression induces the resistance of intestinal epithelial cells to apoptosis after polyamine depletion.45 IAPs are important regulators of intestinal epithelial cell apoptosis. In this study, we hypothesize that bile salts increase the cellular resistance to apoptosis through increased XIAP expression regulated by bile salt-induced NF-κB.
MATERIALS AND METHODS
Materials
Disposable culture ware was purchased from Corning Glass Works (Corning, NY) and Becton Dickinson (Franklin Lakes, NJ). Tissue culture media and fetal bovine serum (FBS) were obtained from GIBCO (Grand Island, NY). Biochemicals were purchased from Sigma (St. Louis, MO). Antibodies against NF-κB and XIAP were from BD Biosciences Clontech (Palo Alto, CA), and antibodies against caspase-3 and IκB were from Santa Cruz Biotechnology (Santa Cruz, CA).
Cell Culture and Experimental Protocol
The IEC-6 cell line was purchased from American Type Culture Collection (Rockville, MD) at passage 13. The IEC-6 cell line was derived from normal rat intestine and was developed and characterized by Quaroni et al.46 IEC-6 cells from jejunal crypt cells were maintained in T-175 flasks in Dulbecco modified Eagle medium (DMEM) supplemented with 5% heat-inactivated FBS, 1% antibiotic, and 0.1 U/mL insulin. They are nontumorigenic and retain the undifferentiated character of epithelial stem cells. Flasks were incubated at 37°C in a humidified atmosphere of 95% air and 5% CO2. Stock cells were subcultured once a week at 1:3. Passages 15–20 were used in all experiments. There were no significant changes of biologic function within these passage numbers. Cells were restarted from frozen stock.
Apoptosis
Apoptosis was quantified by counting the number of dead cells and using terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL). Apoptosis identification was performed using the DeadEnd IEC-6 cells Florometric TUNEL system (Promega, Madison, WI). Cells were fixed in freshly prepared 4% methanol-free formaldehyde for 25 minutes, centrifuged at 300 g for 10 minutes at 4°C, and then washed with PBS. This cell pellet was then resuspended in phosphate-buffered saline (PBS) at a concentration of 2 × 107cells/mL; 100 μL of the cell suspension was smeared onto poly-L-lysine-coated slides. The TUNEL reagent was prepared as directed (Promega). The reactions were terminated by immersing the slides in 2× SSC for 15 minutes at room temperature. The samples were washed 3 times in PBS to remove unincorporated fluorescein-12-dUTP. The samples were stained in 0.1 mg/mL propidium iodide buffer. The samples were analyzed using a fluorescent microscope. Percentage of apoptotic cells was determined by the number of stained cells per all cells by cell counting.
Preparation of Nuclear Protein and Electrophoretic Shift Assays
Nuclear proteins were prepared by the procedure described previously,47 and the protein contents in nuclear preparations were determined by the method described by Bradford.48 Six hours after wounding, cells were harvested for nuclear protein extractions. The double-stranded oligonucleotides used in these experiments included 5′-AGTTGAGGGGACTTTCCCAGGC-3′, which contain a consensus NF-κB binding site that is underscored. These oligonucleotides were radioactively end-labeled with [γ-32P]ATP and T4 polynucleotide kinase. For mobility shift assays, 0.035 pmol of 32P-labeled oligonucleotides (∼30,000 cpm) and 10 μg of nuclear protein were incubated in a total volume of 25 μL in the presence of 10 mmol/L Tris • HCL (pH 7.5), 50 mmol/L NaCl, 1 mmol/L EDTA, 1 mmol/L dithiothreitol, 5% glycerol, and 1 μg of poly(dI-dC). The binding reactions were allowed to proceed at room temperature for 20 minutes. Thereafter, 2 μL of bromphenol blue (0.1% in water) was added, and protein-DNA complexes were resolved by electrophoresis on nondenaturing 5% polyacrylamide gels and visualized by autoradiography. The specificity of binding interactions was assessed by competition with an excess of unlabeled double-stranded oligonucleotide of identical sequence. Gel supershift assays were accomplished by adding 1 μg (in 1 μL) of p65 supershift antibody to the reaction mixture and incubating for an additional 30 minutes at room temperature.
All experiments were repeated in triplicate.
Western Blot Analysis
Cell samples, placed in SDS sample buffer, were sonicated and centrifuged (12,000 rpm) at 4°C for 15 minutes. The supernatant from cell samples was boiled for 5 minutes and then subjected to electrophoresis on 7.5% SDS-PAGE gels according to Laemmli.49 After sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), the gels were transferred to nitrocellulose membranes for 1 hour at 4°C. The blots were blocked with 5% nonfat dry milk in PBS-0.1% Tween 20 (PBS-T) overnight at 4°C. Immunologic evaluation was performed for 1 hour in PBS-T containing specific antibodies against NF-κB (p65), IκBα, XIAP, and caspase-3 proteins. The filters were subsequently washed with PBS-T and incubated with the secondary antibodies conjugated with horseradish peroxidase for 1 hour at room temperature. After extensive washing in PBS-T, the blots were developed for 30 to 60 seconds with enhanced chemiluminescence reagents.
IκB Super-Repressor
The recombinant replication-deficient adenovirus Ad5IκB was constructed by the methods of Graham and Prevec50 and Bett et al51 as performed by Iimuro et al.52 In brief, the plasmid pCMV-IκBαM, which contains a human super-repressor of NF-κB, was subcloned into the XbaI site of the pACCMV.PLPASR (+) plasmid to construct the plasmid pACCMV/IκB, in which IκB is driven by the CMV promoter/enhancer. The plasmid DNA was prepared by the alkaline lysis method and purified by CsCl-tethidium bromide density gradient centrifugation. The recombinant adenovirus IκB was constructed by cotransfection of the 293 embryonic human kidney cell line with the pACCMV/IκB plasmid plus the purified fragment of the ClaI-digested DNA from E1-deleted adenovirus type 5 (Ad5). The presence of the mutant IκB sequence packaged into the recombinant Ad5 virus (Ad5IκB) was confirmed by PCR and Western blotting. Ad5IκB was grown in 293 cells and purified by banding twice on CsCl gradients. Viral titer was determined by optical densitometry (particle per milliliter) and plaque assay. Recombinant virus was stored in 10% glycerol at −20°C.
Measurement of Caspase-3 Activity
Caspase-3 activity was measured by using a caspase-3 colorimetric assay kit (R&D Systems, Minneapolis, MN) and performed according to the protocol recommended by the manufacturer. Briefly, cells were treated with TNF-α and CHX for 4 hours, washed with ice-cold Dulbecco's phosphate-buffered saline (D-PBS), and scraped from the dishes. The collected cells were washed with d-PBS and then lysed in ice-cold cell lysis buffer [50 mmol/L HEPES, pH 7.4, 3-[(3-cholamidopropyl) dimethylammonia]-1-propanesulfonate (CHAPS), 1 mmol/L DTT, 0.1 mmol/L EDTA, and 0.1% Nonidet P-40]. The assay for caspase-3 activity was carried out in a 96-well plate. In each well, there were 50 μL of cell lysate (∼150 μg of total proteins), 50 μL of reaction buffer (50 mmol/L HEPES, pH 7.4, 0.1% CHAPS, 100 mmol/L NaCl, 10 mmol/L DTT, and 1 mmol/L EDTA), 5 μL of caspase-3 colorimetric substrate, and a caspase-specific peptide that was conjugated to a chromogen, p-nitroanilide (p-NA). The 96-well plate was incubated at 37°C for 90 minutes, during which the caspase-3 in the sample presumably cleaved the chromophore p-NA from the substrate molecule. Absorbance at 405 nm was monitored to assess caspase-3 activity. Protein levels of each sample were determined by the method described by Bradford.48
Experimental Design
The purpose of the first series of experiments was to determine whether bile salt exposure was paralleled by increased expression of XIAP proteins in IEC-6 cells. The general protocol of the experiments and methods were similar to those described previously.53,54 Briefly, IEC-6 cells were plated at 6.25 × 104 cells/cm2 and cultured for 6 days in control medium (DMEM + 5% dialyzed FBS + 10 μg/mL insulin and 50 μg/mL gentamicin sulfate) or in DMEM medium containing bile salts. The dishes were placed on ice, and the monolayers were washed 3 times with ice-cold d-PBS. Levels of XIAP mRNA and protein expression were measured by polymerase chain reaction (PCR) and Western blot analyses, respectively.
The purpose behind the second series of experiments was to determine whether the observed increase in XIAP expression following bile salt exposure resulted from NF-κB activation. The increased NF-κB activity in bile salt-exposed cells was specifically prevented by ectopic expression of IκBα super-repressor through the infection with the AdIκBSR vector, and apoptosis was induced by TNF-α in combination with CHX (TNF-α/CHX). The NF-κB binding activity and XIAP expression were measured in cells grown for 6 days in the DMEM medium with either AdIκBSR or control vector during the last 48 hours.
The third series of experiments was designed to define the relationship between increased expression of XIAP and the resistance to TNF-α/CHX-induced apoptosis in bile salt-exposed cells. Functions of IAP proteins were examined by using the specific IAP inhibitor Smac.55,56 Cells were initially grown for 5 days in medium containing bile salt, exposed to Smac for 24 hours, and then treated with TNF-α/CHX. Apoptosis was measured 4 hours after administration of TNF-α/CHX. In addition, the involvement of caspase-3 with XIAP in the resistance to apoptosis was investigated. The levels of procaspase-3 and caspase-3 proteins, as well as the activity of caspase-3 were measured in bile salt-exposed cells in the presence or absence of Smac.
Statistics
Values are mean ± SE from 6 samples. Autoradiography results were repeated 3 times. The significance of the difference between means was determined by ANOVA. The level of significance was determined by using Duncan's multiple-range test.57
RESULTS
Effect of Bile Salts on Tumor Necrosis Factor-α + Cycloheximide (TNF-α/CHX)-Induced Apoptosis
We examined the effect of exposure to the cholate and the conjugated bile salts taurochenodeoxycholate (TCDCA) and TDCA on intestinal epithelial apoptosis.
Exposure of the cells to TNF-α coupled with cycloheximide (CHX), a widely accepted protocol for inducing apoptosis16,21,45 was used in this study. Within 4 hours, TNF-α/CHX induced 75% of the IEC-6 cells to undergo cell death. Exposure in a dose-dependent fashion of the intestinal epithelial cells to TCDCA (0.5–1 mmol/L) and TDCA (0.2–0.5 mmol/L), but not cholate for 6 days prior to induction of apoptosis significantly reduced the number and percentage of cells that underwent apoptosis in the presence of TNF-α/CHX (Fig. 1A). The DeadEnd cells Fluorometric TUNEL system (Promega, Madison, WI) stain was used to show nuclear fragmentation, confirming that the cell death visualized in the presence of the TNF-α/CHX was apoptosis (Fig. 1B). These results indicate that the conjugated bile salts TCDCA and TDCA promote the resistance of IEC-6 cells to apoptotic cell death induced by TNFα/CHX.
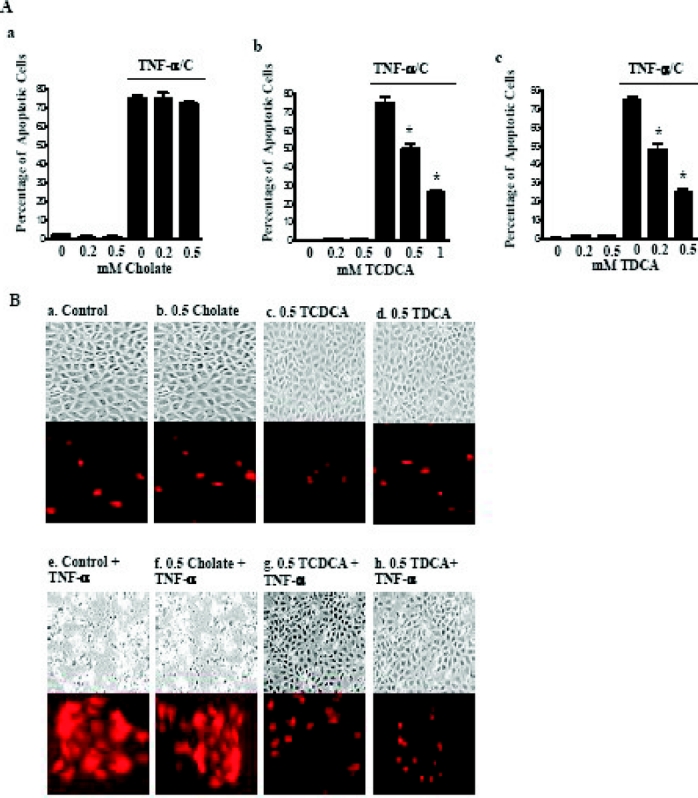
FIGURE 1. Apoptotic response of IEC-6 cells to 20 ng/mL TNF-α and 25 μg/mL cycloheximide (TNF-α/CHX) after 6 days of exposure to bile salts. A, Percentage of apoptotic cells. The effect of (a) cholate, (b) taurochenodeoxycholate (TCDCA), and (c) taurodeoxycholate (TDCA). TNF-α/CHX induced IEC-6 cell apoptosis. IEC-6 cells were grown in TDCA for 6 days and exposed to 20 ng/mL of TNF-α and 25 μg/mL cycloheximide. Apoptosis was quantified with cell count with trypan blue and the TUNEL stain. *P < 0.01 versus TNF-α/CHX alone. B, Representative photomicrographs of TNF-α/CHX induced apoptosis and the TUNEL stain in the presence of bile salts: a, control; b, cells exposed to 0.5 mmol/L cholate; c, cells exposed to 0.5 mmol/L TCDCA; d, cells exposed to 0.5 mmol/L TCDCA; e, cells exposed to TNF-α/CHX for 4 hours; f, cells exposed to TNF-α/CHX for 4 hours in the presence of 0.5 mmol/L cholate; g, cells exposed to TNF-α/CHX for 4 hours in the presence of 0.5 mmol/L TCDCA; h, cells exposed to TNF-α/CHX for 4 hours in the presence of 0.5 mmol/L TDCA.
Bile Salt-Induced Changes in NF-κB Sequence Specific-Binding Activity
We had previously shown that exposure to TDCA significantly increased NF-κB nuclear binding and that this NF-κB nuclear binding was sequence specific.31,34 TCDCA also stimulated NF-κB nuclear binding activity (Fig. 2A). Further, competitive inhibition of NF-κB nuclear binding using unlabeled cold oligonucleotide as a competitive inhibitor confirmed sequence-specific binding (Fig. 2B).

FIGURE 2. Changes in sequence-specific NF-κB binding activity in IEC-6 cells exposed to TCDCA. A, Representative audiograms of NF-κB binding. Cells were grown in DMEM with 5% dialyzed FBS with and without TCDCA for 6 days. Nuclear extracts were prepared, and electrophoretic mobility shift assay (EMSA) was performed by using 10 μg of nuclear proteins and 0.035 pmol of 32P-end-labeled oligonucleotides containing a single NF-κB binding site. Position of specifically bound DNA-protein complex is indicated. B, Effects of unlabeled NF-κB oligonucleotide as a cold competitor on NF-κΒ binding activity in the presence of 0.5 mmol/L TCDCA as measured by EMSA.
Changes in Caspase-3 Activity Following Exposure to Bile Salts
Active caspase-3 not only participates directly as a terminal effector of apoptosis by cleaving various substrate proteins but also activates additional pro-caspases.58 We studied the effect of TCDCA and TDCA on procaspase-3 cleavage as compared with caspase-3 during TNF-α-induced apoptosis. Exposure of control cells to TNF-α/CHX significantly induced caspase-3 activation from procaspase-3, as indicated by the increases in both active caspase-3 protein levels and enzyme activity (Fig. 3). At concentrations that increase XIAP expression and protect the intestinal mucosa against TNF-α/CHX-induced apoptosis, the bile salts TCDCA and TDCA significantly inhibited the cleavage of caspase-3 from procaspase-3 and significantly decreased caspase-3 enzyme activity (Fig. 3).

FIGURE 3. Changes in caspase-3 protein expression and activity in IEC-6 cells after exposure to TNF-α/CHX alone or with incubation with the bile salts TCDCA and TDCA. A, Representative autoradiograms of Western immunoblots: a, TDCA; b, TCDCA. Cells were grown in DMEM with 5% dialyzed FBS in the presence or absence of TCDCA or TDCA for 6 days. Cells were exposed to 20 ng/mL TNF-α and 25 μg/mL cycloheximide for 4 hours. Whole cell lysates were harvested and applied to each lane equally (20 μg), and levels of procaspase-3 (32 kDa) and caspase-3 (17 kDa) were identified by probing nitrocellulose with the specific anticaspase-3 antibody. Actin immunoblotting was performed as an internal control for equal loading. B, Quantitative analysis of the ratio of caspase-3 protein expression to actin derived from densitometric analysis of autoradiograms displayed in A. Values are mean ± SE from 3 separate experiments: a, TDCA (*P < 0.05 vs. control, **P < 0.05 vs. TDCA + TNF-α/CHX); b, TCDCA (*P < 0.05 vs. control, **P < 0.05 vs. TCDCA + TNF-α/CHX); C, changes in caspase-3 activity in cells described in A. Values are mean ± SE from 3 separate experiments: a, TDCA (*P < 0.05 vs. control, **P < 0.05 vs. TDCA + TNF-α/CHX); b, TCDCA (*P < 0.05 vs. control, **P < 0.05 vs. TCDCA + TNF-α/CHX).
Effect of NF-κB Inhibition on Bile Salt-Induced Resistance to TNF-α/CHX-Induced Apoptosis
Previously, we studied the specificity of these effects of NF-κB activation using an IκB super-repressor (IκBSR) to inhibit NF-κB activity. IκBαM contains 2 mutations of serine to alanine (at residues 32 and 36), which prevent phosphorylation of IκBα. Therefore, ubiquination and degradation of IκBα are inhibited, which prevents activation of the NF-κB pathway. We confirmed that transfection of IEC-6 cells with the IκBSR increased expression of IκB and decreased the concentration of free NF-κB.34 The IκBSR transfected into the IEC-6 cells significantly inhibited NF-κB nuclear binding activity in the presence of TDCA versus control or transfection of the vector alone.34 TNF-α/CHX-induced apoptosis was significantly increased in the cells treated with of TDCA and the IκBSR (54% of the cells) versus TDCA alone (24%) or TDCA and vector alone (27%). The protective effect against TNF-α/CHX-induced apoptosis established by the presence of TDCA on the intestinal epithelium was eliminated after transfection with the IκBSR.34 We repeated this experiment using TCDCA, as TCDCA also protected IEC-6 cells against TNF-α/CHX-induced apoptosis as well as activating NFκB. As expected, TNF-α/CHX-induced apoptosis was significantly increased in the cells treated with of TCDCA and the IκBSR (60% of the cells) versus TCDCA alone (24%) or TDCA and vector alone (25%) (Fig. 4).

FIGURE 4. The effect of the IκBSR on enterocyte apoptosis. A, Changes in NF-κB sequence-specific binding activity in IEC-6 cells exposed to TCDCA in the presence of the IκBSR or control vector. Representative autoradiograms from nuclear extracts in control cells and cells 6 hours after exposure to 0.5 mmol/L TDCA. EMSA was performed as described above. Positions of the specifically bound DNA-protein binding complex are indicated. B, The effect of the IκBSR on TNF-α/CHX induced apoptosis in the presence of 0.5 mmol/L TDCA. IEC-6 cells transfected with the IκBSR were grown in the presence and absence of 0.5 mmol/L TDCA for 6 days. Apoptosis was induced using TNF-α/CHX as described above. Displayed on the y-axis is the percentage of apoptotic cells. *P < 0.01 versus control. **P < 0.01 versus TCDCA.
Changes in Expression of XIAP Protein Following Exposure to TDCA
The inhibitor of apoptosis (IAP) family of proteins are potent natural suppressor of apoptosis and function by directly inhibiting the activity of caspases, the principal effectors of apoptotic cell death.40,41 XIAP is a member of this family of proteins. Recently, it has been shown that expression of XIAP is regulated by NF-κB and that NF-κB-mediated XIAP is involved in protecting endothelial cells from TNF-α/CHX-induced apoptosis.44,45 We examined the effect of bile salt exposure to IEC-6 cells on mRNA and protein expression of XIAP using reverse transcription polymerase chain reaction (rtPCR) and Western blot analysis, respectively. Exposure to TCDCA and TDCA at concentrations that protect intestinal epithelial cells against TNF-α/C-induced apoptosis increased both mRNA and protein expression of XIAP (Fig. 5).

FIGURE 5. Expression of XIAP after exposure to bile salts. A, Expression of XIAP mRNA in IEC-6 cells. Cells were grown in DMEM with 5% dialyzed FBS in the presence or absence of 0.5 mmol/L TCDCA and TDCA for 6 days. PCR-amplified products were displayed on agarose gel for XIAP 510 bp, when first-strand cDNA synthesized from total RNA extracted from IEC-6 cells was amplified with the specific sense and antisense primers for XIAP. GAPDH was used for internal control for equal loading. B, Representative autoradiograms of Western immunoblots from IEC-6 cells exposed to 0.1 to 0.5 mmol/L TDCA for 6 days. Cells were grown in DMEM with 5% dialyzed FBS in the presence or absence of TDCA for 6 days. Whole cell lysates were harvested and applied to each lane equally (20 μg), and levels of XIAP (57 kDa) were identified by probing nitrocellulose with the specific anti-XIAP antibody. Actin immunoblotting was performed as an internal control for equal loading. C, Representative autoradiograms of Western immunoblots from IEC-6 cells exposed to 0.1 to 0.5 mmol/L TCDCA for 6 days as described in B.
Effect of the XIAP Inhibitor Smac on TNF-α/CHX-Induced Apoptosis in Bile Salt-Exposed Cells
To further define the role of NF-κB-mediated XIAP protein in the process of apoptosis, we used the specific IAP inhibitor Smac45,55,56 in this study. Zou et al45 have shown that Smac specifically inhibits expression of IAP proteins, such as XIAP, and does not affect IEC-6 cell viability. As shown in Figure 6A, exposure of bile salt-exposed cells to Smac significantly reduced the levels of XIAP protein. When Smac at the concentration of 1 μg/mL was added to the medium for 24 hours, the level of XIAP protein was decreased by approximately 50%.

FIGURE 6. Effect of treatment of Smac on levels of XIAP protein in IEC-6 cells exposed to Smac. A, Representative autoradiograms of Western immunoblots from IEC-6 cells exposed to 0.5 mmol/L TDCA for 6 days and Smac at the concentration of 1 μg/mL during the last 24 hours. Cells were grown in DMEM with 5% dialyzed FBS in the presence or absence of TDCA for 6 days. Whole cell lysates were harvested and applied to each lane equally (20 μg), and levels of XIAP (57 kDa) were identified by probing nitrocellulose with the specific anti-XIAP antibody. Actin immunoblotting was performed as an internal control for equal loading. B, Quantitative analysis of XIAP immunoblots by densitometry from cells described in A. Values are 3 separate experiments. *P < 0.05 versus TDCA alone.
Inhibition of XIAP protein by treatment with Smac not only enhanced programmed cell death in control cells but also prevented the resistance of bile salt-exposed cells to apoptosis after exposure to TNF-α/CHX (Fig. 6B). In control cells, typical morphologic features of apoptosis increased markedly when cells were pretreated with Smac at the concentration of 1 μg/mL for 24 hours. The percentage of TNF-α/CHX-induced apoptotic cells was significantly increased (Fig. 6B). In bile salt-exposed cells, the percentage of TNF-α/CHX-induced apoptotic cells was significantly increased with Smac treatment compared with the percentage of apoptotic cells in the TNF-α/CHX-treated group that was not supplemented with Smac. These findings strongly suggest that NF-κB-mediated XIAP induced by exposure to bile salts plays a critical role in the regulation of susceptibility of intestinal epithelial cells to TNF-α/CHX-induced apoptosis.
Effect of the IκBSR on XIAP Expression and Enterocyte Resistance to Apoptosis
We studied the effect the presence of the IκB super-repressor had on expression in the presence of TDCA. Figure 7 shows that TDCA-induced XIAP expression is suppressed after NF-κB inhibition using the IκBSR. These data show that bile salt-induced expression of XIAP is at least partially mediated through an increase in NF-κB activation.

FIGURE 7. The effects of the IκBSR on XIAP expression. A, IEC-6 cells transfected with the IκBSR were grown in the presence and absence of 0.5 mmol/L TDCA for 6 days. Apoptosis was induced using TNFα/CHX as described above. Representative autoradiograms of Western immunoblots from IEC-6 cells TDCA obtained as described above. Actin immunoblotting was performed as an internal control for equal loading. B, Quantitative analysis of the ratio of XIAP protein expression to actin derived from densitometric analysis of autoradiograms displayed in A. *P < 0.05 versus control + TNF-α/CHX.
DISCUSSION
We have recently reported that the bile salt TDCA induces NF-κB activation and promotes resistance to TNF-α/CHX-induced apoptosis.34 The present study extends our previous observations in that TCDCA, another conjugated bile salt, also induces resistance to TNF-α/CHX-induced apoptosis, while cholate, the unconjugated bile salt, has no effect on enterocyte resistance to apoptosis. Further, TCDCA regulates this effect through NF-κB. Inactivation of NF-κB via the recombinant adenoviral vector containing the IκBα super-repressor (AdIκBSR) blocks the antiapoptotic effect of the bile salts TDCA and TCDCA. Another significant finding is that these bile salts increase the cellular resistance to TNF-α/CHX-induced apoptosis through the antiapoptotic protein XIAP. Activation of NF-κB was associated with an increase in XIAP expression in the presence of TDCA and TCDCA. Furthermore, decreased expression of XIAP by either inactivation of NF-κB through the AdIκBSR or treatment with the IAP inhibitor Smac decreased the resistance of bile salt-exposed cells to TNF-α/CHX-induced apoptosis.
Previously, we have shown that bile salts stimulate intestinal epithelial cell migration after injury through a process that involves NF-κB; this process requires the induction of IκB phosphorylation, ubiquination, and degradation, thereby allowing NF-κB to translocate into the nucleus and regulate gene transcription. This study demonstrates that physiologic concentrations of bile salts stimulate intestinal proliferation and activate NF-κB. These bile salts protect the intestinal mucosa against TNF-α/CHX-induced apoptosis through a NF-κB-dependent process. The finding that NF-κB activation is protective against TNF-α/CHX-induced apoptosis is consistent with other investigators. Li et al21 have shown that polyamine depletion activates NF-κB, which in turn decreases the susceptibility of IEC-6 cells to TNF-α/CHX-induced apoptosis. Others16,20,27 have shown that NF-κB is a cell survival factor, which protects cells from cell death stimuli. In general, NF-κB is thought to inhibit apoptosis through caspase-dependent pathways. Numerous reports document both the antiapoptotic action of NF-κB and the proapoptotic effects of inactivating NF-κB by a variety of cell types.16,20,27,29 Many of the genes that are involved in apoptosis are target genes of NF-κB. Activated NF-κB regulates transcription of its downstream target genes, interacts with other cellular effectors, and plays distinct roles in regulating apoptosis.
The current studies provide new evidence that bile salt-induced NF-κB increases expression of the antiapoptotic factor XIAP, and that bile salt-induced XIAP expression at least partially regulates resistance to TNF-α/CHX-induced apoptosis elicited in the presence of bile salts. Moreover, these data show that bile salts inhibit caspase-3 activation. The IAP family of proteins is a potent natural suppressor of apoptosis and functions by directly inhibiting the activity of caspases, the principal effectors of apoptosis. The IAP proteins are defined as baculovirus IAP repeat (BIR)-containing proteins that inhibit cell death. The BIR motif consists of approximately 70 amino acids of a zinc-binding protein. There are 1 to 3 BIR motifs per IAP, and these BIR motifs are important for the antiapoptotic function of the IAP. In addition to the BIR motif, some IAPs contain another zinc-binding motif called the RING domain.40 IAPs inhibit caspases through 2 distinct mechanisms. The first is through a direct interaction between IAP and caspases, while the second in through RING-dependent ubiquination and proteasomal degradation.
Members of the mammalian IAP family including XIAP, cellular inhibitor of apoptosis protein-1 (cIAP-1), cellular inhibitor of apoptosis protein-2 (cIAP-2), and neuronal apoptosis-inhibitory protein (NAIP) inhibit caspase-3, caspase-7, and capspase-9 by a direct interaction with the caspases.40 BIR 3 is responsible for the inhibition of active caspase-9, while the linker between BIR 1 and BIR 2 inhibits active caspase-3. XIAP also has additional antiapoptotic activities because mutant XIAP proteins unable to inhibit caspase-3 and caspase-9 retain their ability to inhibit apoptosis.40–43 Bile salts increase resistance to apoptosis at least partially through a decrease in activity of caspase-3, which is consistent with an increase in the antiapoptotic protein XIAP. Under physiologic conditions, bile salts have various biologic effects besides their role in the digestion of lipids. TDCA increases esophageal mucosal growth in an explant model,59 whereas deoxycholate (DOC) modulates p53 gene expression,60 and stimulates prostaglandin E2 synthesis by causing abrupt transient increases in cytosolic calcium.61 Bile acid response elements (DNA sequences that contain AGGTCA direct repeats similar to the elements recognized by nuclear receptors) regulate the transcription of target genes.62,63 Bile salts regulate many functions within cells by activating signaling pathways and transcription factors. Because of their varying pKa, solubility, and intracellular and extracellular concentrations, different bile salts have different biologic effects.
The observation that bile salts at physiologic concentrations have beneficial effects on mucosal integrity is not surprising given that bile salts are a normal component of the intestinal luminal content. It is difficult to determine the physiologic concentration of soluble bile salts to which the intestinal mucosa actually is exposed because most bile salts are in micellar form and their concentrations vary widely during the digestive process. Micellar concentrations of bile salts may reach 10 mmol/L during digestion while free bile salts can reach soluble concentrations of up to 2 to 3 mmol/L.64 Preprandial newborns, who possess minimal lipid to form micelles, have intestinal concentration of bile salts around 4 mmol/L.65 Thus, our experimental concentrations of bile salts that stimulated mucosal renewal are within the physiologic range of mucosal exposure.
The intestinal mucosa is known to require the presence of luminal contents to maximize the adaptive response.35–39 In this regard, oral sodium taurocholate was shown to improve anastomotic healing after bile duct ligation.66 While luminal nutrients and trophic gut peptides may participate in this adaptive response, our data suggest a role for bile salts as trophic agents as well. In support of this functional role for bile salts, animals with biliary diversion after small bowel resection demonstrate an impaired adaptive response.37–39
Clearly, bile salts have many purposes beyond fat absorption. The presence of bile salts serves to regulate these trophic functions, whereas the absence of bile salts may be quite detrimental to intestinal function. The mucosal atrophy of starvation and critical illness, while multifactorial in etiology may also be related to cholestasis and diminished luminal bile. Bile salts may play a critical role during necrotizing enterocolitis, ischemia, inflammatory bowel disease, and other conditions in which mucosal injury occurs. Further characterization of the beneficial functions of bile salts on the small intestinal mucosal are warranted.
Footnotes
Supported by the Research Career Development program of the Department of Veterans' Affairs and the C. James Carrico Fellowship of the American College of Surgeons.
Reprints: Douglas J. Turner, MD, Department of Surgery, Baltimore Veterans Affairs Medical Center, 11 North Greene Street, Baltimore, MD 21201. E-mail: DTURNER@smail.umaryland.edu.
REFERENCES
- 1.Johnson LR. Regulation of gastrointestinal mucosal growth. Physiol Rev. 1988;68:456–502. [DOI] [PubMed] [Google Scholar]
- 2.Loeffler M, Birke A, Winton D, et al. Somatic mutation, monoclonality and stochastic-models of stem-cell organization in the intestinal crypt. J Theor Biol. 1993;160:471–491. [DOI] [PubMed] [Google Scholar]
- 3.Potten CS. Epithelial cell growth and differentiation: 2. Intestinal apoptosis. Am J Physiol Gastrointest Liver Physiol. 1997;273:G253–G257. [DOI] [PubMed] [Google Scholar]
- 4.Potten CS, Wilson JW, Booth C. Regulation and significance of apoptosis in the stem cells of the gastrointestinal epithelium. Stem Cells. 1997;15:82–93. [DOI] [PubMed] [Google Scholar]
- 5.Shin CE, Falcone RA, Kemp CJ, et al. Intestinal adaptation and enterocyte apoptosis following small bowel resection is p53 independent. Am J Physiol Gastrointest Liver Physiol. 1999;277:G717–G724. [DOI] [PubMed] [Google Scholar]
- 6.Gavrieli Y, Sherman Y, Bensasson SA. Identification of programmed cell-death in situ via specific labeling of nuclear-DNA fragmentation. J Cell Biol. 1992;119:493–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hall PA, Coates PJ, Ansari B, et al. Regulation of cell number in the mammalian gastrointestinal tract: the importance of apoptosis. J Cell Sci. 1994;107:3569–3577. [DOI] [PubMed] [Google Scholar]
- 8.Jobin C, Sartor RB. The IκB/NF-κB system: a key determinant of mucosal inflammation and protection. Am J Physiol Cell Physiol. 2000;278:C451–C462. [DOI] [PubMed] [Google Scholar]
- 9.Baeuerle P, Henkle T. Function and activation of NF-κB in the immune system. Annu Rev Immunol. 1994;12:141–179. [DOI] [PubMed] [Google Scholar]
- 10.Barnes P, Karin M. Nuclear factor-κB, a pivotal transcription factor in chronic inflammatory diseases. N Engl J Med. 1997;336:1066–1071. [DOI] [PubMed] [Google Scholar]
- 11.Brown K, Gerstberger S, Carlson L, et al. Control of IκB proteolysis by site-specific, signal-induced phosphorylation. Science. 1995;267:1485–1488. [DOI] [PubMed] [Google Scholar]
- 12.Beg A, Finco T, Nantermet P, et al. Tumor necrosis factor and interleukin-1 lead to phosphorylation and loss of IkB, a mechanism for NF-kB activation. Mol Cell Biol. 1993;13:3301–3310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee F, Hagler J, Chen Z, et al. Activation of the IκB kinase complex by MEKK1, a kinase of the JNK pathway. Cell. 1997;88:213–222. [DOI] [PubMed] [Google Scholar]
- 14.Mercurio F, Zhu H, Murray BW, et al. IKK-1 and IKK-2: cytokine-activated IkB kinases essential for NF-kB activation. Science. 1997;278:860–866. [DOI] [PubMed] [Google Scholar]
- 15.Thompson J, Phillips R, Erdjument-Bromage H, et al. IκBβ regulates the persistent response in a biphasic activation of NF-κB. Cell. 1995;80:573–582. [DOI] [PubMed] [Google Scholar]
- 16.Beg AA, Baltimore D. An essential role for NF-κB in preventing TNF-α-induced cell death. Science. 1996;274:782–784. [DOI] [PubMed] [Google Scholar]
- 17.Lin K-I, DiDonato JA, Hoffman A, et al. Suppression of steady-state, but not stimulus induced, NF-κB activity inhibits alphavirus-induced apoptosis. J Cell Biol. 1998;141:1479–1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lin K-I, Lee SH, Narayanan R, et al. Thiol agents and Bcl-2 identify and alphavirus-induced apoptotic pathway that requires activation of the transcription factor NF-κB. J Cell Biol. 1995;131:1149–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Qin ZH, Wang Y, Nakai M, et al. Nuclear factor-κB contributes to excitotoxin-induced apoptosis in rat striatum. Mol Pharmacol. 1998;53:33–42. [DOI] [PubMed] [Google Scholar]
- 20.Van Antwerp DJ, Martin SJ, Kafri TD, et al. Suppression of TNF-α-induced apoptosis by NF-κB. Science. 1996;274:787–789. [DOI] [PubMed] [Google Scholar]
- 21.Li L, Rao JN, Bass BL, et al. NF-κB activation and susceptibility to apoptosis after polyamine depletion in intestinal epithelial cells. Am J Physiol Gastrointest Liver Phsyiol. 2001;280:G992–G1004. [DOI] [PubMed] [Google Scholar]
- 22.Baeuerle PA, Baltimore D. NF-κB, ten years after. Cell. 1996;87:13–20. [DOI] [PubMed] [Google Scholar]
- 23.La Rosa FA, Pierce JP, Sonenshein GE. Differential regulation of the c-myc oncogene promoter by the NF-κB Rel family of transcription factors. Mol Cell Biol. 1994;14:1039–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Qin ZH, Chen RW, Wang Y, et al. Nuclear factor κB nuclear translocation upregulates c-Myc and p53 expression during NMDA receptor-mediated apoptosis in rat striatum. J Neurosci. 1999;19:4023–4033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu H, Lozano G. NF-κB activation of p53. A potential mechanism for suppressing cell growth in response to stress. J Biol Chem. 1994;269:20067–20074. [PubMed] [Google Scholar]
- 26.Beg AA, Sha WC, Bronson RT, et al. Embryonic lethality and liver degeneration in mice lacking the RelA component of NF-κB. Nature. 1995;376:167–170. [DOI] [PubMed] [Google Scholar]
- 27.Wu M, Lee H, Bellas RE, et al. Inhibition of NF-κB/Rel induces apoptosis of murine B cells. EMBO J. 1996;15:4682–4690. [PMC free article] [PubMed] [Google Scholar]
- 28.Opipari AW Jr, Hu HM, Yabknoitz R, et al. The A20 zinc finger protein protects cells from tumor necrosis factor cytotoxicity. J Biol Chem. 1992;267:12424–12427. [PubMed] [Google Scholar]
- 29.Wang CY, Mayo MW, Korneluk RG, et al. NF-κB antiapoptosis: induction of TRAF1 and TRAF2 and c-IAP2 to suppress caspase-8 activation. Science. 1998;281:1680–1683. [DOI] [PubMed] [Google Scholar]
- 30.Strauch ED, Wang J, Bass BL. Bile salt stimulates intestinal epithelial cell migration through TGF-β after wounding. J Surg Res. 2001;97:49–53. [DOI] [PubMed] [Google Scholar]
- 31.Strauch ED, Bass BL, Rao J, et al. NF-κB regulates intestinal epithelial cell and bile salt induced migration after injury. Ann Surg. 2003;237:494–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Strauch ED, Yamaguchi J, Bass BL, et al. Bile salts regulate intestinal epithelial cell migration by NF-B induced expression of TGF- β. J Am Coll Surg. 2003;197:974–984. [DOI] [PubMed] [Google Scholar]
- 33.Yamaguchi J, Toledo A, Bass BL, et al. Taurodeoxycholate increases intestinal epithelial cell proliferation through C-myc expression. Surgery. 2004;135:215–221. [DOI] [PubMed] [Google Scholar]
- 34.Toledo A, Yamaguchi J, Wang J-Y, et al. Taurodeoxycholate stimulates intestinal cell proliferation and protects against apoptotic cell death through activation of NF-kappaB. Dig Dis Sci. 2004;49:1664–1671. [DOI] [PubMed] [Google Scholar]
- 35.Ford WD, de Vries JE, Ross JS, et al. Effect of luminal contents on postresectional longitudinal and mucosal growth in the ileum of suckling rats. Surgery. 1985;98:935–941. [PubMed] [Google Scholar]
- 36.Thompson JS, Ferguson DC. Effect of the distal remnant on ileal adaptation. J Gastrointest Surg. 2000;4:430–434. [DOI] [PubMed] [Google Scholar]
- 37.Roy CC, Laurendeau G, Doyon G, et al. The effect of bile and of sodium taurocholate on the epithelial cell dynamics of the rat small intestine. Proc Soc Exp Biol Med. 1975;149:1000–1004. [DOI] [PubMed] [Google Scholar]
- 38.Williamson RC, Bauer FL, Ross JS, et al. Contributions of bile and pancreatic juice to cell proliferation in ileal mucosa. Surgery. 1978;83:570–576. [PubMed] [Google Scholar]
- 39.Al-Mukhtar MY, Sagor GR, Ghatei MA, et al. The role of pancreatico-biliary secretions in intestinal adaptation after resection, and its relationship to plasma enteroglucagon. Br J Surg. 1983;70:398–400. [DOI] [PubMed] [Google Scholar]
- 40.Leblanc A. Natural cellular inhibitors of caspases C. Prog Neuropsychopharmacol Biol Psychiatry. 2003;27:215–229. [DOI] [PubMed] [Google Scholar]
- 41.Potts PR, Singh S, Knezek M, et al. Critical function of endogenous XIAP in regulating caspase activation during sympathetic neuronal apoptosis. J Cell Biol. 2003;163:789–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Silke J, Ekert PG, Day CL, et al. Direct inhibition of caspase 3 is dispensable for the anti-apoptotic activity of XIAP. EMBO J. 2001;20:3114–3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Silke J, Hawkins CJ, Ekert PG, et al. The anti-apoptotic activity of XIAP is retained upon mutation of both the caspase 3- and caspase 9-interacting sites. J. Cell Biol. 2002;157:115–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stehlik C, de Martin R, Kumabashiri I, et al. Nuclear factor (NF)-κB-regulated X-chromosome-linked IAP gene expression protects endothelial cells from tumor necrosis factor-α induced apoptosis. J Exp Med. 1998;188:211–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zou T, Rao JN, Guo X, et al. NF-kB-mediated IAP expression induces resistance of intestinal epithelial cells to apoptosis after polyamine depletion. Am J Physiol Cell Physiol. 2004;286:1009–1017. [DOI] [PubMed] [Google Scholar]
- 46.Quaroni A, Wands J, Trelstad R, et al. Epithelial cell cultures from rat small intestine. J Cell Biol. 1979;80:248–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ye ZS, Samuels HH. Cell- and sequence-specific binding of nuclear protein to 5′-flanking DNA of the rat growth hormone gene. J Biol Chem. 1987;262:6313–6317. [PubMed] [Google Scholar]
- 48.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. [DOI] [PubMed] [Google Scholar]
- 49.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. [DOI] [PubMed] [Google Scholar]
- 50.Graham FL, Prevec L. Manipulation of adenovirus vectors. In: Murray EJ, ed. Methods in Molecular Biology: Gene Transfer and Expression Protocols. Clifton, NJ: Humana Press, 109–128. [DOI] [PubMed] [Google Scholar]
- 51.Bett AJ, Haddara L, Prevec L, et al. An efficient and flexible system for construction of adenovirus vectors with insertion of deletions in regions 1 and 3. Proc Natl Acad Sci USA. 1994;191:8802–8806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Iimuro Y, Nishiura T, Hellerbrand C, et al. NF-κB prevents apoptosis and liver dysfunction during liver regeneration. J Clin Invest. 1998;101:802–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li L, Li J, Rao JN, et al. Inhibition of polyamine synthesis induces p53 gene expression but not apoptosis. Am J Physiol Cell Physiol. 1999;276:C946–C954. [DOI] [PubMed] [Google Scholar]
- 54.Li L, Liu L, Rao JN, et al. JunD stabilization results in inhibition of normal intestinal epithelial cell growth through p21 after polyamine depletion. Gastroenterology. 2002;123:764–779. [DOI] [PubMed] [Google Scholar]
- 55.Arnt CR, Chiorean MV, Heldebrant MV, et al. Synthetic Smac/DIABLO peptides enhance the effects of chemotherapeutic agents by binding XIAP and cIAP1 in situ. J Biol Chem. 2002;277:44236–44243. [DOI] [PubMed] [Google Scholar]
- 56.MacFarlane M, Merrison W, Bratton SB, et al. Proteasome-mediated degradation of Smac during apoptosis: XIAP promotes Smac ubiquitination in vitro. J Biol Chem. 2002;277:36611–36616. [DOI] [PubMed] [Google Scholar]
- 57.Harter JL. Critical values for Duncan's new multiple range test. Biometrics. 1960;16:671–685. [Google Scholar]
- 58.Reed JC, Paternostro G. Postmitochondrial regulation of apoptosis during heart failure. Proc Natl Acad Sci USA. 1999;96:7614–7616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.D'Addio V, Sayles J, Alvarez C, et al. Bile: a trophic factor for rabbit esophageal epithelium in vitro. Surg Forum. 1996;647:125–127. [Google Scholar]
- 60.Palmer D, Paraskeva C, Williams A. Modulation of p53 expression in cultured colonic adenoma cell lines by the naturally occurring lumenal factors butyrate and deoxycholate. Int J Cancer. 1997;73:702–706. [DOI] [PubMed] [Google Scholar]
- 61.Zhu Y, Hua P, Rafiq S, et al. Ca2+ and PKC-dependent stimulation of PGE2 synthesis by deoxycholic acid in human colonic fibroblasts. Am J Physiol Gastrointest Liver Physiol. 2002;283:G503–G510. [DOI] [PubMed] [Google Scholar]
- 62.Chiang JYL, Kimmel R, Weinberger C, et al. Farnesoid X receptor responds to bile acids and represses cholesterol 7α-hydroxylase gene (CYP7A1) transcription. J Biol Chem. 2000;275:10918–10924. [DOI] [PubMed] [Google Scholar]
- 63.Chen W, Owsley E, Yang Y, et al. Nuclear receptor-mediated repression of human cholesterol 7α-hydroxylase gene transcription by bile acids. J Lipid Res. 2001;42:1402–1412. [PubMed] [Google Scholar]
- 64.Hoffman AF. Intestinal absorption of bile acids and biliary constituents. Physiol Gastrointest Tract. 1994;2:1845–1865. [Google Scholar]
- 65.Boehm G, Braun W, Moro G, et al. Bile acid concentrations in serum and duodenal aspirates of healthy and preterm infants: Effects of gestational and postnatal age. Biol Neonate. 1997;71:207–214. [DOI] [PubMed] [Google Scholar]
- 66.Sayan M, Alponat A, Yavus N, et al. The effect of oral sodium taurocholate on endotoxemia and intestinal anastomotic wound healing in rats with obstructive jaundice. Surg Today. 1997;27:953–957. [DOI] [PubMed] [Google Scholar]