

Abstract
Objectives:
Coagulopathy following major trauma is conventionally attributed to activation and consumption of coagulation factors. Recent studies have identified an acute coagulopathy present on admission that is independent of injury severity. We hypothesized that early coagulopathy is due to tissue hypoperfusion, and investigated derangements in coagulation associated with this.
Methods:
This was a prospective cohort study of major trauma patients admitted to a single trauma center. Blood was drawn within 10 minutes of arrival for analysis of partial thromboplastin and prothrombin times, prothrombin fragments 1+2, fibrinogen, thrombomodulin, protein C, plasminogen activator inhibitor-1, and d-dimers. Base deficit (BD) was used as a measure of tissue hypoperfusion.
Results:
A total of 208 patients were enrolled. Patients without tissue hypoperfusion were not coagulopathic, irrespective of the amount of thrombin generated. Prolongation of the partial thromboplastin and prothrombin times was only observed with an increased BD. An increasing BD was associated with high soluble thrombomodulin and low protein C levels. Low protein C levels were associated with prolongation of the partial thromboplastin and prothrombin times and hyperfibrinolysis with low levels of plasminogen activator inhibitor-1 and high d-dimer levels. High thrombomodulin and low protein C levels were significantly associated with increased mortality, blood transfusion requirements, acute renal injury, and reduced ventilator-free days.
Conclusions:
Early traumatic coagulopathy occurs only in the presence of tissue hypoperfusion and appears to occur without significant consumption of coagulation factors. Alterations in the thrombomodulin-protein C pathway are consistent with activated protein C activation and systemic anticoagulation. Admission plasma thrombomodulin and protein C levels are predictive of clinical outcomes following major trauma.
Acute traumatic coagulopathy, present immediately on arrival in the emergency department, is a consequence of tissue hypoperfusion and may be due to systemic anticoagulation modulated through the protein C pathway.
Trauma remains the leading cause of death and disability in adults, eclipsing ischemic heart disease, cerebrovascular disease, and HIV/AIDS.1 Worldwide, 1 in 7 deaths is due to injury, and this is expected to rise to 1 in 5 in the next 15 years, despite advances in resuscitation, trauma surgery, and critical care.2 Coagulation abnormalities are common following major trauma, and are associated with poor outcomes.3 Classically, traumatic coagulopathy is thought to be due to the consumption of coagulation factors and dilution from intravenous blood and fluid therapy.4 Coagulopathy is therefore currently managed by coagulation factor replacement such as with fresh frozen plasma and cryoprecipitate.5 Consistent with this concept, recombinant factor VIIa has recently been investigated as another therapy for traumatic coagulopathy.6
Two recent studies have identified an acute coagulopathy that is present on arrival in the emergency department in approximately one quarter of major trauma patients.7,8 Patients arriving with a coagulopathy were 4 times more likely to die than those with normal coagulation. This effect was independent of injury severity and hence unlikely to be exclusively due to tissue injury and coagulation factor consumption. Apart from the severity of injury, shock with tissue hypoperfusion is also known to be an independent predictor of mortality and morbidity in severe trauma. Elevated admission base deficit (BD) levels are associated with an increase in transfusion requirements, acute lung injury, multiple organ failure, and mortality.9–13 We hypothesized that acute traumatic coagulopathy is primarily caused by tissue hypoperfusion resulting in activation of the anticoagulant protein C pathway, not the consumption of coagulation factors. An increase in thrombomodulin induced by hypoperfusion would divert thrombin from fibrin generation to the activation of protein C, leading to anticoagulation and inhibition of further thrombin generation.14
The first aim of this study was therefore to identify the mechanisms responsible for the acute coagulopathy that is evident early in the clinical course of victims of major trauma. Since coagulopathy is an independent predictor of mortality after trauma,7,8 the second aim was to identify the prognostic significance of early alterations in the thrombomodulin-protein C pathway.
METHODS
Study Design
This was a single center, prospective, cohort study of major trauma patients presenting directly to a level 1 trauma center between March 2003 and June 2004.
Patient Selection
All adult trauma patients who met criteria for full trauma team activation were eligible for enrollment into the study. Patients younger than 18 years or transferred from other hospitals were immediately excluded. Patients were retrospectively excluded if they were found to be taking anticoagulant medications, had moderate or severe liver disease,15 or a known bleeding diathesis. The Institutional Review Board of the University of California at San Francisco approved the research protocol and granted a waiver of consent for the blood sampling as a minimal risk intervention.
Sampling Technique
As part of standard trauma management, 1 member of the trauma team is designated to gain intravenous access and draw blood for laboratory analyses as soon as the patient arrives in the trauma room. Access is via a 14- or 16-gauge cannula in the forearm or via a 7-French line in the femoral vein. A 10 mL research sample of blood was drawn from this line or as a separate venepuncture along with the standard trauma laboratory tests within 10 minutes of arrival in the emergency department. The sample was placed in a citrated tube and sent immediately to the hospital's central laboratory where it was spun down, plasma extracted, and stored in a −80°C freezer.
Sample Analysis
Samples were analyzed at the conclusion of the study by researchers who were blinded to all patient data. Normal ranges of 2 standard deviations from the mean were determined by testing 50 normal samples of the George King Biomedical Core Set 0070. The following assays were used: prothrombin fragments (Enzygnost F1+2 EIA, Dade Behring, Germany; normal range 0.09–0.40 nM); fibrinogen (AssayPro, USA; normal range 2.3–3.3 g/L); soluble thrombomodulin (Asserachrom Thrombomodulin EIA, Diagnostica Stago, USA; normal range 18–53 ng/mL); protein C activity (Staclot Protein C clot-based activity assay, Diagnostica Stago; normal range 64–164%); plasminogen activator inhibitor 1 (PAI-1) (Stachrom PAI chromogenic activity assay, Diagnostica Stago; normal range 4–14 AU/mL), and d-dimers (STA Liatest D-DI, Diagnostica Stago; normal range 0–39 ng/mL).
Data Collection
Data were collected prospectively on patient demographics, the injury time, mechanism (blunt or penetrating) and severity, prehospital fluid administration, the time of arrival in the trauma room, and admission vital signs. The Injury Severity Score (ISS) was used as a measure of the degree of tissue injury.16 Prothrombin time and an arterial blood gas were drawn at the same time as the research sample as part of the standard management of major trauma patients. The BD was used as a measure of the degree of tissue hypoperfusion. Admission BD is a clinically useful early marker of tissue hypoperfusion in trauma patients and an admission BD greater than 6 mEq/L has previously been identified as predictive of worse outcome in trauma patients.10,11,17,18
Outcome Measures
Patients were followed until hospital discharge or death. For mortality analysis, patients surviving to hospital discharge were assumed to still be alive. Outcome measures were also recorded for 28-day ventilator-free days, acute lung injury (American-European consensus conference definition19) and acute renal injury (Acute Dialysis Quality Initiative consensus conference definition20), and blood transfusions required in the first 24 hours.
Statistical Analysis
Data analysis was performed by the investigators. Normal-quartile plots were used to test for normal distribution. Parametric data are expressed as mean ± 95% confidence intervals. Two-group analysis was performed with a 2-tailed unequal variance Student t test. Correlation was assessed by Pearson method. The χ2 test was used for dichotomous data analysis. A P value of 0.05 was chosen to represent statistical significance.
RESULTS
There were 208 patients enrolled into the study over a 15-month period. There were no retrospective exclusions. Patients, injury type, and admission physiology data are shown in Table 1. Median time from injury to blood sampling was 32 minutes, there was no vasopressor or colloid use and patients received an average of 150 ± 100 mL of intravenous crystalloid before specimen collection. The platelet count was normal in all patients (average 274 ± 86 × 109/mL).
TABLE 1. Clinical Characteristics of Major Trauma Patients
As tissue hypoperfusion increased (as measured by the BD) both partial thromboplastin time and the prothrombin time increased (Figs. 1A,B). Prothrombin fragments 1+2 (PF1+2) levels were assayed as a measure of total thrombin generation and increased as injury severity increased as measured by both the ISS (PF1+2: ISS <16, 2.2 nM; ISS 16–30, 3.8 nM; ISS >30, 6.3 nM; P < 0.001) and the New Injury Severity Score (NISS) (PF1+2: NISS <16, 2.1 nM; NISS 16–30, 3.1 nM; NISS >30, 5.4 nM; P < 0.001). Thrombin generation was unchanged by the degree of hypoperfusion (PF1+2: BD ≤6 mEq/L, 3.6 nM; BD >6 mEq/L, 4.0 nM; P = 0.20). Coagulopathy was present when thrombin generation occurred with a raised BD (Figs. 1C, D; black bars) but was normal in the absence of hypoperfusion, regardless of the amount of thrombin generated (Figs. 1C, D; white bars). Fibrinogen levels were normal in all patients (2.6 ± 0.9 g/L).

FIGURE 1. Effects of tissue hypoperfusion on coagulation. A: Increasing partial thromboplastin time with increasing BD. BD in quartiles. *P = 0.003 comparing BD ≤2.1 mEq/L with BD ≥7.7 mEq/L. B: Increasing prothrombin time with increasing BD. BD in quartiles. *P = 0.01 comparing BD ≤2.1 mEq/L with BD ≥7.7 mEq/L. C: Partial thromboplastin time is prolonged only in the presence of a raised BD. While the BD remains low (white bars), increasing thrombin generation has no effect on the prothrombin time. With a raised BD (black bars), prolongation of the prothrombin time is seen with increasing thrombin generation. PF1+2 in tertiles. *P = 0.01 comparing PF1+2 >4.0 nM with PF1+2 <2.0 nM for BD >6 mEq/L. +P = 0.03 comparing BD ≤6 and >6 mEq/L for PF1+2 >4.0 nM. D: Prothrombin time is prolonged only in the presence of a raised BD (as C). PF1+2 in tertiles. *P = 0.002 comparing PF1+2 >4.0 nM with PF1+2 <2.0 nM for BD >6 mEq/L. +P = 0.008 comparing BD ≤6 and >6 mEq/L for PF1+2 >4.0 nM.
These data suggest that prolongation of functional clotting assays at this acute postinjury stage were not primarily due to consumption. We next sought to identify variations with the BD in other coagulation proteins. Increasing BD was associated with a rise in the levels of soluble thrombomodulin (Fig. 2A) and a fall in protein C levels (Fig. 2B). There was a close inverse relationship between soluble thrombomodulin and protein C levels (TM <28: 85%, TM >43: 68%; P = 0.02, upper-lower quartile comparison). Protein C levels fell with increasing thrombin generation but only in the presence of an increased BD (Fig. 2C) and the associated raised thrombomodulin levels (Fig. 2D).

FIGURE 2. Changes in thrombomodulin and protein C. A: Increased plasma thrombomodulin levels with increasing BD. BD in quartiles. *P = 0.01 compared with BD ≤2.1 mEq/L. B: Reduced plasma protein C levels with increasing BD. BD in quartiles. *P < 0.001 compared with BD ≤2.1 mEq/L. C: Reduced plasma protein C levels with increasing prothrombin fragment levels occur only in the presence of an increased BD (>6 mEq/L). Prothrombin fragments in tertiles. *P = 0.002 comparing PF1+2 <2 nM to PF1+2 2–4 nM, P < 0.001 to PF1+2 >4 nM for BD >6 mEq/L. +P = 0.014 comparing BD ≤6 mEq/L and BD >6 mEq/L for PF1+2 >4 nM. D: Reduced plasma protein C levels with increasing prothrombin fragment levels occurs only in the presence of increased thrombomodulin levels (>53 ng/mL). Prothrombin fragments in tertiles. *P = 0.03 comparing thrombomodulin ≤53 ng/mL and thrombomodulin >53 ng/mL for PF1+2 >4 nM.
These data suggest activation of protein C during hypoperfusion by increased formation of the thrombin–thrombomodulin complex. However, we were unable to measure activated protein C during this study and thus we present indirect evidence of protein C activation. Falling protein C levels were associated with anticoagulation and prolongation of the partial thromboplastin and prothrombin times (Figs. 3A, B). Activated protein C in excess consumes PAI-1 and we were able to identify a dose-dependent reduction in PAI-1 as protein C levels reduced (Fig. 3C), leading to increased fibrinolysis with a rise in tissue plasminogen activator (tPA) levels (protein C ≥102: tPA 18.3 ± 2.5 ng/mL; protein C ≤57: tPA 32.5 ± 7.9 ng/mL; P = 0.001) and an associated increase in d-dimers (Fig. 3D).

FIGURE 3. Indirect evidence for activation of protein C. A: Partial thromboplastin time is prolonged as protein C falls. Protein C in quartiles. *P < 0.001 compared with protein C ≥102%. B: Prothrombin time is prolonged as protein C falls. Protein C in quartiles. *P < 0.001 compared with protein C ≥102%. C: Plasma plasminogen activator inhibitorI-1 (PAI-1) levels fall as protein C falls. Protein C in quartiles. *P = 0.004 compared with protein C ≥102%. D: Plasma d-dimer levels rise as protein C levels decrease. Protein C in quartiles. *P = 0.01 for protein C 58–78%, P < 0.001 for protein C ≤57% compared with protein C ≥102%.
Outcomes
The mortality rate of the trauma patients in this study was 12%. Low protein C and high thrombomodulin levels were both significantly associated with increased mortality (Fig. 4A, B; Table 2). An elevated thrombomodulin was associated with a 27% mortality rate compared with 11% when thrombomodulin was normal (P = 0.02). A low protein C level carried a 30% mortality, compared with a 5% mortality with normal protein C levels (P < 0.001).
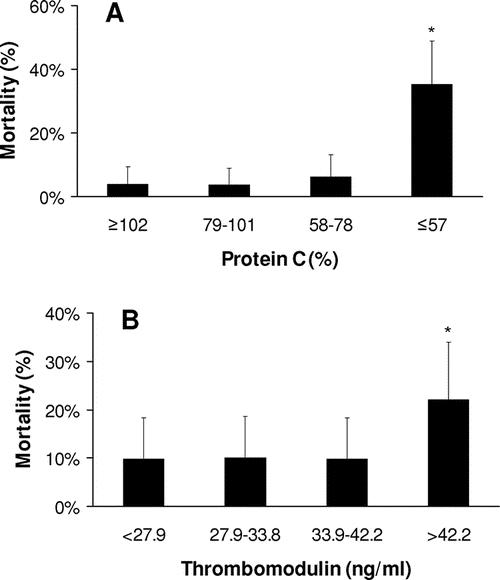
FIGURE 4. Mortality associated with early activation of the thrombomodulin-protein C pathway. A: Low plasma protein C levels on admission are associated with increased mortality. Protein C in quartiles. *P < 0.001 compared with protein C ≥102%. B: High plasma thrombomodulin levels on admission are associated with increased mortality. Thrombomodulin in quartiles. *P < 0.001 compared with thrombomodulin >42.2 ng/mL.
TABLE 2. The Relationship of Low Protein C and High Thrombomodulin Levels to Clinical Outcomes

The incidence of organ injury was increased with activation of the thrombomodulin-protein C pathway. Acute renal injury was present in 10% of patients with normal thrombomodulin levels but 31% of those with a high thrombomodulin (P = 0.002). Similarly, 7% of patients with a normal protein C developed acute renal injury but the incidence was 25% in those with a low protein C (P < 0.001) (Table 2). There was a trend towards increased incidence of acute lung injury with elevated thrombomodulin levels (23% vs. 14%, P = 0.09). The number of ventilator-free days at 28 days was significantly reduced in patients with high thrombomodulin levels (17 ± 5 vs. 23 ± 1 day, P = 0.03) and low protein C levels (18 ± 3 vs. 24 ± 1 day, P < 0.001) (Table 2). Blood transfusion requirements were increased in patients with high thrombomodulin levels (6 ± 3 vs. 2 ± 1 units, P = 0.002) and low protein C levels (6 ± 3 vs. 1 ± 1 units, P = 0.003) on admission.
DISCUSSION
This study represents a very early investigation of the coagulation system following major traumatic injury in humans. There was no significant fluid resuscitation or other potentially confounding medical therapy before blood sampling, and therefore alterations of the coagulation system represent the effects of the injury and its initial physiological sequelae.
Our results suggest that clinical coagulopathy at this early postinjury time point is not due to insufficient amounts of coagulation factors. Prothrombin levels are normally in the 1–2 μM range21 and prolongation of the prothrombin time is not usually seen until levels fall significantly below normal. However PF1+2 levels were in the low nanomolar range, suggesting that utilization of prothrombin is not the cause of this measurable coagulopathy. This is supported by the normal fibrinogen levels seen in all patients.
Acute traumatic coagulopathy as measured by the prothrombin and partial thromboplastin times only occurred in the presence of tissue hypoperfusion. In the presence of an increased BD soluble thrombomodulin levels increased, and increasing thrombin generation produced a decrease in protein C levels and anticoagulation. With a normal BD, partial thromboplastin and prothrombin times were always normal.
Protein C is cleaved by thrombin when it is complexed with thrombomodulin at the endothelial protein C receptor. Activated protein C inhibits coagulation cofactors V and VIII, reducing further thrombin generation.14 We were not able to measure activated protein C levels in this study because of its rapid breakdown by plasma proteases and thus we could only investigate the sequelae of protein C activation. Conversion to activated protein C is supported by increasing anticoagulation with decreasing protein C levels. If protein C was being inhibited or otherwise reduced this should produce a procoagulant state. Further support is given by the associated increase in fibrinolysis. When present in excess, activated protein C depletes PAI-1 thus reducing tPA inhibition and accelerating the conversion of plasminogen to plasmin.22
Both high thrombomodulin and low protein C levels were associated with increased mortality. Systemic anticoagulation and hyperfibrinolysis may increase mortality initially by increasing blood loss and exacerbating the shock state. Coagulopathy will also worsen traumatic brain injury by increasing the size of extra-axial hematomas and cerebral contusions, thus exacerbating any secondary brain injury.23 Similar effects may be observed for pulmonary contusions leading to worsening oxygenation and hypoxic injury to other organs.24,25
In contrast to this and previous studies of an early anticoagulant state,7,8 some studies have described a procoagulant state following injury. A study identifying low protein C levels early after injury assumed that this represented a procoagulant state rather than anticoagulation due to the activation of protein C.26 However, there are studies investigating trauma patients several hours or days after injury which have identified low protein C levels27,28 and trauma patients are known to have an increased risk of venous thromboembolism.29 These findings are not necessarily contradictory to those of the current study. After a severe or prolonged episode of shock and activated protein C production, protein C may be exhausted and further generation of activated protein C would be impaired. This would result in a procoagulant, thrombogenic state and contribute to the increased incidence of organ failure as well as the predisposition to venous thromboembolism. In support of this, admission coagulopathy has previously been identified as a strong predictor for the subsequent development of thromboembolic complications.30 Further studies are required to investigate late changes in coagulation resulting from this early anticoagulant state.
There are several limitations to this study in addition to the absence of direct measurement of activated protein C. First, in the absence of a biochemical marker of the volume of tissue injury, the Injury Severity Score (ISS) was used as a surrogate. Although worsening ISS scores do reflect more severe injuries, the ISS is calculated from Abbreviated Injury Scale scores31 which are assigned according to how likely an injury is to contribute to mortality, rather than how much tissue has been injured. However, in most cases the ISS will reflect the degree of tissue trauma. Second, we used the BD alone as the measure of tissue hypoperfusion and did not measure plasma lactate levels. However, the BD has been shown to be closely correlated to lactate levels, a clinically useful marker of tissue hypoperfusion and a better predictor of outcome in trauma and hemorrhagic shock.9,18,32 Third, we measured soluble thrombomodulin levels as a surrogate for endothelial membrane-bound thrombomodulin. The exact relationship between endothelial and soluble thrombomodulin has never been elucidated and some authors have suggested that soluble thrombomodulin, while a good marker of endothelial injury, may impair the activation of protein C.33 We have found that increasing soluble thrombomodulin levels are associated with decreasing protein C levels with evidence of increased activated protein C activity. This is consistent with either soluble thrombomodulin levels reflecting membrane-bound activity, or soluble thrombomodulin retaining some ability to convert protein C, as some studies have suggested.34,35 Further laboratory research will be needed to definitively answer this question. Finally, we did not measure the other anticoagulant proteins antithrombin III and tissue factor pathway inhibitor. However, antithrombin III is produced by the liver and will be cleared as thrombin-antithrombin complexes, and so activity is unlikely to rise in this early postinjury phase. No change in tissue factor pathway inhibitor levels were identified in a previous study in trauma patients.36
The findings of this study have important implications on several levels. Currently the management of traumatic coagulopathy is almost entirely directed at augmenting thrombin generation with blood component therapy or recombinant Factor VIIa. However, if the primary derangement is anticoagulation from activation of the thrombomodulin–protein C pathway, augmenting thrombin generation in the presence of hypoperfusion may cause further activation of anticoagulant and fibrinolytic pathways. Subsequently, once protein C is exhausted, increasing thrombin generation further may result in clot formation in underperfused vascular beds, microvascular thrombosis, and subsequent organ dysfunction. Therefore, management of acute traumatic coagulopathy should focus on limiting the degree and duration of shock and tissue hypoperfusion.
Finally, the observation that low protein C levels are associated with hypoperfusion is interesting in light of the importance of the protein C pathway in other pathologic states. Depletion of protein C is well described in severe sepsis.33,37 A recent study has also identified activated protein C generation immediately following cardiac arrest.38 We found that a reduction in protein C is detectable very early in the evolution of traumatic shock. This suggests that hypoperfusion itself may be the common denominator that leads to early protein C activation and subsequently the protein C depletion observed in severe sepsis, cardiac arrest, and major trauma.
ACKNOWLEDGMENTS
The authors thank Meghan Levee (San Francisco General Hospital) and Justin Parekh (University of California, San Francisco Medical School) for their assistance in conducting this study, David Glidden (Department of Biostatistics and Epidemiology, University of California at San Francisco) for statistical advice, Peter MacCallum (Department of Hematology, the Royal London Hospital) for reviewing the manuscript, and S. Betty Yan and Suzane Um (Eli Lilly and Company) for measuring plasma levels of prothrombin fragments 1+2, thrombomodulin, protein C, tissue plasminogen activator and plasminogen activator inhibitor-1.
Footnotes
Supported in part by Grant R01 GM62188 from the National Institutes of Health.
Reprints: Karim Brohi, Trauma Service, Department of Surgery, The Royal London Hospital, Whitechapel Road, London E1 1BB, UK. E-mail: karim@trauma.org.
REFERENCES
- 1.Mathers CD, Lopez A, Stein C, et al. Deaths and disease burden by cause: global burden of disease estimates for 2001 by World Bank country groups. Fogarty International Center, National Institutes of Health, 2003. Available at http://www.fic.nih.gov/dcpp/wps/wp18.pdf. Accessed December 16, 2005.
- 2.Injury Chart Book. Geneva: World Health Organization; 2002. Available at http://www.who.int/violence_injury_prevention/injury/chartbook/chartb/en/. Accessed December 16, 2005.
- 3.Gando S, Nanzaki S, Kemmotsu O. Disseminated intravascular coagulation and sustained systemic inflammatory response syndrome predict organ dysfunctions after trauma: application of clinical decision analysis. Ann Surg. 1999;229:121–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Armand R, Hess JR. Treating coagulopathy in trauma patients. Transfus Med Rev. 2003;17:223–231. [DOI] [PubMed] [Google Scholar]
- 5.College of American Pathologists. Practice parameters for the use of fresh frozen plasma, cryoprecipitate and platelets. JAMA. 1994;271:777–781. [PubMed] [Google Scholar]
- 6.Boffard KD, Riou B, Warren B, et al. Recombinant factor VIIa as adjunctive therapy for bleeding control in severely injured trauma patients: two parallel randomized, placebo-controlled, double-blind clinical trials. J Trauma. 2005;59:8–18. [DOI] [PubMed] [Google Scholar]
- 7.Brohi K, Singh J, Heron M, et al. Acute traumatic coagulopathy. J Trauma. 2003;54:1127–1130. [DOI] [PubMed] [Google Scholar]
- 8.Macleod JBA, Lynn M, McKenney MG, et al. Early coagulopathy predicts mortality in trauma. J Trauma. 2003;55:39–44. [DOI] [PubMed] [Google Scholar]
- 9.Siegel JH, Rivkind AI, Dalal S, et al. Early physiologic predictors of injury severity and death in blunt multiple trauma. Arch Surg. 1990;125:498–508. [DOI] [PubMed] [Google Scholar]
- 10.Rutherford EJ, Morris JA Jr, Reed GW, et al. Base deficit stratifies mortality and determines therapy. J Trauma. 1992;33:417–423. [DOI] [PubMed] [Google Scholar]
- 11.Davis JW, Parks SN, Kaups KL, et al. Admission base deficit predicts transfusion requirements and risk of complications. J Trauma. 1996;41:769–774. [DOI] [PubMed] [Google Scholar]
- 12.Eberhard LW, Morabito DJ, Matthay MA, et al. Initial severity of metabolic acidosis predicts the development of acute lung injury in severely traumatized patients. Crit Care Med. 2000;28:125–131. [DOI] [PubMed] [Google Scholar]
- 13.Durham RM, Moran JJ, Mazuski JE, et al. Multiple organ failure in trauma patients. J Trauma. 2003;55:608–616. [DOI] [PubMed] [Google Scholar]
- 14.Jakubowski HV, Owen WG. Macromolecular specificity determinants on thrombin for fibrinogen and thrombomodulin. J Biol Chem. 1989;264:11117–11121. [PubMed] [Google Scholar]
- 15.Pugh RN, Murray-Lyon IM, Dawson JL, et al. Transection of the oesophagus for bleeding oesophageal varices. Br J Surg. 1973;60:646–649. [DOI] [PubMed] [Google Scholar]
- 16.Baker SP, O'Neill B, Haddon W, et al. The injury severity score: a method for describing patients with multiple injuries and evaluating emergency care. J Trauma. 1974;14:187–196. [PubMed] [Google Scholar]
- 17.Davis JW, Shackford SR, Holbrook TL. Base deficit as a sensitive indicator of compensated shock and tissue oxygen utilization. Surg Gynecol Obstet. 1991;173:473–476. [PubMed] [Google Scholar]
- 18.Davis JW. The relationship of base deficit to lactate in porcine hemorrhagic shock and resuscitation. J Trauma. 1994;36:168–172. [DOI] [PubMed] [Google Scholar]
- 19.Bernard GR, Artigas A, Brigham KL. The American-European Consensus Conference on ARDS. Definitions, mechanisms, relevant outcomes, and clinical trial coordination. Am J Respir Crit Care Med. 1994;149:818–824. [DOI] [PubMed] [Google Scholar]
- 20.Bellomo R, Ronco C, Kellum JA, et al. Acute renal failure—definition, outcome measures, animal models, fluid therapy and information technology needs: the Second International Consensus Conference of the Acute Dialysis Quality Initiative (ADQI) Group. Crit Care. 2004;8:R204–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.von Ahsen N, Lewczuk P, Schutz E, et al. Prothrombin activity and concentration in healthy subjects with and without the prothrombin G20210A mutation. Thromb Res. 2000;99:549–556. [DOI] [PubMed] [Google Scholar]
- 22.Rezaie AR. Vitronectin functions as a cofactor for rapid inhibition of activated protein C by plasminogen activator inhibitor-1. Implications for the mechanism of profibrinolytic action of activated protein C. J Biol Chem. 2001;276:15567–15570. [DOI] [PubMed] [Google Scholar]
- 23.Oertel M, Kelly DF, McArthur D. Progressive hemorrhage after head trauma: predictors and consequences of the evolving injury. J Neurosurg. 2002;96:109–116. [DOI] [PubMed] [Google Scholar]
- 24.Ratliff JL. The risk of anticoagulation with experimental pulmonary contusion. Arch Surg. 1974;109:802–804. [DOI] [PubMed] [Google Scholar]
- 25.Tyburski JG, Collinge JD, Wilson RF, et al. Pulmonary contusions: quantifying the lesions on chest X-ray films and the factors affecting prognosis. J Trauma. 1999;46:833–838. [DOI] [PubMed] [Google Scholar]
- 26.Engelman DT, Gabram SG, Allen L, et al. Hypercoagulability following multiple trauma. World J Surg. 1996;20:5–10. [DOI] [PubMed] [Google Scholar]
- 27.Boldt J, Papsdorf M, Rothe A, et al. Changes of the hemostatic network in critically ill patients—is there a difference between sepsis, trauma, and neurosurgery patients? Crit Care Med. 2000;28:445–450. [DOI] [PubMed] [Google Scholar]
- 28.Gando S. Serial studies of protein C in trauma patients. Jpn J Thromb Hemost. 1996;7:312–318. [Google Scholar]
- 29.Geerts WH, Code KI, Jay RM, et al. A prospective study of venous thromboembolism after major trauma. N Engl J Med. 1994;331:1601–1606. [DOI] [PubMed] [Google Scholar]
- 30.Knudson MM, Collins JA, Goodman SB, et al. Thromboembolism following multiple trauma. J Trauma. 1992;32:2–11. [DOI] [PubMed] [Google Scholar]
- 31.Association for the Advancement of Automotive Medicine. The Abbreviated Injury Scale, 1990 Revision. Des Plaines, IL: Association for the Advancement of Automotive Medicine; 1990. [Google Scholar]
- 32.Weiskopf RB, Fairley HB. Anesthesia for major trauma. Surg Clin North Am. 1982;62:31–45. [DOI] [PubMed] [Google Scholar]
- 33.Faust SN, Levin M, Harrison OB, et al. Dysfunction of endothelial protein C activation in severe meningococcal sepsis. N Engl J Med. 2001;345:408–416. [DOI] [PubMed] [Google Scholar]
- 34.Xu J, Esmon NL, Esmon CT. Reconstitution of the human endothelial cell protein C receptor with thrombomodulin in phosphatidylcholine vesicles enhances protein C activation. J Biol Chem. 1999;274:6704–6710. [DOI] [PubMed] [Google Scholar]
- 35.Conway EM, Van de Wouwer M, Pollefeyt S, et al. The lectin-like domain of thrombomodulin confers protection from neutrophil-mediated tissue damage by suppressing adhesion molecule expression via nuclear factor κB and mitogen-activated protein kinase pathways. J Exp Med. 2002;196:561–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gando S, Nanzaki S, Morimoto Y, et al. Tissue factor pathway inhibitor response does not correlate with tissue factor-induced disseminated intravascular coagulation and multiple organ dysfunction syndrome in trauma patients. Crit Care Med. 2001;29:262–266. [DOI] [PubMed] [Google Scholar]
- 37.Esmon CT. Protein C pathway in sepsis. Ann Med. 2002;34:598–605. [DOI] [PubMed] [Google Scholar]
- 38.Adrie C, Monchi M, Laurent I, et al. Coagulopathy after successful cardiopulmonary resuscitation following cardiac arrest. Implication of the protein C anticoagulant pathway. J Am Coll Cardiol. 2005;46:21–28. [DOI] [PubMed] [Google Scholar]