

Abstract
E-cadherin function leads to the density-dependent contact inhibition of cell growth. Because cadherins control the overall state of cell contact, cytoskeletal organization, and the establishment of many other kinds of cell interactions, it remains unknown whether E-cadherin directly transduces growth inhibitory signals. To address this question, we have selectively formed E-cadherin homophilic bonds at the cell surface of isolated epithelial cells by using functionally active recombinant E-cadherin protein attached to microspheres. We find that E-cadherin ligation alone reduces the frequency of cells entering the S phase, demonstrating that E-cadherin ligation directly transduces growth inhibitory signals. E-cadherin binding to β-catenin is required for cell growth inhibition, but β-catenin/T-cell factor transcriptional activity is not involved in growth inhibition resulting from homophilic binding. Neither E-cadherin binding to p120-catenin nor β-catenin binding to α-catenin, and thereby the actin cytoskeleton, is required for growth inhibition. E-cadherin ligation also inhibits epidermal growth factor (EGF) receptor-mediated growth signaling by a β-catenin–dependent mechanism. It does not affect EGF receptor autophosphorylation or activation of ERK, but it inhibits transphosphorylation of Tyr845 and activation of signal transducers and activators of transcription 5. Thus, E-cadherin homophilic binding independent of other cell contacts directly transduces growth inhibition by a β-catenin–dependent mechanism that inhibits selective signaling functions of growth factor receptors.
INTRODUCTION
Cell–cell adhesion mediated by cadherins is fundamental for the differentiation and integrity of most adult tissues (Gumbiner, 1996, 2005). E-cadherin is a major constituent of polarized epithelial cell junctions, and it mediates cell adhesion through Ca2+-dependent homophilic interaction of its extracellular domain and interaction of its cytoplasmic domain with catenins. E-cadherin is also a tumor suppressor protein, because its loss of expression or function has been shown to be associated with tumorigenesis and tumor progression (Takeichi, 1993). Restoration of E-cadherin expression in cancer cells results in decreased invasiveness, growth suppression, and terminal differentiation (Behrens et al., 1989; Vleminckx et al., 1991; Perl et al., 1998; Gottardi et al., 2001; Wong and Gumbiner, 2003).
Cadherins have also been postulated to be responsible for the phenomenon of contact inhibition of cell growth. Contact inhibition is a widely acknowledged property of cells in tissue (Fagotto and Gumbiner, 1996), but the mechanisms that are responsible are not well understood. Although, there is evidence that cadherin expression can influence cell growth rates (Watabe et al., 1994; Caveda et al., 1996; St Croix et al., 1998; Mueller et al., 2000; Motti et al., 2005), their exact roles in contact inhibition are not well understood. A density-dependent inhibition of cell proliferation could result from many other factors that are indirectly influenced by the establishment of many kinds of cell interactions (Perez-Moreno et al., 2003) that can affect cell growth. For example, tight junctions may limit access of growth factors or nutrients to their receptors at the cell surface. Also, several signaling proteins are associated with tight junctions, and they could potentially respond to the state of cell junctions (Gibson and Perrimon, 2003; Funke et al., 2005; Matter et al., 2005). Furthermore, the facilitation of other types of molecular cell interactions, including gap junctions, juxtacrine ligand–receptor interactions (such as transforming growth factor-α and epidermal growth factor receptor [EGFR] or Notch and Delta, and receptor tyrosine phosphatases), all depend on intimate cell contacts (Bosenberg and Massague, 1993; Fagotto and Gumbiner, 1996). Even some growth factors (including Wnts, fibroblast growth factor [FGF], and bone morphogenetic proteins) are known to act over extremely short distances or only when cells form contacts, presumably because they diffuse poorly through the extracellular matrix.
Nevertheless, the observed associations of cadherins and adherens junctions with signaling proteins and growth factor receptors raises the possibility that cadherins directly generate growth related signals. For example, by manipulating overall cell adhesion and junction assembly, several studies have shown recruitment of phosphatidylinositol 3-kinase (PI3K) to adhesive contacts through a tyrosine kinase activity (Pece et al., 1999; Shinohara et al., 2001) and to signal to the Rho family of GTPase (Braga et al., 1999). Some studies used a functional cadherin ligand to specifically engage cadherins to show that E-cadherin homophilic ligation signals directly through PI3K, Rac, and Src activity (Noren et al., 2001; Kovacs et al., 2002; Lambert et al., 2002; Pang et al., 2005). Moreover, cadherins interact with growth factor receptors at the cell surface, and cell adhesion can modulate growth factor signaling activities (Takahashi and Suzuki, 1996; Pece and Gutkind, 2000; Qian et al., 2004).
In addition to its role in adhesion, β-catenin is involved in Wnt signal transduction, and it interacts with transcription factors of the leukocyte enhancer factor (LEF)/T-cell factor (TCF) family to regulate transcription of target genes implicated in cell growth control, such as cyclin D1 and c-myc (van Noort and Clevers, 2002). By sequestering β-catenin at the cell surface, cadherins have been shown to antagonize β-catenin signaling pathways and to induce growth inhibition (Heasman et al., 1994; Fagotto et al., 1996; Orsulic et al., 1999; Shtutman et al., 1999; Gottardi et al., 2001).
Although cadherins have been implicated in growth inhibition and signaling, their roles in contact inhibition relative to other cell–cell interactions and other surface receptors are not well understood. The main purpose of this article was to determine whether engagement of E-cadherin in a homophilic adhesive bond independent of all other cell interactions is capable of transducing a growth inhibitory signal. Because the molecular interactions present at a normal epithelial cell–cell contact are so diverse and complex, we have selectively engaged the E-cadherin interaction at the cell surface of isolated epithelial cells to determine its role in growth regulation.
MATERIALS AND METHODS
Cell Culture and Antibodies
The human colon carcinoma cell lines HT29 and SW480 and the human mammary adenocarcinoma cell line MCF-7 were obtained from American Type Culture Collection (Manassas, VA). HT29 and MCF-7 cells were maintained in 1:1 ratio of DMEM and Ham's F-12 nutrient medium (Invitrogen, Carlsbad, CA), and SW480 cells were maintained in DMEM. Each medium contains 10% fetal bovine serum (FBS), 1% l-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin. The human mammary epithelial cells (HMEC; Cambrex Bio Science Walkersville, Walkersville, MD) were maintained in serum-free mammary epithelial cell growth medium (MEGM; Cambrex Bio Science Walkersville) supplemented with growth factors (MEGM singlequots; Cambrex Bio Science Walkersville). The human epidermal keratinocytes (HEK; Cell Applications, San Diego, CA) were maintained in HEK growth medium (Cell Applications). The stably transfected Chinese hamster ovary (CHO) cells expressing secreted and wild-type form of human E-cadherin and the stably transfected SW480 and MDA-MB-231 cells expressing wild-type and mutant forms of E-cadherin were cultured as described previously (Gottardi et al., 2001; Niessen and Gumbiner, 2002, Wong and Gumbiner, 2003). The human epidermoid carcinoma cell line A431 was maintained in DMEM high-glucose medium. In epidermal growth factor (EGF) studies, cells were grown in 0.5% FBS-containing medium before treated with 10 ng/ml EGF (Sigma-Aldrich, St. Louis, MO).
The following antibodies were used to perform the experiments: anti-β2 microglobulin (HLA; Santa Cruz Biotechnology, Santa Cruz, CA), anti-Na+K+-ATPase β1 subunit (Upstate Biotechnology, Lake Placid, NY), anti-5-bromodeoxyuridine (BrdU) (Upstate Biotechnology), anti-human E-cadherin (BD Biosciences Transduction Laboratories, Lexington, KY), monoclonal anti-human β-catenin (BD Transduction Laboratories), polyclonal anti-β-catenin (McCrea et al., 1993), anti-actin (Sigma-Aldrich, St. Louis, MO), anti-phospo-EGFR (Tyr1173) (Santa Cruz Biotechnology), anti-phospho-EGFR (Tyr845) (BioSource International, Camarillo, CA), anti-phospho-extracellular signal-regulated kinase (ERK) (Cell Signaling Technology, Danvers, MA), and anti-phospho-signal transducers and activators of transcription 5 (p-STAT5) (Tyr 694) (Cell Signaling Technology).
Preparation of Protein-coated Microspheres and Protein-coated Glass Coverslips
Protein A-coated polystyrene microspheres (1 μM carboxylate modified; either dyed fluorescent red or not) were obtained from Bangs Laboratories (Fishers, IN). They were washed twice in 1 mM sodium acetate, pH 3.9, and twice in 20 mM HEPES, 50 mM NaCl, and 1 mM CaCl2, pH 7.2. Then, either a rabbit anti-mouse antibody (The Jackson Laboratory, Bar Harbor, ME) plus the anti-HLA or Na+K+-ATPase monoclonal antibodies, or the Fc-hE recombinant protein (Chappuis-Flament et al., 2001), was bound to the microspheres at a ratio of 100 μg of protein per 100 μl of microspheres suspension in 20 mM HEPES, 50 mM Nacl, 1 mM CaCl2, pH 7.2, shaking for 1 h at 4°C. The coated beads were washed twice in 20 mM HEPES, 50 mM NaCl, and 1 mM CaCl2, pH 7.2, and incubated with 1% bovine serum albumin (BSA) for 1 h at 4°C to block nonspecific binding. They were then washed and resuspended in 1 ml of sterile phosphate-buffered saline (PBS) supplemented with 1 mM CaCl2 and 0.5 mM MgCl2 (PBS++), and 10 μl per milliliter of medium was added to coat the surface of the cells. For each experiment, protein-coated microspheres were freshly prepared.
The experiments were also done using protein-coated glass coverslips, in which coverslips were coated overnight at 4°C with maximum amount of protein: 10 μg of fibronectin along with either 10 μg of Fc-hE or 10 μg of anti-HLA antibody. Coverslips were washed in PBS++ and coated with 1% BSA in PBS++ for 1 h at 4°C before plating cells.
Cell Proliferation Assay
Cells growing under standard conditions were harvested in a way to preserve E-cadherin molecules at the cell surface (Chappuis-Flament et al., 2001). Cell monolayers were washed twice in PBS++ and incubated with 0.01% trypsin in PBS++. Cells were then washed and resuspended in complete media culture for plating.
Five hundred cells/well were plated on 24-well plates containing glass coverslips coated with 10 μg of fibronectin and incubated for 2 h to allow cell attachment. Then, 10 μl of Fc-hE or anti-HLA or anti-Na+K+-ATPase–coated beads suspension was added per well, and the 24-well plates were incubated with gentle agitation for 24 and 48 h at 37°C and 5% CO2 atmosphere. In other cases, cells were plated directly on fibronectin/Fc-hE or fibronectin/anti-HLA or anti-Na+K+-ATPase 1 subunit antibodies-coated coverslips. To evaluate cell proliferation, at 16 h before fixing cells, 50 μM BrdU, a marker of DNA replication, was added per well. Cells were fixed in 4% paraformaldehyde 24 and 48 h after beads were added to cells. Coverslips were washed, and BrdU incorporation was detected by immunofluorescence, by using an anti-BrdU monoclonal antibody (Upstate Biotechnology), whereas nuclei were detected by staining with 4,6-diamidino-2-phenylindole (DAPI). The BrdU-labeled cells were counted from population of completely isolated cells present on the coverslips, and the percentage of BrdU incorporation in this population was calculated.
Each experiment was performed in duplicate and repeated at least three times. Three independent clones of each of the E-cadherin constructs expressing SW480 cells were used, but only results for one clone are shown. Data are expressed as mean ± SEM. Statistical significance was determined by unpaired Student's t test; p < 0.05 was considered significant.
Small Interfering RNA (siRNA) Transfection
siRNA duplex oligonucleotides (Dharmacon RNA Technologies, Lafayette, CO) (13.5 μg/100-mm plate) targeting β-catenin mRNAs (β-cat#1, 5′-AAGUCCUGUAUGAGUGGGAAC-3′; β-cat#2, 5′-AAAGCUGAUAUUGAUGGACAG-3′; or β-cat#3, 5′-AACAGUUGUGGUUAAGCUCUU-3′) (Deng et al., 2002, Verma et al., 2003) or green fluorescent protein (GFP) mRNA (5′-GGCTACGTCCAGGAGCGCACC-3′) as a negative control was transfected using Oligofectamine (Invitrogen). After 24 h, transfected cells were harvested, and cell proliferation assay was performed as described previously.
Cell Growth Assays
Cells (1 × 105) previously transfected with siRNA-targeting β-catenin (β-cat#1) or GFP mRNAs were plated on six-well plates in regular media. At regular intervals, cells were harvested and counted using a Malasez cell. Data are expressed as mean ± SEM.
Transient Transfection
Cells (15 × 103 cells/well) were seeded in glass coverslips in 24-well plates. They were transfected using Effectene (QIAGEN, Valencia, CA) with 0.5 μg of the β-catenin–engrailed repressor fusion construct, 0.5 μg of the dominant-negative XTCF-3, and 0.5 μg of the activated VP16–TCF fusion construct. They were cotransfected with 0.5 μg of pEGFP vector expressing green fluorescent protein (Clontech, Palo Alto, CA) to detect transfected cells. After 24 h, transfected cells were harvested, and cell proliferation was performed as described previously. BrdU-labeled cells were quantified from the total number of transfected GFP-positive isolated cells.
To determine TCF activity, cells were transiently transfected with 0.5 μg of the TOPFLASH or FOPFLASH reporter plasmids by using Effectene (QIAGEN). Transfection efficiencies were determined by cotransfection of the pRL-TK reporter construct that contained the Renilla luciferase cDNA. Activities of firefly and Renilla luciferases were measured sequentially from a single sample by using the dual-luciferase reporter assay system (Promega, Madison, WI). Results are expressed as relative luciferase units normalized to Renilla luciferase. Data are expressed as mean ± SEM.
Quantification of Apoptotic Cells
The terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) assay was performed using the terminal transferase and biotin-16-dUTP system as described in the manufacturer's instruction manual (Roche Diagnostics, Indianapolis, IN). Data are expressed as mean ± SEM.
Immunofluorescence
A431 cells were serum starved and treated with beads coated with either anti-HLA or Fc-hE-cadherin overnight. Then, cells were stimulated with 10 ng/ml EGF for the indicated time. For phospho-EGFR staining, cells were fixed with methanol at −20°C for 5 min; for phospho-ERK staining, they were fixed with 4% paraformaldehyde for 15 min; and for phospho-STAT5 staining, fixation was with 4% paraformaldehyde for 15 min followed by methanol at −20°C for 5 min. Coverslips were blocked with 5% milk/PBS and incubated with primary antibodies. Samples were examined on an upright immunofluorescence microscope by using 63× oil immersion lens (Carl Zeiss, Thornwood, NY). To determine the percentage of positive cells for the various antibodies, isolated cells were identified by DAPI staining of nuclei. Positive cells were those that exhibited detectable immunofluorescence staining over background; examples are shown in Figures 8B and 9A. The negative cells were scored as for those with DAPI nuclear staining, but no detectable immunofluorescence staining with the antibody was used.
Figure 8.

E-cadherin ligation selectively inhibits EGF receptor Tyr845 phosphorylation. (A) E-cadherin ligation inhibits EGF-induced increase in growth of A431 cells. A431 cells plated in low serum (0.5%) were incubated with 10 ng/ml EGF, and beads were coated with either anti-HLA or Fc-hE for 24 h. The percentage of cells entering S phase was determined by the cell proliferation assay. (B) EGF treatment induces the immunofluorescence staining of phosphorylated-EGFR (Tyr1173) and phosphorylated-EGFR (Tyr845) in A431 cells. DAPI staining of nuclei is shown at bottom. (C) E-cadherin ligation has no effect on EGFR autophosphorylation at Tyr1173. A431 cells were serum starved and incubated with either anti-HLA or Fc-hE–coated beads overnight, and then they were stimulated with 10 ng/ml EGF for the indicated times and stained with antibody to phosphorylated-EGFR (Tyr1173). The number of p-EGFR (Tyr1173)-positive cells was counted and percentage of positive cells was calculated. (D) E-cadherin ligation selectively inhibited EGFR Tyr845 phosphorylation. A431 cells were treated as in C and stained with antibody to phosphorylated-EGFR (Tyr845). The number of p-EGFR (Tyr845)-positive cells was counted, and percentage of positive cells was calculated. Data are expressed as mean ± SEM (*p < 0.05).
Figure 9.

Effect of E-cadherin ligation on ERK and STAT5 activation. (A) EGF treatment induces the immunofluorescence staining of phosphorylated-ERK and phosophorylated-STAT5 in A431 cells. DAPI staining of nuclei is shown at bottom. (B) E-cadherin ligation caused only a very small and transient inhibition of ERK phosphorylation. A431 cells were serum starved and incubated with either anti-HLA– or Fc-hE–coated beads overnight, and then they were stimulated with 10 ng/ml EGF for the indicated times and stained with antibody to phospho-ERK. The number of p-ERK–positive cells was counted, and the percentage of positive cells was calculated. (C) E-cadherin ligation inhibited STAT5 phosphorylation. A431 cells were treated as described in B and stained with antibody to phospho-STAT5. The number of p-STAT5–positive cells was counted and percentage of positive cells was calculated. #, no p-STAT5–positive cells observed without EGF treatment. Data are expressed as mean ± SEM (* p < 0.05).
RESULTS
E-Cadherin Ligation Reduces Entry of Epithelial Cells into S Phase
To measure the direct effect of E-cadherin on cell proliferation, we have used a specially designed experimental system. Epithelial cells expressing E-cadherin were harvested under conditions that minimize removal of the surface E-cadherin (Chappuis-Flament et al., 2001). Because epithelial cells have a tendency to form cell clusters, we have grown them at very low density to avoid establishment of any cell–cell interactions and any contact-dependent signaling. Although almost half of cells still formed clusters, we focused our attention only on isolated cells devoid of all cell–cell contacts to ensure that they did not already receive growth inhibitory signals from endogenous junctions. Cells were grown under conditions in which they received strong growth stimulatory signals, from serum in the medium, and from attachment to fibronectin on the substrates (Figure 1A). Cadherin molecules present at the cell surface were specifically engaged using purified and functionally active chimeric extracellular domain of E-cadherin fused to the immunoglobulin (IgG) Fc domain (Fc-hE), developed previously in our laboratory (Niessen and Gumbiner, 2002; Figure 1, A and B). Fc-hE was presented to cells either attached to microspheres or by cocoating them with fibronectin on the coverslips. Antibodies against two other cell surface proteins, class I MHC (HLA β2 microglobulin) and the Na+K+-ATPase β1 subunit, were used as controls for ligand specificity of bead attachment to cell surface (Figure 1, A and B). After 24 and 48 h, the percentage of isolated proliferating cells was determined by measuring the incorporation of BrdU by using an indirect immunofluorescence microscopy.
Figure 1.

E-cadherin ligation inhibits proliferation of isolated primary cells. (A) Schematic model of the approach used to selectively engage E-cadherin onto homophilic adhesive bonds at the cell surface. Cells were grown under conditions that stimulate cell growth from serum in the medium and plating on fibronectin. E-cadherin ligation was activated at the cell surface either by Fc-hE–coated microspheres or by cocoating Fc-hE with fibronectin on the coverslips. Antibodies directed against HLA or Na+K+-ATPase-1 subunit molecules were used as controls for ligand specificity. (B) Binding of anti-HLA–, anti-Na+K+-ATPase-1–, and Fc-hE–coated microspheres to the surface of HEMC. Microspheres were added in sufficient quantities to cover the surface of cells plated under sparse condition to avoid other types of cell–cell interactions. Blue, DAPI-stained nuclei and red, coated beads. (C and D) Activation of E-cadherin ligation by using Fc-hE–coated microspheres triggers a strong and specific growth inhibitory signal by reducing the number of isolated HMEC (C) and HEK (D) cells entering into S phase as determined by BrdU incorporation compared with ligation control. (E and F) Activation of E-cadherin ligation by using Fc-hE–coated coverslips specifically reduces the number of isolated HMEC (E) and HEK (F) entering into S phase as determined by BrdU incorporation compared with ligation control. Data are expressed as mean ± SEM (*p < 0.05).
In initial experiments, primary cells were used because they are not transformed, and we could expect that they retain contact inhibition properties. Early passages of two human primary cells from different origins, HMEC and HEK, were used in our assays. When E-cadherin homophilic ligation was activated at the cell surface with Fc-hE–coated microspheres, levels of BrdU incorporation in HMEC and HEK cells were specifically and substantially reduced compared with anti-HLA and anti-Na+K+-ATPase ligation controls (Figure 1, C and D). To rule out that the observed growth inhibitory effect mediated by Fc-hE–coated microspheres was due to engulfment and phagocytic signaling rather than specific homophilic engagement of E-cadherin at the cell surface, we presented E-cadherin to cells by plating them onto Fc-hE–coated coverslips. Fc-hE was still able to inhibit cell growth compared with ligation controls in HMEC cells (Figure 1E) and HEK cells (Figure 1F). We also asked whether these results were secondary to an increase in the rate of apoptosis by using the TUNEL assay. As shown in Table 1, the apoptosis rate was very low in HMEC cells, and the engagement of E-cadherin onto the cell surface (Fc-hE) did not alter the number of apoptotic cells, compared with ligation controls (anti-HLA). Thus, homophilic ligation of E-cadherin at the cell surface can directly transduce a growth inhibitory signal to the cells, which does not involve changes in apoptosis.
Table 1.
Apoptosis rate of different cell lines after E-cadherin ligation onto homophilic adhesive bonds by TUNEL assay
| Cell type | Time (h) | Anti-HLA (% TUNEL-positive cells) | Fc-hE (% TUNEL-positive cells) |
|---|---|---|---|
| HUMEC | 24 | 1.60 ± 1.50 | 1.08 ± 0.87 |
| 48 | 1.50 ± 0.10 | 1.35 ± 0.15 | |
| HT29 | 24 | 1.67 ± 0.03 | 1.22 ± 0.87 |
| 48 | 1.92 ± 0.05 | 1.7 ± 0.02 | |
| MCF-7 | 24 | 2.40 ± 0.64 | 1.13 ± 0.36 |
| 48 | 2.20 ± 0.05 | 2.19 ± 0.09 |
Because established cell lines offer many experimental advantages, we have also determined whether E-cadherin homophilic binding was capable of reducing the proliferation of epithelial cell lines. We selected the human breast adenocarcinoma MCF-7 and the human colon carcinoma HT29 cell lines because they are known to be well differentiated in culture, and they express endogenous E-cadherin. The engagement of E-cadherin into homophilic adhesive contacts at the cell surface by using Fc-hE–coated microspheres reduced the percentage of HT29 (Figure 2A) and MCF-7 cells (Figure 2B) in S phase compared with anti-HLA and anti-Na+K+-ATPase ligation controls. Moreover, there was no significant effect on apoptosis rate, which was very low in the different cell lines used in this study (Table 1). In contrast, CHO cells expressing hE-cadherin did not exhibit reduced growth rates upon engagement of E-cadherin at the cell surface (Figure 2C). This could be explained by the fact that this is a highly transformed cell line that has lost contact inhibition, and expression of E-cadherin alone is not enough to inhibit cell growth.
Figure 2.
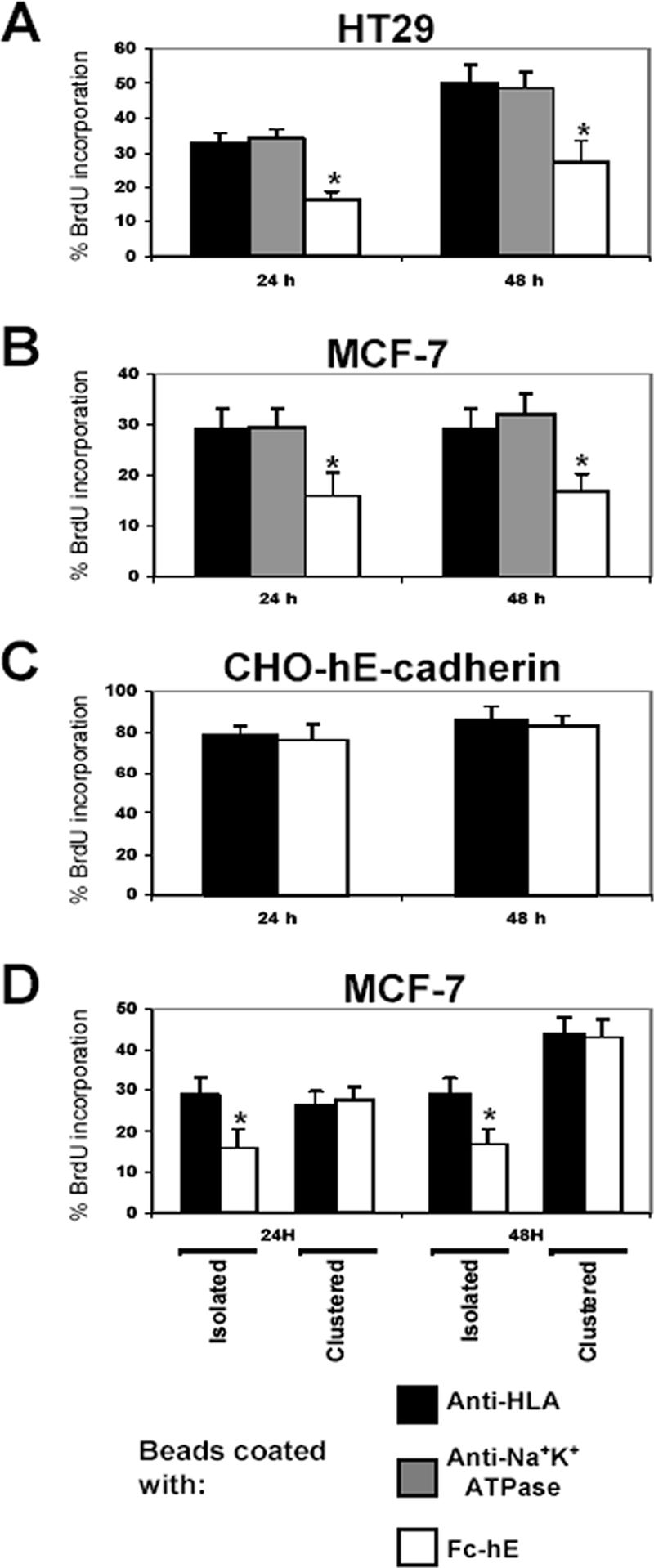
E-cadherin ligation inhibits the proliferation of isolated cells of certain cell lines. (A and B) E-cadherin ligation inhibits cell proliferation in isolated HT29 colorectal cancer cells (A) and MCF-7 breast cancer cells (B), respectively, as determined by percentage of BrdU incorporation compared with anti-HLA ligation control. (C) E-cadherin engagement at cell surface had no effect on cell growth in E-cadherin–expressing CHO cells compared with anti-HLA ligation control. (D) E-cadherin ligation inhibits the proliferation of isolated MCF-7 cells, whereas no effect is observed on clustered cells. Data are expressed as mean ± SEM (*p < 0.05).
To test the specificity of our cellular model, we then determined the percentage of BrdU-positive cells from two different cell populations: isolated cells versus clustered cells that already have extensive E-cadherin–mediated contacts. Binding of Fc-hE–coated microspheres promoted a decrease in the proliferation of MCF-7 (Figure 2D) and HT29 (data not shown) isolated cells, compared with HLA ligation control. In clustered cells, beads coated with Fc-hE had no effect on cell growth compared with beads coated with HLA. Presumably, in clustered cells, E-cadherin–E-cadherin interactions (cell–cell contacts) have occurred, and signaling pathways(s) involved in cell growth inhibition have probably already been stimulated.
The E-Cadherin Growth Inhibitory Effect Is Dependent on β-Catenin
The cytoplasmic domain of E-cadherin binds directly to p120ctn and β-catenin, each of which could play a role in cell growth suppression. To analyze the potential roles of these proteins in the inhibition of cell growth mediated by E-cadherin, wild-type E-cadherin and various E-cadherin mutant and chimera constructs were expressed in the SW480 colon cancer cell line and the MDA-MB-231 breast cancer cell line (Gottardi et al., 2001; Figure 3A). The re-expression of E-cadherin in SW480 (SW480/E-cadherin full length [FL]) cells causes the binding of Fc-hE–coated beads at the cell surface (Figure 3B) and the cell growth inhibition mediated by E-cadherin beads compared with SW480 parental cells (compare Figure 3D with C). The E-cadherin/α-catenin fusion chimera and E-cadherin Δ β-catenin mutant lack the ability to bind β-catenin, but they can still mediate physical cell adhesion (Gottardi et al., 2001). Fc-hE–coated beads bound to the cell surface of SW480 cells expressing E-cadherin Δ β-catenin mutant (data not shown) but had no significant effect on their proliferating rate, when compared with the anti-HLA ligation control (Figure 3E). In SW480 E-cadherin/α-catenin fusion-expressing cells, Fc-hE beads bound to the cell surface (data not shown), but they had no significant effect on cell proliferation (Figure 3F). The E-cadherin Δ p120ctn construct, which is not capable of binding to p120ctn, mediated cell growth inhibition induced by E-cadherin beads (Figure 3G), indicating that direct binding of p120ctn to E-cadherin is not necessary for the growth inhibitory effect. Similar results were observed using the MDA-MB-231 cell line, which expressed the different E-cadherin constructs by using a tetracycline-inducible system (Supplemental Figure 1S). The differences in cell growth inhibition mediated by E-cadherin ligation do not likely result from differences in the levels of expression of E-cadherin constructs (Figure 3H). Only the E-cadherin–α-catenin fusion protein (which is bigger ∼220 kDa) is expressed at somewhat lower levels. However, a previous study showed that this cell line expressing the E-cadherin–α-catenin fusion exhibited adhesive activity as good as the wild-type cadherin, even though it is less well expressed (Gottardi et al., 2001). Moreover, the E-cadherin Δ β-catenin–expressing cell line exhibited even stronger adhesive activity than the wild-type cadherin–expressing cells. Furthermore, each construct was localized to the region of cell contacts in confluent cells (Figure 3I). These data strongly suggest that the β-catenin binding domain of E-cadherin is involved in cell growth inhibition mediated by E-cadherin ligation.
Figure 3.

β-Catenin binding domain of E-cadherin is necessary for inhibition of SW480 cell proliferation by E-cadherin ligation. (A) Schematic diagram of E-cadherin constructs used in this study (Gottardi et al., 2001). (B) Microspheres coated with antibody directed against HLA bind SW480 parental and SW480/E-cadherin FL cells, whereas microspheres coated with Fc-hE bind only SW480/E-cadherin FL cells. (C–G) Effects of Fc-hE–coated microspheres binding to cell surfaces on cell proliferation of isolated SW480 parental and SW480 cells expressing wild-type and mutant forms of E-cadherin as determined by BrdU incorporation compared with anti-HLA ligation control. (C) SW480 parental cells. (D) SW480/E-cadherin FL cells. (E) SW480/E-cadherin Δ β-catenin cells. (F) SW480/E-cadherin/α-catenin cells. (G) SW480/E-cadherin Δ p120ctn cells. (H) Western blot of E-cadherin in lysates from SW480 cells expressing wild-type E-cadherin and various E-cadherin mutant and chimera constructs. (I) Indirect immunofluorescence staining of E-cadherin in SW480 cells expressing wild-type E-cadherin and various E-cadherin mutant and chimera constructs. Data are expressed as mean ± SEM (*p < 0.05).
The observation that the β-catenin binding domain is required for E-cadherin induced inhibition of cell growth suggests that β-catenin may be involved in the growth inhibition signaling pathway. To test this hypothesis, we depleted β-catenin levels with siRNA, by using three distinct specific siRNAs shown previously to deplete β-catenin (Deng et al., 2002; Verma et al., 2003). Treatment of cells with β-catenin siRNAs (β-cat#1, β-cat#2, and β-cat#3) resulted in a decrease in β-catenin levels in MCF-7 and SW480/E-cadherin FL cells (Figure 4A). Treatment with siRNA targeting the GFP sequence, as negative control, had no effect. β-Catenin depletion lasted 4–5 d for both cell lines (data not shown). Immunofluorescence studies showed β-catenin staining only at cell contacts in MCF-7 cells transfected with siRNA-targeting GFP (control), and no or very weak staining with siRNAs targeting β-catenin (Figure 4B). In SW480/E-cadherin FL cells, β-catenin is highly expressed in cytosol and nucleus (Figure 4B). After transfection with siRNAs against β-catenin, β-catenin expression is significantly reduced overall (Figure 4, A and B). By western blotting (Figure 4A) and immunofluorescence (Figure 4B), we showed that E-cadherin expression and localization are not affected by β-catenin depletion.
Figure 4.

β-Catenin depletion eliminates E-cadherin–mediated growth inhibition and increases cell proliferation. (A) Western blot of β-catenin and E-cadherin in lysates from cells transfected with or without siRNA against GFP or β-catenin (three different targets inside β-catenin coding sequence: β-cat#1, β-cat#2, and β-cat#3; see Materials and Methods). (B) Indirect immunofluorescence staining of β-catenin and E-cadherin in MCF-7 and SW480/E-cadherin FL cells transfected with siRNAs against GFP or β-catenin. (C and D) Binding of Fc-hE–coated microspheres promote a specific decrease in the proliferation of cells transfected with siRNA targeting GFP protein in MCF-7 (C) and SW480/E-cadherin FL (D) cells, respectively. These effects were lost when cells were transfected with siRNA targeting β-catenin. (E and F) MCF-7 and SW480/E-cadherin FL cells transfected with siRNA targeting GFP (circles) or β-catenin (squares) were plated at 105 cells/well, and then they were harvested at the indicated times, and the number of viable cells was counted. Data are expressed as mean ± SEM (*p < 0.05).
Depletion of β-catenin by any of the three siRNAs resulted in the loss of cell growth inhibition mediated by E-cadherin ligation in MCF-7 cells (Figure 4C) and SW480/E-cadherin FL cells (Figure 4D). siRNA against GFP had no effect on cell growth inhibition. To test the specificity of the siRNAs, we then performed rescue experiments by using three different siRNA-resistant β-catenin expression vectors that encode β-catenin containing silent mutations in the regions targeted by the siRNAs (Supplemental Figure 2SA). β-Catenin encoded by these cDNAs was expressed in the presence of siRNAs targeting β-catenin (Supplemental Figure 2S, B–E). Moreover these constructs rescued the cell growth inhibition mediated by E-cadherin in an siRNA-specific manner in MCF-7 and SW480/E-cadherin FL cells (Supplemental Figure 2S, F and G). Therefore, β-catenin is required for cell growth inhibition signaling mediated by E-cadherin ligation at the cell surface. To confirm the role of β-catenin under standard cell culture conditions, we examined the effects of β-catenin depletion on the growth rate in normal cultures. After β-catenin depletion, we observed an increase of cell growth rate in MCF-7 (Figure 4E) and SW480/E-cadherin FL cells (Figure 4F). These data suggest that β-catenin plays a role in the inhibition of cell proliferation, perhaps as a mediator of the E-cadherin growth inhibitory effects.
We then examined whether certain binding domain(s) of β-catenin are required for cell growth inhibition mediated by E-cadherin. Three different vectors expressing β-catenin mutants were tested for their ability to rescue siRNA-depleted β-catenin (all either lacking of siRNA target sequence): 1) ΔN89 lacks N-terminal regulation domain involved in β-catenin degradation, 2) ΔN132 lacks the α-catenin binding domain, and 3) ΔC695 lacks the transcription transactivation domain and the regulatory domain that can inhibit binding to cadherin (Figure 5A). These deleted forms of β-catenin are all known to interact with cadherin (Funayama et al., 1995; Fagotto et al., 1996). In MCF-7 (Figure 5B) and also in SW480/E-cadherin FL cells (data not shown), transient cotransfection of these three constructs in the presence of siRNA β-cat#1 rescued cell growth inhibition mediated by E-cadherin ligation, compared with control cells transfected with pcDNA3 empty vector. We also tested a β-catenin construct deleted for the last C-terminal 314 amino acids (ΔC468), but this construct did not rescue cell growth inhibition mediated by E-cadherin ligation (data not shown). However, the ΔC468 construct did not localize to cell contacts (data not shown). This is not surprising because all 12 armadillo repeats have been shown to be involved in the interaction of β-catenin with E-cadherin (Huber and Weis, 2001); therefore, no other construct with deletion inside armadillo region have been tested in this experiment. These data suggest that β-catenin–α-catenin interactions, N-terminal regulatory and C terminal regulatory and transactivation domains of β-catenin are not required for the transduction of cell growth inhibition signals induced by E-cadherin ligation.
Figure 5.
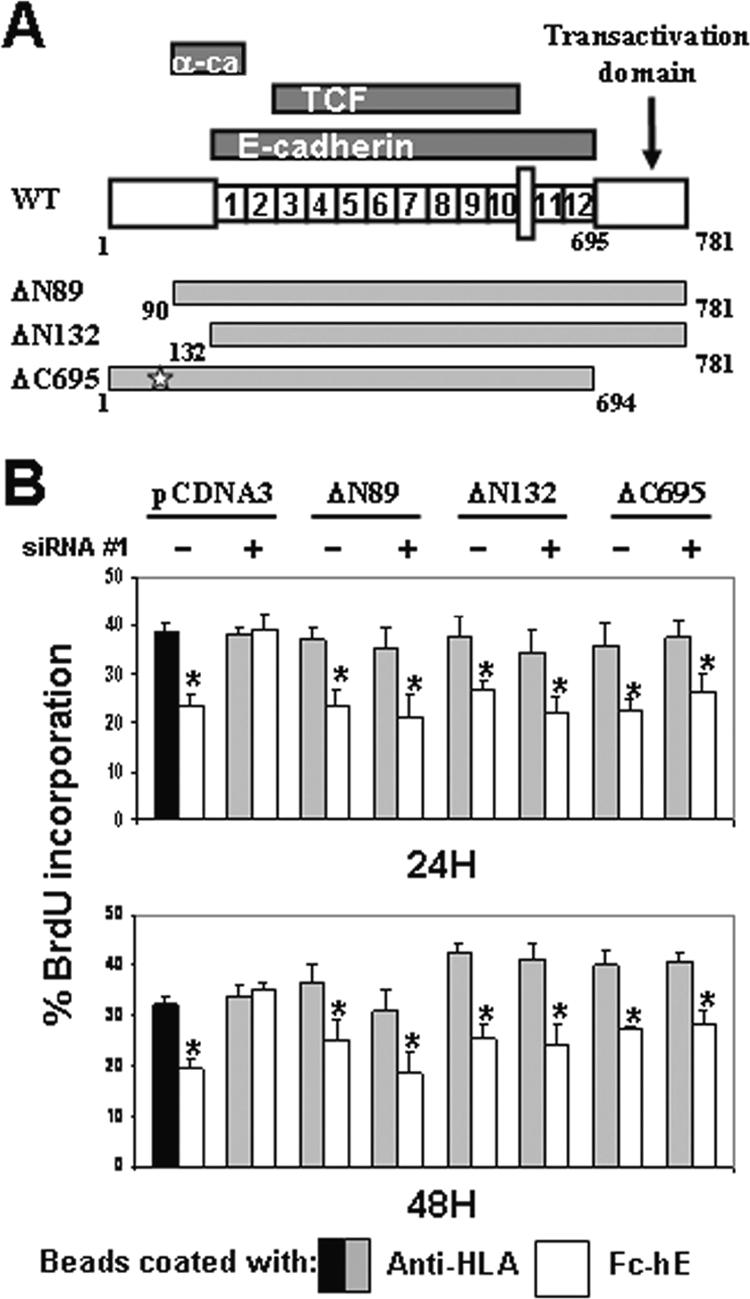
N-terminal and transactivation domains of β-catenin are not involved in cell growth inhibition signaling. (A) Schematic diagram shows where α-catenin, E-cadherin, and TCF interact with β-catenin (Kolligs et al., 1999). In addition to β-catenin FL, the structures of deleted β-catenin constructs are indicated. The star indicates sites of mutation introduced into the β-catenin DNA sequence in the siRNA#1 target region. (B) MCF-7 cells were transiently transfected with siRNA#1 or control siRNA and cotransfected with either empty pcDNA3 or expression vectors encoding deleted β-catenin forms (ΔN89, ΔN132, or ΔC695). Expression of ΔN89, ΔN132, and ΔC695 β-catenin forms is able to rescue cell growth inhibition mediated by E-cadherin in presence of siRNA targeting β-catenin. Data are expressed as mean ± SEM (*p < 0.05).
Specific Engagement of E-Cadherin Inhibits Cell Growth by a TCF-independent Mechanism
As a mediator of the Wnt signaling pathway, β-catenin can regulate cell growth by its interaction with TCF/LEF transcription factors to modulate the gene expression (van Noort and Clevers, 2002). E-cadherin expression can inhibit nuclear β-catenin signaling and cell growth in an adhesion-independent manner (Fagotto et al., 1996; Orsulic et al., 1999; Shtutman et al., 1999; Gottardi et al., 2001), but it is not known whether E-cadherin homophilic ligation alters β-catenin nuclear signaling. Our finding that the C-terminal transcription activation domain of β-catenin is not required for E-cadherin ligation-induced growth inhibition (Figure 5B) indicates that Wnt/β–catenin signaling pathway may not be involved.
To test directly whether the inhibition of cell growth by E-cadherin beads is mediated by β-catenin/TCF signaling, we used two constructs that have been used to inhibit or activate β-catenin/TCF transcriptional activity. β-Catenin fused to the engrailed repressor domain chimera (β-cat/engrailed) potently inhibits β-catenin/TCF–mediated transcription and a form of TCF fused to the potent VP16 transactivation domain (VP16/TCF) is constitutively active (Montross et al., 2000; Vonica et al., 2000). Neither of the constructs had any effect on the growth of HT29 or MCF-7 cells or on the cell growth inhibition induced by E-cadherin ligation at the surface of HT29 and MCF-7 cells (Figure 6A). Therefore, β-catenin/TCF transcriptional activity does not seem to have a role in transducing the E-cadherin growth inhibitory signal in HT29 and MCF-7 cell lines.
Figure 6.

No role for β-catenin/TCF-dependent gene activity in E-cadherin–mediated inhibition of cell growth. (A) HT29, MCF-7, and SW480/E-cadherin FL cells were transiently transfected with GFP alone or with either β-cat/engrailed (dominant-negative) or VP16/TCF (constitutively active TCF) expression vectors. Isolated transfected cells were identified by GFP expression and proliferation assayed by BrdU incorporation. (B) SW480/E-cadherin FL cells were transfected with siRNAs targeting β-catenin, dominant-negative β-catenin–engrailed or dominant-negative TCF (DN TCF). At 24 h posttransfection, the cells were transfected transiently with either TOPFLASH or FOPFLASH reporter to determine TCF activity. (C) E-cadherin was immunoprecipitated (IP) from detergent lysates of subconfluent MCF-7 or SW480/E-cadherin FL cells treated with (lane 2 and 4) and without (lane 1 and 3) siRNA against β-catenin for 24 h. The total immunoprecipitates and 4% of the supernatant (Sup) were analyzed by western blotting for β-catenin and E-cadherin. Data are expressed as mean ± SEM (*p < 0.05).
Alternatively, with SW480/E-cadherin FL cells, inhibition of β-catenin/TCF nuclear signaling with β-cat/engrailed significantly inhibited the number of cells entering the S phase even without E-cadherin ligation (Figure 6A). This is consistent with earlier reports showing that β-catenin/TCF transcription controls SW480 cell proliferation which is inhibited by β-cat/engrailed (Gottardi et al., 2001). However, this result differed from those obtained in the siRNA experiments in which β-catenin depletion blocked E-cadherin ligation induced growth inhibition but not basal cell growth (Figure 4D and 2S). Because β-catenin depletion by siRNA was incomplete, we hypothesize that β-cat/engrailed differs because it causes a more complete inhibition of β-catenin/TCF transcriptional activity in SW480 cells than β-catenin siRNA. To test this, we assayed β-catenin/TCF-dependent transcription directly in SW480 cells using the TOP/FOPFLASH reporter assay. siRNAs targeting specifically β-catenin caused 40–60% inhibition of TCF activity (Figure 6B), whereas β-cat/engrailed and dominant-negative TCF had much stronger effects (>90% inhibition of TCF activity). Thus, siRNA depletion of β-catenin seems to selectively inhibit E-cadherin ligation-driven inhibition of cell growth with relatively less effect on β-catenin/TCF transcriptional regulation of growth.
The difference between the effects of siRNA and dominant-negative β-cat/engrailed constructs on SW480/E-cadherin FL cell growth suggests that they affect different pools of β-catenin. We therefore asked whether siRNA depletion affected the E-cadherin–β-catenin interactions in MCF-7 and in SW480/E-cadherin FL cells by using coimmunoprecipitation assays (Figure 6C). The E-cadherin/β-catenin association was greatly reduced in both cell lines (95% of decreased in MCF-7 cells and 85% in SW480/E-cadherin FL cells) when the cells were treated with siRNA (compare lane 2 with lane 1). In MCF-7 cells, there is almost no cytosolic β-catenin even before siRNA treatment; therefore, β-catenin depletion occurs entirely in the E-cadherin–associated pool. In SW480/E-cadherin FL cells, the β-catenin cytosolic pool (supernatant) was also decreased proportionately, but the total amount of cytosolic β-catenin remained significant; note that only 4% of supernatant was loaded in the gel lane. These results suggest that in this cell line, siRNAs against β-catenin inhibited the growth inhibition mediated by E-cadherin ligation by strongly decreasing the amount of E-cadherin–associated β-catenin. In MCF-7 cells, there is no cytosolic pool of β-catenin to contribute to TCF-driven cell proliferation, whereas in SW480/E-cadherin FL cells, the cytosolic pool of β-catenin may not be sufficiently depleted to significantly reduce β-catenin/TCF–driven cell proliferation.
In conclusion, our data show that cell growth inhibition mediated by E-cadherin ligation is independent of β-catenin/TCF/Lef signaling pathway and by siRNA technique, β-catenin depletion decreases significantly E-cadherin–β-catenin interaction.
Specific Engagement of E-Cadherin Inhibits EGF-mediated Signaling and Cell Growth
The EGFR, a tyrosine kinase, is known to colocalize with E-cadherin to basolateral areas of epithelial cells and to form complex with E-cadherin (Hoschuetsky et al., 2004; Pece and Gutkind, 2000; Qian et al., 2004). Cell–cell contact has been found to either inhibit EGF-dependent activation of EGFR (Takahashi and Suzuki, 1996; Qian et al., 2004) or transiently activate EGFR signaling (Pece and Gutkind, 2000), and these effects have been attributed to the interaction of E-cadherin with EGFR. These studies did not distinguish between E-cadherin homophilic binding and the complex process of cell–cell contact formation. Therefore, we tested the direct role of E-cadherin ligation on EGF signaling, because in our cellular model involving isolated cells, only E-cadherin interactions are engaged at the cell surface.
First, we determined that EGF was able to stimulate cell proliferation in MCF-7 (Figure 7A) and SW480/E-cadherin FL cells (Figure 7B) grown in low serum (0.5%) in the presence of beads coated with anti-HLA. However, in the presence of beads coated with Fc-hE, EGF treatment did not increase BrdU incorporation. Depletion of β-catenin by siRNA was able to restore cell proliferation in response to EGF in presence of Fc-hE beads in both cell lines (Figure 7, A and B), indicating that E-cadherin ligation inhibits EGF-stimulated cell growth through a β-catenin–dependent mechanism. Moreover, in SW480/E-cadherin Δ β-catenin (Figure 7C) and SW480/E-cadherin/α-catenin fusion cells (Figure 7D), in which β-catenin cannot interact with E-cadherin, E-cadherin homophilic interactions had no effect on cell proliferation induced by EGF. These data suggest that the E-cadherin–β-catenin complex inhibits EGF-induced cell growth just as it inhibits serum-dependent growth.
Figure 7.
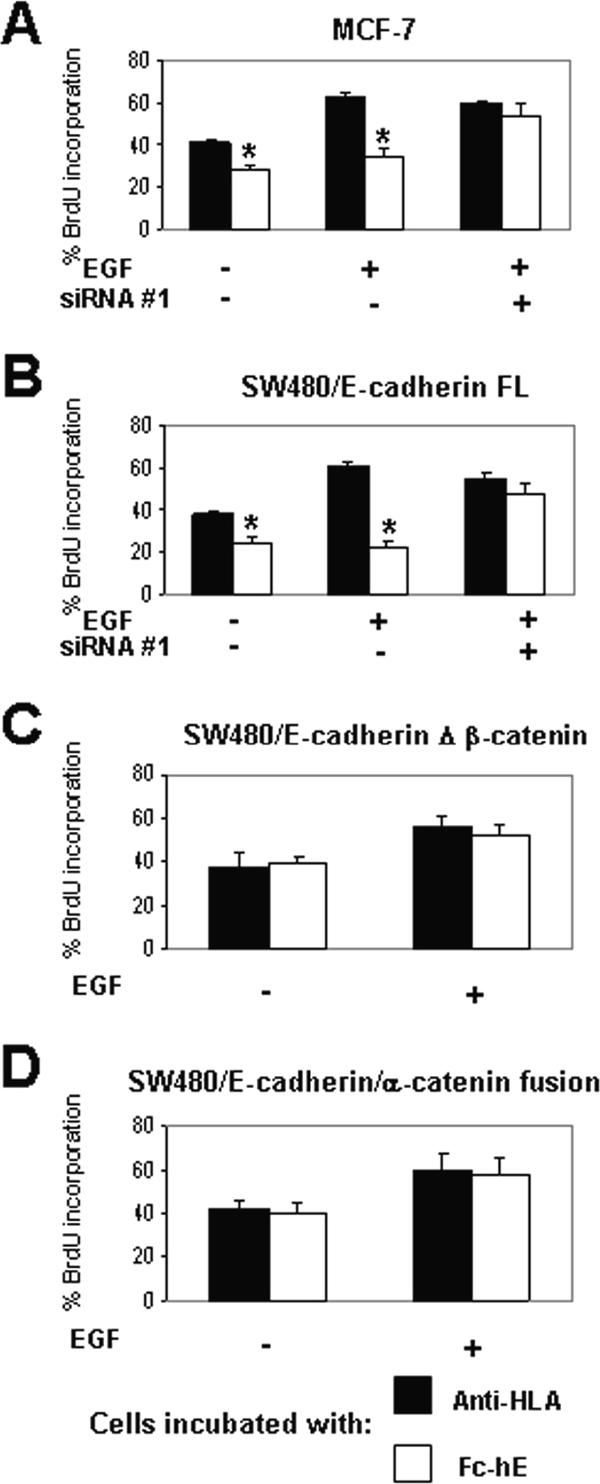
E-cadherin ligation inhibits EGF-stimulated cell growth via a β-catenin–dependent mechanism. Cells were grown in 0.5% FBS and incubated with 10 ng/ml EGF for 24 h. (A and B) Binding of Fc-hE–coated microspheres to MCF-7 (A) and SW480/E-cadherin FL (B) cells, respectively, inhibits cell growth mediated by EGF treatment. EGF signaling is not affected by E-cadherin ligation when β-catenin is depleted by siRNA. (C) No effect of E-cadherin ligation when the β-catenin binding domain of E-cadherin is deleted. (D) No effect of E-cadherin ligation when E-cadherin–α-catenin chimera is expressed. Data are expressed as mean ± SEM (*p < 0.05).
To determine how E-cadherin ligation might inhibit EGFR-mediated growth signaling, we asked whether E-cadherin ligation could inhibit EGFR phosphorylation, the first step in the transduction of the EGFR signal (Figure 8). For these experiments, we used the A431 epithelial cell line, because it expresses high levels of EGFR, making it better for detecting specific protein modifications by immunofluorescence microscopy. A431 cells, like the other epithelial cell lines used in this study, respond to E-cadherin ligation at the cell surface, which reduces cell proliferation stimulated by addition of EGF (Figure 8A). Addition of EGF to A431 cells stimulated EGFR phosphorylation at Tyr1173 (Figure 8B), a well-known autophosphorylation site that leads to activation of Ras/MAP kinase signaling cascade and subsequent cell proliferation (Downward et al., 1984). Autophosphorylation of EGFR at Tyr1173 was detected within few minutes and continued for at least 1 h, but Fc-hE beads had no effect on Tyr1173 phosphorylation relative to control anti-HLA beads (Figure 8C).
We also examined phosphorylation of EGFR at Tyr845, a site that is transphosphorylated by c-Src after it is recruited and activated by EGFR autophosphorylation (Boerner et al., 2005). Phosphorylation of EGFR on Tyr845 is known to mediate the phosphorylation and activation of STAT5b, which leads to ERK-independent DNA synthesis (Olayioye et al., 1999; Kloth et al., 2003; Boerner et al., 2005). EGF stimulated EGFR phosphorylation at Tyr845 (Figure 8B), and EGFR Tyr845 phosphorylation was significantly inhibited by the presence of Fc-hE beads compared with the control anti-HLA beads (Figure 8D). Therefore, E-cadherin homophilic ligation selectively inhibits EGFR Tyr845 phosphorylation in response to EGF.
We then asked whether EGFR downstream signaling is affected by E-cadherin homophilic ligation. We first examined EGF-stimulated ERK signaling, because it is a major mitogenic pathway downstream of EGFR activation. In A431 cells, the immunofluorescence staining of cytoplasmic phosphorylated ERK (Figure 9A) is present in ∼30% of the cells before addition of EGF, but the number of positive cells increased transiently, lasting for <30 min. We observed only a very small inhibition of ERK phosphorylation by Fc-hE beads compared with control beads at 2 and 5 min, but no effect at 10 and 30 min of EGF stimulation (Figure 9B). Thus, E-cadherin ligation had minimal effect on ERK activation by EGFR.
We also examined the activation of an alternative mitogenic signaling pathway, the STAT5-dependent pathway that results from phosphorylation of Tyr845 of the EGFR. EGF treatment of A431 cells stimulated a large increase in the number of cells with cytoplasmic staining of p-STAT5 (Figure 9A), and p-STAT5 staining persisted for an hour (Figure 9C). The appearance of p-STAT5 in response to EGF treatment was significantly inhibited by incubation with Fc-hE–cadherin beads at all times compared with the control anti-HLA beads (Figure 9C). Together, these data suggest that E-cadherin homophilic ligation inhibits the EGF-induced cell growth by inhibiting the EGFR(Tyr845)/STAT5 pathway.
DISCUSSION
E-cadherin is known to be a tumor suppressor protein. Re-expression of E-cadherin in epithelial cells that have lost E-cadherin leads to an inhibition of cell proliferation (Perl et al., 1998; Gottardi et al., 2001; Wong and Gumbiner, 2003). In some cases, E-cadherin expression has been found to inhibit cell growth by a mechanism that is independent of cell adhesion, by its binding to β-catenin and subsequent inhibition of Wnt signaling and β-catenin/TCF transcriptional activity (Gottardi et al., 2001; Conacci-Sorrell et al., 2002). Nonetheless, E-cadherin has also been proposed to mediate contact inhibition of cell growth via its adhesive function at the cell surface. However, it has been difficult to determine whether E-cadherin directly transduces a growth inhibitory signal upon homophilic binding, because of the complex cellular consequences of adhesion. E-cadherin could signal indirectly, because it controls the state of the entire epithelial junctional complex and cell polarity, and it strongly influences the ability of cells to physically interact and signal through juxtacrine mechanisms, i.e., other receptors that can inhibit cell growth (Bosenberg and Massague, 1993; Fagotto and Gumbiner, 1996). Therefore, we developed an experimental approach to isolate the response to E-cadherin homophilic binding independently of all other cell interactions, and we demonstrated that E-cadherin ligation directly transduces a cell growth inhibitory signal in several epithelial cells.
Several criteria showed that the effect of E-cadherin ligation on cell growth is specific. Its effects were not simply due to the binding of beads nonspecifically to cell surface glycoproteins; in all experiments, bead binding via antibodies to other cell surface proteins, HLA antigen, or the Na+/K+-ATPase served as negative controls. Nonspecific phagocytosis of the E-cadherin beads could not have caused the growth inhibition, because E-cadherin specifically inhibited cell growth when it was presented as a substrate attached to the glass coverslip. Moreover E-cadherin beads did not inhibit the growth of cells that were present in clusters, presumably because they already have endogenous cell adhesions transducing a growth inhibitory signal. Finally, the E-cadherin beads did not inhibit the growth of all cell types, because they had no effect on the growth of the highly transformed mesenchymal CHO cell line transfected to express E-cadherin. Thus, E-cadherin beads inhibit cell growth by mimicking a normal cell contact rather than via a general nonselective mechanism.
E-cadherin required the β-catenin binding domain to mediate growth inhibition, and β-catenin depletion by siRNA blocked this growth inhibitory signal and increased cell proliferation rate. In contrast to growth suppression resulting from E-cadherin overexpression or re-expression in previous studies, homophilic ligation did not inhibit cell growth through antagonism of the Wnt pathway via inhibition of β-catenin/TCF transcriptional activity. In MCF7 cells and HT29 cells, perturbation of β-catenin/TCF transcriptional activity had no effect on cell growth in our assay. Moreover, the C-terminal transactivation domain of β-catenin required for TCF-dependent transcriptional activity was not required for the growth inhibitory signal. Strong inhibition of β-catenin/TCF signaling in SW480 cells by dominant-negative inhibitors reduced growth even independently of E-cadherin ligation, consistent with previous findings (Gottardi et al., 2001). However, siRNA-mediated depletion of β-catenin selectively inhibited E-cadherin ligation-induced inhibition of cell growth, most likely because the siRNA depleted the functional pool of β-catenin required for the ligation-induced signal more significantly than the cytoplasmic/nuclear pool required for TCF-dependent transcription. Thus, β-catenin mediates the growth inhibitory signal resulting from E-cadherin homophilic ligation independently of its well-known role in Wnt signaling.
Although it is difficult to identify the pool of β-catenin responsible for the transduction of the ligation-induced signal definitively, β-catenin bound to E-cadherin at the plasma membrane is probably responsible. Treatment with siRNA strongly depleted the cadherin-bound fraction. Also, the β-catenin binding domain of E-cadherin was required for growth inhibition. It is unlikely that E-cadherin acts by depleting a cytosolic pool of β-catenin, as has been observed with cadherin overexpression (Shtutman et al., 1999; Gottardi et al., 2001), because homophilic binding is not known to alter the amount of β-catenin associated with cadherins. Yet, E-cadherin–associated β-catenin must have a function in signaling growth inhibition that is separate from its role in the cell adhesive function of E-cadherin. The E-cadherin–α-catenin chimera, which is known to mediate strong adhesion (Nagafuchi et al., 1994; Gottardi et al., 2001), was unable to transduce the growth inhibitory signal, whereas an E-cadherin construct with a mutated p120-binding domain, which interferes with cell adhesion (Thoreson et al., 2000), is still capable of transducing the growth inhibitory signal. Furthermore, a mutant form of β-catenin lacking the N-terminal α-catenin binding domain and unable to form the full complex linked to the actin cytoskeleton is still capable of transducing the growth inhibitory signal. Together, these findings suggest that E-cadherin–associated β-catenin acts to transduce a homophilic ligation induced growth inhibitory signal by coupling E-cadherin to other signaling molecules at the plasma membrane.
Numerous studies have reported that cadherins interact with growth factor receptors, including VE-cadherin with vascular endothelial growth factor (VEGF) receptor, N-cadherin with FGF receptor, and E-cadherin with EGFR (Williams et al., 2001; Lampugnani et al., 2003; Qian et al., 2004). Cadherins have been found to influence growth factor receptor signaling, either activating (Pece and Gutkind, 2000) or inhibiting (Takahashi and Suzuki, 1996; Qian et al., 2004) signaling. The EGFR has been shown to interact with E-cadherin via β-catenin, and this interaction occurs through the core armadillo repeat domain of β-catenin (Hoschuetzky et al., 1994), the same region we found to be required to transduce the E-cadherin ligation-induced signal for inhibition of cell growth. Therefore, we asked whether E-cadherin homophilic ligation, independently of other cell contacts, can regulate EGFR signaling.
We found that E-cadherin ligation inhibits EGF-stimulated cell growth by a β-catenin–dependent process. Inhibition of EGFR signaling could occur at a very early stage of EGFR signal transduction at the plasma membrane. E-cadherin ligation did not interfere with EGFR autophosphorylation at Tyr1173 or ensuing activation of the mitogen-activated protein kinase pathway, as measured by phospho-ERK. However, it did inhibit a secondary receptor activation step, the transphosphorylation of Tyr845. Phosphorylation of EGFR on Tyr845 is thought to be dependent on the recruitment and activation of c-Src, and it is known to mediate the phosphorylation and activation of STAT5b in a pathway leading to ERK-independent DNA synthesis (Olayioye et al., 1999; Kloth et al., 2003; Boerner et al., 2005). Notably, we found that E-cadherin ligation selectively inhibits the EGF stimulation of phospho-STAT5, consistent with the selective inhibition of phosphorylation at Tyr845 of the EGFR. Inhibition of STAT5 phosphorylation and activation may account, at least in part, for the inhibition of EGF-dependent cell proliferation by E-cadherin ligation. Therefore, E-cadherin homophilic ligation does not seem to block receptor activation itself, but instead it selectively inhibits a subset of downstream signal transduction steps.
The detailed mechanism by which E-cadherin homophilic ligation selectively inhibits a subset of EGFR signaling events is unclear, but it may involve a poorly understood complex of interacting proteins. In endothelial cells, β-catenin and the DEP-1/CD148 phosphatase are required for VE-cadherin–mediated inhibition of VEGF-induced proliferation (Lampugnani et al., 2003). Furthermore, VE-cadherin limits cell proliferation by retaining VEGF receptor at the membrane, and it decreased its internalization into signaling compartments (Lampugnani et al., 2006). In mouse embryo fibroblasts, the tumor suppressor protein merlin/NF2 has been shown to be required for contact inhibition of cell growth mediated by N-cadherin adhesion (Lallemand et al., 2003). Interestingly, merlin/NF2 inhibits EGFR signaling at an early step after receptor activation (Curto and McClatchey, unpublished), similar to E-cadherin ligation. Furthermore, E-cadherin and N-cadherin have been shown to enhance growth factor signaling in some cell types (Pece and Gutkind, 2000; Suyama et al., 2002), indicating that the mechanism of coupling between cadherins and receptors varies in different cells.
Contact inhibition is a complex phenomenon and many different signaling mechanisms may be involved. Our findings provide evidence for a direct signaling mechanism through E-cadherin as a result of homophilic ligation. Nonetheless, other cell–cell interactions indirectly influenced by cadherins, such as other cell junctions or juxtacrine signaling receptors, are likely to have roles in regulation of cell growth. A pathway involving p21-activated kinase, merlin/NF-2, and Rac has been found to mediate contact inhibition of growth in endothelial cells (Okada et al., 2005), and other junctional proteins such as discs large and scribble have been shown to suppress cell proliferation in Drosophila epithelia (Bilder et al., 2000). Furthermore, contact inhibition is not a constitutive property of all epithelial cells; there are times in development when tightly adherent cells undergo very rapid proliferation. Elucidation of all the pathways regulating contact inhibition of growth will be needed to fully understand this phenomenon.
Supplementary Material
ACKNOWLEDGMENTS
We thank K. Schwartz for technical assistance and H. Clevers (Hubrecht Institute, The Netherlands), J. Massague (Memorial Sloan-Kettering Cancer Center, New York, NY), U. Muller (The Scripps Research Institute, La Jolla, CA), P. McCrea (M. D. Anderson Cancer Center, Houston, TX), and E. Fearon (University of Michigan, Ann Arbor, MI) for gifts of reagents. This work was supported by National Institutes of Health grant GM-037432 (to B.M.G.) and fellowships by “Fondation pour la Recherche Médicale” and “La Ligue Nationale Contre le Cancer” (to M.P.).
Abbreviations used:
- BrdU
5-bromodeoxyuridine
- EGF
epidermal growth factor
- EGFR
epidermal growth factor receptor
- ERK
extracellular signal-regulated kinase
- Fc-hE
Fc-hE–cadherin
- FL
full length
- LEF
leukocyte enhancer factor
- siRNA
small interfering RNA
- STAT
signal transducers and activators of transcription
- TCF
T cell factor.
Footnotes
This article was published online ahead of print in MBC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E06-04-0348) on March 28, 2007.
 The online version of this article contains supplemental material at MBC Online (http://www.molbiolcell.org).
The online version of this article contains supplemental material at MBC Online (http://www.molbiolcell.org).
REFERENCES
- Behrens J., Mareel M. M., Van Roy F. M., Birchmeier W. Dissecting tumor cell invasion: epithelial cells acquire invasive properties after the loss of uvomorulin-mediated cell-cell adhesion. J. Cell Biol. 1989;108:2435–2447. doi: 10.1083/jcb.108.6.2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilder D., Li M., Perrimon N. Cooperative regulation of cell polarity and growth by Drosophila tumor suppressors. Science. 2000;289:113–116. doi: 10.1126/science.289.5476.113. [DOI] [PubMed] [Google Scholar]
- Boerner J. L., Biscardi J. S., Silva C. M., Parsons S. J. Transactivating agonists of the EGF receptor require Tyr845 phosphorylation for induction of DNA synthesis. Mol. Carcinog. 2005;44:262–273. doi: 10.1002/mc.20138. [DOI] [PubMed] [Google Scholar]
- Bosenberg M. W., Massague J. Juxtacrine cell signaling molecules. Curr. Opin. Cell Biol. 1993;5:832–838. doi: 10.1016/0955-0674(93)90032-l. [DOI] [PubMed] [Google Scholar]
- Braga V. M., Del Maschio A., Machesky L., Dejana E. Regulation of cadherin function by Rho and Rac: modulation by junction maturation and cellular context. Mol. Biol. Cell. 1999;10:9–22. doi: 10.1091/mbc.10.1.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caveda L., Martin-Padura I., Navarro P., Breviario F., Corada M., Gulino D., Lampugnani M. G., Dejana E. Inhibition of cultured cell growth by vascular endothelial cadherin (cadherin-5/VE-cadherin) J. Clin. Invest. 1996;98:886–893. doi: 10.1172/JCI118870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chappuis-Flament S., Wong E., Hicks L. D., Kay C. M., Gumbiner B. M. Multiple cadherin extracellular repeats mediate homophilic binding and adhesion. J. Cell Biol. 2001;154:231–243. doi: 10.1083/jcb.200103143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conacci-Sorrell M., Zhurinsky J., Ben-Ze'ev A. The cadherin-catenin adhesion system in signaling and cancer. J. Clin. Invest. 2002;109:987–991. doi: 10.1172/JCI15429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng J., Miller S. A., Wang H. Y., Xia W., Wen Y., Zhou B. P., Li Y., Lin S. Y., Hung M. C. beta-Catenin interacts with and inhibits NF-kappa B in human colon and breast cancer. Cancer Cell. 2002;2:323–334. doi: 10.1016/s1535-6108(02)00154-x. [DOI] [PubMed] [Google Scholar]
- Downward J., Parker P., Waterfield M. D. Autophosphorylation sites on the epidermal growth factor receptor. Nature. 1984;311:483–485. doi: 10.1038/311483a0. [DOI] [PubMed] [Google Scholar]
- Fagotto F., Funayama N., Gluck U., Gumbiner B. M. Binding to cadherins antagonizes the signaling activity of beta-catenin during axis formation in Xenopus. J. Cell Biol. 1996;132:1105–1114. doi: 10.1083/jcb.132.6.1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagotto F., Gumbiner B. M. Cell contact-dependent signaling. Dev. Biol. 1996;180:445–454. doi: 10.1006/dbio.1996.0318. [DOI] [PubMed] [Google Scholar]
- Funayama N., Fagotto F., McCrea P., Gumbiner B. M. Embryonic axis induction by the armadillo repeat domain of beta-catenin: evidence for intracellular signaling. J. Cell Biol. 1995;128:959–968. doi: 10.1083/jcb.128.5.959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funke L., Dakoji S., Bredt D. S. Membrane-associated guanylate kinases regulate adhesion and plasticity at cell junctions. Annu. Rev. Biochem. 2005;74:219–245. doi: 10.1146/annurev.biochem.74.082803.133339. [DOI] [PubMed] [Google Scholar]
- Gibson M. C., Perrimon N. Apicobasal polarization: epithelial form and function. Curr. Opin. Cell Biol. 2003;15:747–752. doi: 10.1016/j.ceb.2003.10.008. [DOI] [PubMed] [Google Scholar]
- Gottardi C. J., Wong E., Gumbiner B. M. E-cadherin suppresses cellular transformation by inhibiting beta-catenin signaling in an adhesion–independent manner. J. Cell Biol. 2001;153:1049–1060. doi: 10.1083/jcb.153.5.1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumbiner B. M. Cell adhesion: the molecular basis of tissue architecture and morphogenesis. Cell. 1996;84:345–357. doi: 10.1016/s0092-8674(00)81279-9. [DOI] [PubMed] [Google Scholar]
- Gumbiner B. M. Regulation of cadherin-mediated adhesion in morphogenesis. Nat. Rev. Mol. Cell Biol. 2005;6:622–634. doi: 10.1038/nrm1699. [DOI] [PubMed] [Google Scholar]
- Heasman J., Crawford A., Goldstone K., Garner-Hamrick P., Gumbiner B., McCrea P., Kintner C., Noro C. Y., Wylie C. Overexpression of cadherins and underexpression of beta-catenin inhibit dorsal mesoderm induction in early Xenopus embryos. Cell. 1994;79:791–803. doi: 10.1016/0092-8674(94)90069-8. [DOI] [PubMed] [Google Scholar]
- Huber A. H., Weis W. I. The structure of the beta-catenin/E-cadherin complex and the molecular basis of diverse ligand recognition by beta-catenin. Cell. 2001;105:391–402. doi: 10.1016/s0092-8674(01)00330-0. [DOI] [PubMed] [Google Scholar]
- Hoschuetzky H., Aberle H., Kemler R. beta-Catenin mediates the interaction of the cadherin-catenin complex with epidermal growth factor receptor. J. Cell Biol. 1994;127:1375–1380. doi: 10.1083/jcb.127.5.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloth M. T., Laughlin K. K., Biscardi J. S., Boerner J. L., Parsons S. J., Silva C. M. STAT5b, a mediator of synergism between c-Src and the epidermal growth factor receptor. J. Biol. Chem. 2003;278:1671–1679. doi: 10.1074/jbc.M207289200. [DOI] [PubMed] [Google Scholar]
- Kolligs F. T., Hu G., Dang C. V., Fearon E. R. Neoplastic transformation of RK3E by mutant beta-catenin requires deregulation of Tcf/Lef transcription but not activation of c-myc expression. Mol. Cell Biol. 1999;19:5696–5706. doi: 10.1128/mcb.19.8.5696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs E. M., Ali R. G., McCormack A. J., Yap A. S. E-cadherin homophilic ligation directly signals through Rac and phosphatidylinositol 3-kinase to regulate adhesive contacts. J. Biol. Chem. 2002;277:6708–6718. doi: 10.1074/jbc.M109640200. [DOI] [PubMed] [Google Scholar]
- Lallemand D., Curto M., Saotome I., Giovannini M., McClatchey A. I. NF2 deficiency promotes tumorigenesis and metastasis by destabilizing adherens junctions. Genes Dev. 2003;17:1090–1100. doi: 10.1101/gad.1054603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert M., Choquet D., Mege R. M. Dynamics of ligand-induced, Rac1-dependent anchoring of cadherins to the actin cytoskeleton. J. Cell Biol. 2002;157:469–479. doi: 10.1083/jcb.200107104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampugnani G. M., Orsenigo F., Gagliani M. C., Tacchetti C., Dejana E. Vascular endothelial cadherin controls VEGFR-2 internalization and signaling from intracellular compartments. J. Cell Biol. 2006;174:593–604. doi: 10.1083/jcb.200602080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampugnani G. M. Contact inhibition of VEGF-induced proliferation requires vascular endothelial cadherin, beta-catenin, and the phosphatase DEP-1/CD148. J. Cell Biol. 2003;161:793–804. doi: 10.1083/jcb.200209019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCrea P. D., Brieher W. M., Gumbiner B. M. Induction of a secondary body axis in Xenopus by antibodies to beta-catenin. J. Cell Biol. 1993;123:477–484. doi: 10.1083/jcb.123.2.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matter K., Aijaz S., Tsapara A., Balda M. S. Mammalian tight junctions in the regulation of epithelial differentiation and proliferation. Curr. Opin. Cell Biol. 2005;17:453–458. doi: 10.1016/j.ceb.2005.08.003. [DOI] [PubMed] [Google Scholar]
- Montross W. T., Ji H., McCrea P. D. A beta-catenin/engrailed chimera selectively suppresses Wnt signaling. J. Cell Sci. 2000;113:1759–1770. doi: 10.1242/jcs.113.10.1759. [DOI] [PubMed] [Google Scholar]
- Motti M. L., Califano D., Baldassarre G., Celetti A., Merolla F., Forzati F., Napolitano F., Tavernise B., Fusco A., Viglietto G. Reduced E-cadherin expression contributes to the loss of p27kip1-mediated mechanism of contact inhibition in thyroid anaplastic carcinomas. Carcinogenesis. 2005;26:1021–1034. doi: 10.1093/carcin/bgi050. [DOI] [PubMed] [Google Scholar]
- Mueller S., Cadenas E., Sconthal A. H. p21WAF1 regulates anchorage-independent growth of HCT116 colon carcinoma cells via E-cadherin expression. Cancer Res. 2000;60:156–163. [PubMed] [Google Scholar]
- Nagafuchi S., Ishihara S., Tsukita S. The roles of catenins in the cadherin-mediated cell adhesion: functional analysis of E-cadherin-alpha catenin fusion molecules. J. Cell Biol. 1994;127:235–245. doi: 10.1083/jcb.127.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niessen C. M., Gumbiner B. M. Cadherin-mediated cell sorting not determined by binding or adhesion specificity. J. Cell Biol. 2002;156:389–399. doi: 10.1083/jcb.200108040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noren N. K., Niessen C. M., Gumbiner B. M., Burridege K. Cadherin engagement regulates Rho family GTPases. J. Biol. Chem. 2001;276:33305–33308. doi: 10.1074/jbc.C100306200. [DOI] [PubMed] [Google Scholar]
- Okada T., Lopez-Lago M., Giancotti F. G. Merlin/NF-2 mediates contact inhibition of growth by suppressing recruitment of Rac to the plasma membrane. J. Cell Biol. 2005;171:361–371. doi: 10.1083/jcb.200503165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olayioye M. A., Beuvink I., Horsch K., Daly J. M., Hynes N. E. ErbB receptor-induced activation of stat transcription factors is mediated by Src tyrosine kinases. J. Biol. Chem. 1999;274:17209–17218. doi: 10.1074/jbc.274.24.17209. [DOI] [PubMed] [Google Scholar]
- Orsulic S., Huber O., Aberle H., Arnold S., Kemler R. E-cadherin binding prevents beta-catenin nuclear localization and beta-catenin/LEF-1-mediated transactivation. J. Cell Sci. 1999;112:1237–1245. doi: 10.1242/jcs.112.8.1237. [DOI] [PubMed] [Google Scholar]
- Pang J. H., Kraemer A., Stehbens S. J., Frame M. C., Yap A. S. Recruitment of phosphoinositide 3-kinase defines a positive contribution of tyrosine kinase signaling to E-cadherin function. J. Biol. Chem. 2005;280:3043–3050. doi: 10.1074/jbc.M412148200. [DOI] [PubMed] [Google Scholar]
- Pece S., Chiariello M., Murga C., Gutkind J. S. Activation of the protein kinase Akt/PKB by the formation of E-cadherin-mediated cell-cell junctions. Evidence for the association of phosphatidylinositol 3-kinase with the E-cadherin adhesion complex. J. Biol. Chem. 1999;274:19347–19351. doi: 10.1074/jbc.274.27.19347. [DOI] [PubMed] [Google Scholar]
- Pece S., Gutkind J. S. Signaling from E-cadherins to the MAPK pathway by the recruitment and activation of epidermal growth factor receptors upon cell-cell contact formation. J. Biol. Chem. 2000;275:41227–41233. doi: 10.1074/jbc.M006578200. [DOI] [PubMed] [Google Scholar]
- Perez-Moreno M., Jamora C., Fuchs E. Sticky business: orchestrating cellular signals at adherens junctions. Cell. 2003;112:535–548. doi: 10.1016/s0092-8674(03)00108-9. [DOI] [PubMed] [Google Scholar]
- Perl A. K., Wilgenbus P., Dahl U., Semb H., Christofori G. A causal role for E-cadherin in the transition from adenoma to carcinoma. Nature. 1998;392:190–193. doi: 10.1038/32433. [DOI] [PubMed] [Google Scholar]
- Qian X., Karpova T., Sheppard A. M., McCally J., Lowy D. R. E-cadherin-mediated adhesion inhibits ligand-dependent activation of diverse receptor tyrosine kinases. EMBO J. 2004;23:1739–1748. doi: 10.1038/sj.emboj.7600136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinohara M., Kodama A., Matozaki T., Fukuhara A., Tachibana K., Nakanishi H., Takai Y. Roles of cell-cell adhesion-dependent tyrosine phosphorylation of Gab-1. J. Biol. Chem. 2001;276:18941–18946. doi: 10.1074/jbc.M100909200. [DOI] [PubMed] [Google Scholar]
- Shtutman M., Zhurinsky J., Simcha I., Albanese C., D'Amico M., Pestell R., Ben-Ze'ev A. The cyclin D1 gene is a target of the beta-catenin/LEF-1 pathway. Proc. Natl. Acad. Sci. USA. 1999;96:5522–5527. doi: 10.1073/pnas.96.10.5522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St Croix B., Sheehan C., Rak J. W., Florenes V. A., Slingerland J. M., Kerbe R. S. E-Cadherin-dependent growth suppression is mediated by the cyclin-dependent kinase inhibitor p27(KIP1) J. Cell Biol. 1998;142:557–571. doi: 10.1083/jcb.142.2.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suyama K., Shapiro I., Guttman M., Hazan R. B. A signaling pathway leading to metastasis is controlled by N-cadherin and the FGF receptor. Cancer Cell. 2002;2:301–314. doi: 10.1016/s1535-6108(02)00150-2. [DOI] [PubMed] [Google Scholar]
- Takeichi M. Cadherins in cancer: implications for invasion and metastasis. Curr. Opin. Cell Biol. 1993;5:806–811. doi: 10.1016/0955-0674(93)90029-p. [DOI] [PubMed] [Google Scholar]
- Takahashi K., Suzuki K. Density-dependent inhibition of growth involves prevention of EGF receptor activation by E-cadherin-mediated cell-cell adhesion. Exp. Cell Res. 1996;226:214–222. doi: 10.1006/excr.1996.0221. [DOI] [PubMed] [Google Scholar]
- Thoreson M. A., Anastasiadis P. Z., Daniel J. M., Ireton R. C., Wheelock M. J., Johnson K. R., Hummingbird D. K., Reynolds A. B. Selective uncoupling of p120(ctn) from E-cadherin disrupts strong adhesion. J. Cell Biol. 2000;148:189–202. doi: 10.1083/jcb.148.1.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Noort M., Clevers H. TCF transcription factors, mediators of Wnt-signaling in development and cancer. Dev. Biol. 2002:1–8. doi: 10.1006/dbio.2001.0566. [DOI] [PubMed] [Google Scholar]
- Verma U. N., Surabhi R. M., Schmaltieg A., Becerra C., Gaynor R. B. Small interfering RNAs directed against beta-catenin inhibit the in vitro and in vivo growth of colon cancer cells. Clin. Cancer Res. 2003;9:1291–1300. [PubMed] [Google Scholar]
- Vleminckx K., Vakaet L., Jr, Mareel M., Fiers W., van Roy F. Genetic manipulation of E-cadherin expression by epithelial tumor cells reveals an invasion suppressor role. Cell. 1991;66:107–119. doi: 10.1016/0092-8674(91)90143-m. [DOI] [PubMed] [Google Scholar]
- Vonica A., Weng W., Gumbiner B. M., Venuti J. M. TCF is the nuclear effector of the beta-catenin signal that patterns the sea urchin animal-vegetal axis. Dev. Biol. 2000;217:230–243. doi: 10.1006/dbio.1999.9551. [DOI] [PubMed] [Google Scholar]
- Watabe M., Nagafuchi A., Tsukita S., Takeichi M. Induction of polarized cell-cell association and retardation of growth by activation of the E-cadherin-catenin adhesion system in a dispersed carcinoma line. J. Cell Biol. 1994;127:247–256. doi: 10.1083/jcb.127.1.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams E. J., Williams G., Howell F. V., Skaper F. D., Walsh F. S., Doherty P. Identification of an N-cadherin motif that can interact with the fibroblast growth factor receptor and is required for axonal growth. J. Biol. Chem. 2001;276:43879–43886. doi: 10.1074/jbc.M105876200. [DOI] [PubMed] [Google Scholar]
- Wong A. S., Gumbiner B. M. Adhesion-independent mechanism for suppression of tumor cell invasion by E-cadherin. J. Cell Biol. 2003;161:1191–1203. doi: 10.1083/jcb.200212033. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.