

Abstract
Objective:
This study summarizes our initial experience with prospective, single-amplicon (mutation-specific) A636P testing in Ashkenazi Jewish patients at risk for Hereditary Nonpolyposis Colorectal Cancer (HNPCC).
Summary Background Data:
We previously described a founder mutation, MSH2*1906G >C (A636P) that causes HNPCC in 8/1345 (0.59%) of Ashkenazim with colorectal cancer. The mutation was more common in Ashkenazim diagnosed at ≤40 years (7%).
Methods:
Twenty-seven Ashkenazi probands at risk for HNPCC were ascertained. Single-amplicon A636P testing was performed on 21 by polymerase chain reaction of exon 12 of MSH2, followed by direct DNA sequencing. Mutational analysis of the entire open reading frame of MLH1 and MSH2 was performed on 7 by PCR of each exon, followed by heteroduplex analysis using denaturing high-performance liquid chromatography and direct sequencing of exons with variant chromatographs. One patient received both studies,
Results:
The A636P mutation was detected in 3/21 (14%) prospectively evaluated patients using single amplicon testing. In 6 patients, the entire open reading frame of MLH1 and MSH2 was analyzed, and 1 additional A636P carrier and 2 carriers of previously unrecognized mutations were identified. The A636P mutation was present in 2 patients who met Amsterdam criteria and in 2 patients who did not.
Conclusions:
Although rare in the general population, A636P mutations are found at increased frequency in Ashkenazim with a personal or family history of colorectal or other HNPCC-associated cancers. This inexpensive and rapid approach may be useful preoperatively in helping determine the extent of colon resection for a subset of patients.
This report summarizes the initial Memorial Sloan-Kettering Cancer Center experience with prospective, single-amplicon (mutation-specific) A636P testing in Ashkenazi Jewish patients at high or intermediate risk for developing colorectal cancer. Our results support using this inexpensive, rapid test preoperatively for a broader segment of this population with colorectal cancer.
Hereditary nonpolyposis colorectal cancer (HNPCC) is an autosomal dominant syndrome characterized by an increased risk to develop colorectal cancer (CRC) at an early age, as well as other cancers including cancers of the uterus, small bowel, stomach, ovary, pancreas, hepatobiliary tract, and ureter.1 HNPCC can be caused by mutations in one of several DNA mismatch repair (MMR) genes, predominantly MLH1, MSH2, and MSH6.2–5 Identification of disease-causing mutations in the MMR genes usually requires analysis of each individual exon of each of the genes. However, in certain populations, a single founder mutation may account for a significant fraction of the disease-causing mutations segregating in the population.6–8 Such founder mutations can be assayed by analysis of a single exon (referred to as single-amplicon testing). Examples of founder mutations in the MMR genes include the following: MLH1*del exon 16 and MLH1*IVS5-1G>A in Finland,9,10 MSH2*IVS5+3A>T in Newfoundland,11 MLH1*2141G>A in the Valais region of Switzerland,12,13 MSH2*1452-1455delAATG in the Southern region of China,14 the MLH1*415G>C in Ashkenazim,15 and a recently described 16 kb deletion of MSH2 in the United States.16
We previously described an HNPCC-associated founder mutation in the Eastern European Jewish (Ashkenazi) population, MSH2*1906G>C, accounting for two thirds of cases in which disease-causing MMR gene mutations were identified.17 The MSH2*1906G>C nucleotide substitution is a missense mutation that results in an alteration of the amino acid sequence from alanine to proline at codon 636 (A636P). In a combined consecutive series from Israel, New York and Toronto, the A636P mutation was found in 8 of 1345 (0.59%) Ashkenazi Jewish CRC cases versus 0 of 1588 healthy Ashkenazi Jewish controls (P = 0.00095).17 Another study detected a similar prevalence in cases (0.4%) versus controls (0%).18 In a series of New York Ashkenazi Jewish patients who had a CRC diagnosed at age 40 or younger, the A636P mutation was detected in 3 of 41 tested (7%).19
In this report, we describe our initial experience with prospective single-amplicon A636P testing of Ashkenazi Jewish patients with familial HNPCC-associated cancers and explore how this approach may be used preoperatively to identify colon cancer patients eligible for a total colectomy.
MATERIALS AND METHODS
Subjects
Between December 2002 and April 2004, all patients with a personal or family history suggestive of HNPCC who presented for genetic counseling at the Clinical Genetics Service of MSKCC were offered genetic testing for HNPCC. Of these, there were a total of 27 Ashkenazi Jewish patients who completed genetic testing. The medical records of these patients were carefully reviewed and the results tabulated. Twenty patients had family histories which met the Amsterdam20,21 or Bethesda22 criteria, and 7 had family histories of cancer that did not meet these criteria, 2 of which had no family history of cancer at all. Twenty persons received single-amplicon A636P testing only, 6 received full-gene MLH1/MSH2 analysis only, and one received both tests. The strategy used for HNPCC genetic testing in Ashkenazi CRC patients at MSKCC is presented in Figure 1. To simplify counseling, tissue acquisition for MSI and A636P tests was usually initiated at the time of the initial consultation for individuals meeting Bethesda criteria (Table 1). However, since A636P results were typically available before tissue had been obtained and prepared for MSI analysis, MSI testing would have been canceled in the event of an A636P-positive result. Also, when single-amplicon testing detected an A636P mutation, full-gene testing was no longer necessary. When the A636P mutation was not present, microsatellite instability (MSI) testing was continued. In certain cases where tissue was not available or the patient declined to participate, MSI testing was not performed. In cases where A636P test results were negative, the plan was to perform full-gene MLH1 and MSH2 mutational analysis if MSI had been present. In addition, for selected individuals not meeting the Bethesda criteria and therefore not qualifying for any genetic testing, A636P testing was nonetheless performed because of a suggestive family history. It should be noted that the Bethesda criteria were revised in February 2004, close to the end of our current series; therefore, the majority of these patients were clinically evaluated using the original Bethesda criteria which were more restrictive.
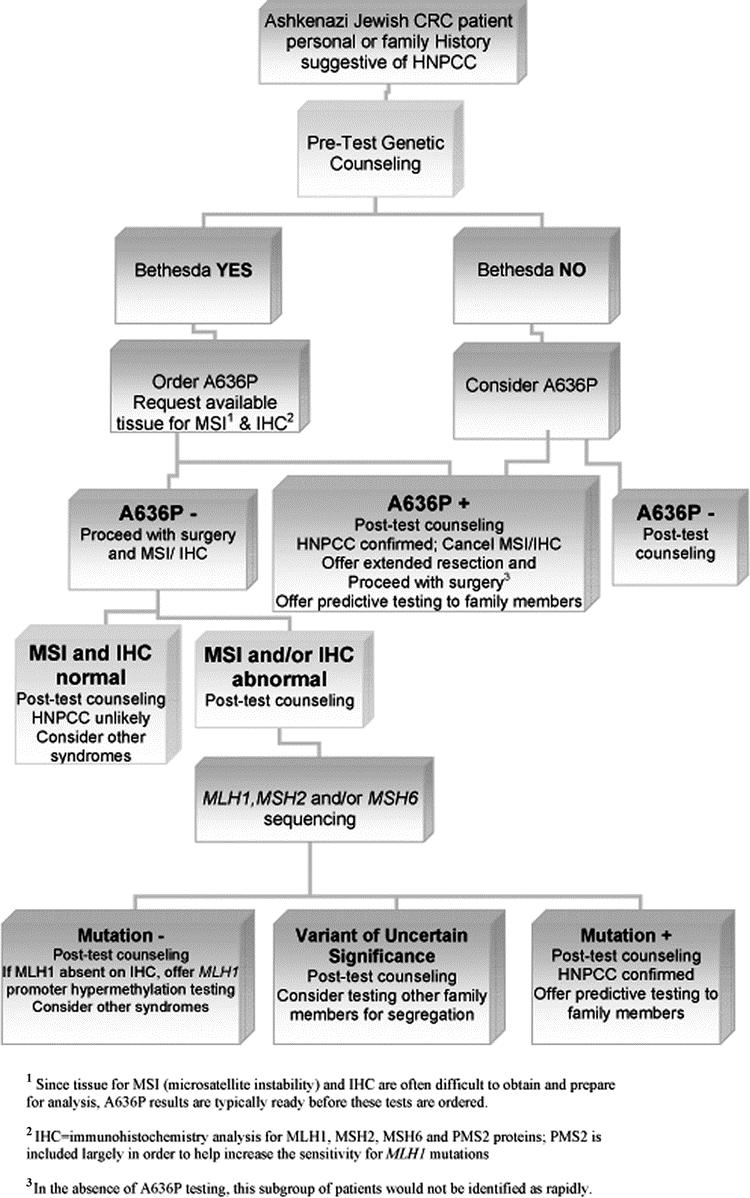
FIGURE 1. The Bethesda criteria are used as a guide to screen patients for genetic testing, which is offered in the context of genetic counseling. For those who meet these criteria and decide to pursue testing, A636P and MSI (with or without IHC) are ordered simultaneously, when possible. Since tissue for MSI (microsatellite instability) and IHC are often difficult to obtain and prepare for analysis, A636P results are typically ready before the results of these tests are available. In the case of an A636P-positive result, MSI and IHC analyses are not considered necessary. Since an A636P-negative result does not exclude the possibility of other MMR mutations, in these cases MSI and IHC analysis should be undertaken. If these are abnormal, sequencing and Southern blot should then be performed.
TABLE 1. The Revised Bethesda Guidelines for Testing Colorectal Tumors for Microsatellite Instability (MSI)

Mutation Analysis
DNA was extracted from whole blood utilizing the Puregene kit (Gentra Systems). For the single-amplicon A636P testing, exon 12 of MSH2 was amplified by polymerase chain reaction (PCR) to generate a 327-bp fragment using forward primer S/E12F 5′-ATTCAGTATTCCTGTGTAC-3′ and reverse primer S/E12R 5′-CGTTACCCCCACAAAGC-3′. The PCR amplification was performed in a final volume of 50 μL, containing 50 ng of DNA, 2.5 mM dNTPs, 15 pmoles of each primer, and 1.25 U of DNA polymerase (AmpliTaq Gold, Applied Biosystems) in Buffer II. The PCR amplification consisted of 10 minutes at 95°C, followed by 35 cycles of 30 seconds at 94°C, 30 seconds at 55°C, and 45 seconds at 72°C; a final extension of 7 minutes ended the amplification reaction. PCR products were purified with the QIAquick PCR purification kit (Qiagen) and quantified by agarose gel electrophoresis. The purified PCR products were sequenced using the forward and reverse PCR primers in separate sequencing reactions performed with ABI Big Dye Terminator chemistry. The products were analyzed by electrophoresis on an ABI 377 DNA sequencer (Applied Biosystems). For the 7 cases that underwent full-gene MLH1 and MSH2 mutation analysis, each of the exons of MLH1 (19 exons) and MSH2 (16 exons) was amplified by PCR, and heteroduplex analyses were performed using denaturing high-performance liquid chromatography (d-HPLC) by modification of the method described for MSH6 gene analysis23 The oligonucleotides used for PCR, PCR conditions, and d-HPLC conditions are available upon request. DNA fragments that displayed an abnormal chromatogram by d-HLPC at any one of the several melting temperatures tested were sequenced directly with Big Dye Terminator chemistry and the reaction products were analyzed using an ABI377 sequencer. The sensitivity of this method in the detection of point mutations has been estimated to be >95%. MSI testing was performed as previously described.24
RESULTS
Of the 27 patients studied, 6 carried a disease-causing MMR gene mutation. Of these, 4 carried the A636P mutation, 3 of which were identified by single-amplicon testing (Table 2). The mean age of first cancer diagnosis for the 4 patients who carried the A636P mutation was 56 years. The pedigrees of all 6 mutation carriers are shown in Figure 2. The patient from Family 1697, whose family history met Amsterdam II criteria and who carried the A636P mutation, was diagnosed with pancreatic cancer at age 50. The patient from Family 5085 did not meet original Bethesda criteria. Nobody in this family was diagnosed with cancer under age 50. The patient from Family 5177, whose family history met Amsterdam criteria, was diagnosed with colon cancer at age 70. The proband from Family 5289, who met Bethesda criteria, was diagnosed with uterine cancer at age 50, a cancer of the duodenum at age 71, and a CRC at age 75, but otherwise had no family history of HNPCC-associated cancers. Full-gene mutation analysis of MLH1 and MSH2 detected 2 families with mutations other than A636P (MSH2*1216C>T and MLH1*3G>A). These 2 mutations have not been previously reported. The patient from Family 5370, whose family history met Amsterdam II criteria and who carried the MSH2*1216C>T mutation, was diagnosed with CRC at age 44. The patient from Family 4709, whose family history met Amsterdam II criteria and who carried the MLH1*3G>A mutation, was diagnosed with uterine cancer and ovarian cancer at age 35.
TABLE 2. Clinicopathologic Characteristics of Ashkenazi Jewish Patients Evaluated for A636P Mutations Between December 2002 and April 2004

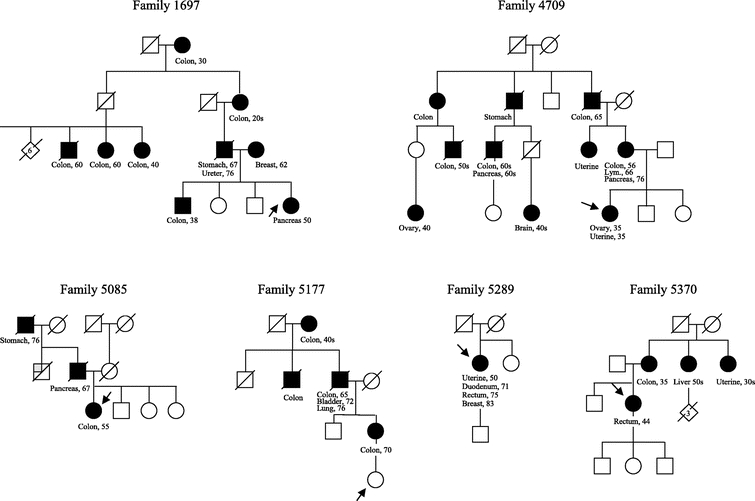
FIGURE 2. Abridged pedigrees of the 6 families identified with a mismatch repair (MMR) gene mutation in this study. Squares designate males, circles designate females, and diamonds designate individuals whose sex is not specified in the pedigree. A number within the symbol indicates multiple persons. A diagonal strikethrough of these symbols indicates the person is known to be deceased. The arrow points to the proband of the family. Filled-in symbols indicate the person was diagnosed with cancer. The type of cancer and age of diagnosis, when known, are indicated below the symbol. Lym, lymphoma.
MSI testing was performed on cancer specimens from 13 of the 27 individuals who underwent mutational analysis. The A636P mutation was present in only 1 of the 13 persons tested (Table 2). In the one A636P carrier tested for MSI, the colon cancer specimen exhibited MSI, as expected. In the 12 A636P-negative cases, MSI was not present, strongly suggesting that a different germline, disease-causing mutation in an MMR gene was not present. Of the 12 MSI-negative cases, only 1 case had a family history that met the Amsterdam criteria. These results are consistent with results from previous studies indicating that MMR gene mutations are infrequent in patients whose families do not meet the Amsterdam criteria.
In our broader overall experience with genetic testing, we have identified 14 Ashkenazi Jewish MMR mutation carriers out of a total of approximately 80 Ashkenazi Jewish probands tested for MMR mutations in the history of the Clinical Genetics Service. Of the 14 carriers, 9 had the A636P mutation and 5 had a different mutation. Thirteen of these 14 carriers met the Bethesda criteria or Amsterdam criteria. One proband carried the A636P mutation but did not meet either Bethesda criteria or Amsterdam criteria; however, there was a notable cluster of colon cancer in third- and fourth-degree relatives. This individual was diagnosed with a gynecologic cancer of uncertain primary, and also had a family history of breast and ovarian cancer.
In summary, during a 17-month period, HNPCC mutations were detected in 6 of 27 (22%) Ashkenazi Jewish individuals tested. Two thirds of these mutations were A636P. These results are consistent with our overall experience between 1998 and 2005, where we found 14 mutation carriers in approximately 80 Ashkenazi Jewish individuals tested (18%). Of the detected mutations, 9 of 14 (64%) were A636P.
DISCUSSION
The results from our prospective, single-amplicon testing approach corroborate previous retrospectively based observations of an increased incidence of the A636P mutation in an Ashkenazi Jewish patient population enriched for a family history of HNPCC-associated cancers. Since 2 of the 6 mutation carriers in our study, both of whom had an A636P mutation, met revised Bethesda but not the Amsterdam criteria, our results suggest that A636P testing should be offered to all Jewish families meeting the revised Bethesda criteria even if they do not meet the Amsterdam criteria. Since the method of determination is simple and inexpensive, this testing should not be overly burdensome. Also, since the testing can be provided in a timely manner (within 1 week), determination of A636P status preoperatively should have minimal to no impact on the scheduling of a semi-elective surgical procedure. Therefore, it seems prudent to consider A636P testing presurgically for all Ashkenazi Jews who meet the revised Bethesda criteria. Without single-amplicon testing, preoperative identification of patients with a MMR mutation but without an obvious family history (such as family 5085 and 5289 in Fig. 1) would not be possible in a timely manner due to time involved in standard diagnostic molecular genetic workup.
Patient interest and the technical feasibility of utilizing presurgical genetic testing in high-risk individuals have already been demonstrated in breast cancer patients.25,26 Consequently, mutation carriers would have the opportunity to consider prophylactic subtotal colectomy over conventional segmental resection because of the increased risk of second primaries in the colon. Indeed, it has been estimated that individuals with an MMR mutation27 have an approximate 40% risk of developing a metachronous CRC within 10 years1,28,29 after segmental colectomy. In addition, female carriers are at increased risk for endometrial and ovarian cancers and may wish to consider prophylactic hysterectomy and bilateral salpingo-oophorectomy at the time of colon resection.30,31
Although presurgical genetic testing is feasible, it is important to recognize that the pros and cons of genetic testing have to be discussed with the patient along with the risks and benefits of the surgical options (segmental resection vs. a subtotal colectomy with or without prophylactic hysterectomy and bilateral salpingo-oophorectomy) within a short period of time. Although the concern for “emotional overload” by offering genetic testing peri-diagnostically has been raised,32 we think that the benefits of this approach outweigh the drawbacks, particularly in patients who plan to use genetic test results to choose between a segmental resection and more extended surgery. Genetic counseling plays an important role in both pretest and posttest settings by facilitating patient education, psychologic adjustment, and complex patient-based decision making.
CONCLUSION
The A636P mutation is a major cause of HNPCC in the Ashkenazi Jewish CRC population. Our experience suggests that two thirds of Ashkenazi Jewish individuals with HNPCC can be identified using the A636P single-amplicon test alone, and that most of these carriers will have personal and family histories meeting the revised Bethesda criteria. Testing for the A636P mutation alone is less expensive and can provide a diagnosis in a shorter period of time, relative to a standard genetic workup for HNPCC. We think that Jewish patients who meet the Bethesda criteria should be offered A636P single-amplicon testing as part of their initial genetic investigation, continuing with MSI and/or IHC in the A636P-negative cases. We think that this targeted genetic testing approach can be performed preoperatively, in a timely manner, and can help direct the extent of colon resection. Larger studies will be necessary to calculate the costs, benefits, and optimal timing of A636P genotyping as a part of CRC risk assessment for these families.
ACKNOWLEDGMENTS
The authors thank the following individuals for their input: Robert Finch, MS, Crystal Palmer, BS, Jesse Hansen, BS, Johanna Lee, MPH, Kelly LaFaro, and other members of the Clinical Genetics Service at Memorial Sloan-Kettering Cancer Center; and Drs. W. Douglas Wong, Philip Paty, Martin Weiser and Larissa Temple of the Memorial Sloan-Kettering Cancer Center Colorectal Service.
Footnotes
Supported by the Lymphoma Foundation, the Koodish Fellowship, the Tavel-Reznik Foundation, the Cancer Research Foundation of America, the Frankel Fellowship Fund, the Grace Fippinger Foundation, the Bianco Family Fund, the Weissenbach-Southworth Family Research Fund, the Edith Nathanson Fund, and the Ferdinand Koch Fund.
Reprints will not be available. Correspondence: Jose G. Guillem, MD, MPH, Memorial Sloan-Kettering Cancer Center, 1275 York Avenue, Room C-1077, New York, NY 10021. E-mail: guillemj@mskcc.org.
REFERENCES
- 1.Guillem JG, Smith AJ, Puig-La Calle J, et al. Gastrointestinal polyposis syndromes. Curr Probl Surg. 1999;36:217–324. [DOI] [PubMed] [Google Scholar]
- 2.Fishel R, Lescoe MK, Rao MR, et al. The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer. Cell. 1993;75:1027–1038. [DOI] [PubMed] [Google Scholar]
- 3.Bronner CE, Baker SM, Morrison PT, et al. Mutation in the DNA mismatch repair gene homologue hMLH1 is associated with hereditary non-polyposis colon cancer. Nature. 1994;368:258–261. [DOI] [PubMed] [Google Scholar]
- 4.Akiyama Y, Sato H, Yamada T, et al. Germ-line mutation of the hMSH6/GTBP gene in an atypical hereditary nonpolyposis colorectal cancer kindred. Cancer Res. 1997;57:3920–3923. [PubMed] [Google Scholar]
- 5.Miyaki M, Konishi M, Tanaka K, et al. Germline mutation of MSH6 as the cause of hereditary nonpolyposis colorectal cancer. Nat Genet. 1997;17:271–272. [DOI] [PubMed] [Google Scholar]
- 6.Barchana M, Lipshitz I, Rozen P. Trends in colorectal cancer incidence and mortality in the Israeli Jewish ethnic population. Fam Cancer. 2004;3:207–214. [DOI] [PubMed] [Google Scholar]
- 7.Thomas MG, Weale ME, Jones AL, et al. Founding mothers of Jewish communities: geographically separated Jewish groups were independently founded by very few female ancestors. Am J Human Genet. 2002;70:1411–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Behar DM, Garrigan D, Kaplan ME, et al. Contrasting patterns of Y chromosome variation in Ashkenazi Jewish and host non-Jewish European populations. Hum Genet. 2004;114:354–365. [DOI] [PubMed] [Google Scholar]
- 9.Moisio AL, Sistonen P, Weissenbach J, et al. Age and origin of two common MLH1 mutations predisposing to hereditary colon cancer. Am J Hum Genet. 1996;59:1243–1251. [PMC free article] [PubMed] [Google Scholar]
- 10.Nystrom-Lahti M, Kristo P, Nicolaides NC, et al. Founding mutations and Alu-mediated recombination in hereditary colon cancer. Nat Med. 1995;1:1203–1206. [DOI] [PubMed] [Google Scholar]
- 11.Spirio L, Green J, Robertson J, et al. The identical 5′ splice-site acceptor mutation in five attenuated APC families from Newfoundland demonstrates a founder effect. Hum Genet. 1999;105:388–398. [DOI] [PubMed] [Google Scholar]
- 12.Salvaara R, Loukola A, Kristo P, et al. Population-based molecular detection of hereditary nonpolyposis colorectal cancer. J Clin Oncol. 2000;18:2193–2200. [DOI] [PubMed] [Google Scholar]
- 13.Hutter P, Courturier A, Scott RJ, et al. Complex genetic predisposition to cancer in an extended HNPCC family with an ancestral hMLH1 mutation. J Med Genet. 1996;33:636–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chan TL, Wai Chan Y, Ho JW, et al. MSH2 c. 1452–1455de1AATG is a founder mutation and an important cause of Hereditary Nonpolyposis Colorectal Cancer in the southern Chinese population. Am J Hum Genet. 2004;74:1035–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lipkin SM, Rozek LS, Rennert G, et al. The MLH1 D132H variant is associated with susceptibility to sporadic colorectal cancer. Nat Genet. 2004;36:694–699. [DOI] [PubMed] [Google Scholar]
- 16.Lynch HT, Coronel SM, Okimoto R, et al. A founder mutation of the MSH2 gene and hereditary nonpolyposis colorectal cancer in the United States. JAMA. 2004;291:718–724. [DOI] [PubMed] [Google Scholar]
- 17.Foulkes WD, Thiffault I, Gruber SB, et al. The founder mutation MSH*1906G–>C is an important cause of hereditary nonpolyposis colorectal cancer in the Ashkenazi Jewish population. Am Hum Genet. 2002;71:1395–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zauber NP, Sabbath-Solitare M, Marotta S, et al. Clinical and genetic findings in an Ashkenazi Jewish population with colorectal neoplasms. Cancer. 2005;104:719–729. [DOI] [PubMed] [Google Scholar]
- 19.Guillem JG, Rapaport BS, Kirchhoff T, et al. A636P is associated with early-onset colon cancer in Ashkenazi Jews. J Am Coll Surg. 2003;196:222–225. [DOI] [PubMed] [Google Scholar]
- 20.Vasen HF, Mecklin JP, Khan PM, et al. The International Collaborative Group on Hereditary Non-Polyposis Colorectal Cancer (ICG-HNPCC). Dis Colon Rectum. 1991;34:424–425. [DOI] [PubMed] [Google Scholar]
- 21.Vasen HF, Watson P, Mecklin JP, et al. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative Group on HNPCC. Gastroenterology. 1999;116:1453–1456. [DOI] [PubMed] [Google Scholar]
- 22.Umar A, Boland CR, Terdiman JP, et al. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst. 2004;96:261–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Peterlongo P, Nafa K, Lerman GS, et al. MSH6 germline mutations are rare in colorectal cancer families. Int J Cancer. 2003;107:571–579. [DOI] [PubMed] [Google Scholar]
- 24.Shia J, Klimstra D, Nafa K, et al. Value of immunohistochemical detection of DNA mismatch repair gene proteins in predicting germline mutation in hereditary colorectal neoplasms. Am J Surg Pathol. 2005;29:96–104. [DOI] [PubMed] [Google Scholar]
- 25.Weitzel JN, McCaffrey SM, Nedelcu R, et al. Effect of genetic cancer risk assessment on surgical decisions at breast cancer diagnosis. Arch Surg. 2003;138:1323–1328. [DOI] [PubMed] [Google Scholar]
- 26.Schwartz MD, Lerman C, Brogan B, et al. Impact of BRCA1/BRCA2 counseling and testing on newly diagnosed breast cancer patients. J Clin Oncol. 2004;22:1823–1829. [DOI] [PubMed] [Google Scholar]
- 27.Guillem JG, Wood WC, Moley J, et al. ASCO/SSO review of current role of risk-reducing surgery in common hereditary cancer syndromes. J Clin Oncol. 2006;24:4642–4660. [DOI] [PubMed] [Google Scholar]
- 28.Lynch HT, de la Chapelle A. Hereditary colorectal cancer. N Engl J Med. 2003;348:919–932. [DOI] [PubMed] [Google Scholar]
- 29.Church J, Simmang C. Practice parameters for the treatment of patients with dominantly inherited colorectal cancer (familial adenomatous polyposis and hereditary nonpolyposis colorectal cancer). Dis Colon Rectum. 2003;46:1001–1012. [DOI] [PubMed] [Google Scholar]
- 30.Lynch HT, Lynch J. Lynch syndrome: genetics, natural history, genetic counseling, and prevention. J Clin Oncol. 2000;18(suppl 21):19–31. [PubMed] [Google Scholar]
- 31.Schmeler KM, Lynch HT, Chen LM, et al. Prophylactic surgery to reduce the risk of gynecologic cancers in the Lynch syndrome. N Engl J Med. 2006;354:261–269. [DOI] [PubMed] [Google Scholar]
- 32.Ardern-Jones A, Kenen R, Eeles R. Too much, too soon? Patients and health professionals’ view concerning the impact of genetic testing at the time of breast cancer diagnosis in women under the age of 40. Eur J Cancer Care. 2005;14:272–281. [DOI] [PubMed] [Google Scholar]