

Abstract
The β1 subunit of BK (large conductance Ca2+ and
voltage-activated K+) channels is essential for many key
physiological processes, such as controlling the contraction of smooth muscle
and the tuning of hair cells in the cochlea. Although it is known that the
β1 subunit greatly increases the open probability of BK channels, little
is known about its mechanism of action. We now explore this mechanism by using
channels in which the Ca2+- and
Mg2+-dependent activating mechanisms have been disrupted
by mutating three sites to remove the Ca2+ and
Mg2+ sensitivity. We find that the presence of the
β1 subunit partially restores Ca2+ sensitivity to
the triply mutated channels, but not the Mg2+
sensitivity. We also find that the β1 subunit has no effect on the
Mg2+ sensitivity of WT BK channels, in contrast to its
pronounced effect of increasing the apparent Ca2+
sensitivity. These observations suggest that the β1 subunit increases
open probability by working through the Ca2+-dependent,
rather than Mg2+-dependent, activating mechanisms, and
that the action of the β1 subunit is not directly on the
Ca2+ binding sites, but on the allosteric machinery
coupling the sites to the gate. The differential effects of the β1
subunit on the Ca2+ and Mg2+
activation of the channel suggest that these processes act separately.
Finally, we show that
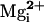 inhibits,
rather than activates, BK channels in the presence of the β1 subunit for
intermediate levels of
inhibits,
rather than activates, BK channels in the presence of the β1 subunit for
intermediate levels of
 . This
Mg2+ inhibition in the presence of the β1 subunit
provides an additional regulatory mechanism of BK channel activity.
. This
Mg2+ inhibition in the presence of the β1 subunit
provides an additional regulatory mechanism of BK channel activity.
BK channels (Slo1) are large conductance K+ channels that are
activated in a highly synergistic manner by membrane depolarization,
intracellular Ca2+
( ), and intracellular
Mg2+
(
), and intracellular
Mg2+
( )
(1–12).
When BK channels are open, K+ flows out of cells through the open
channels, driving the membrane potential more negative. This change in
membrane potential allows BK channels to modulate many important physiological
processes, such as smooth muscle contraction
(13–15),
frequency tuning of sensory hair cells
(16), and regulation of
neurotransmitter release (17,
18). BK channels can be
comprised of four α subunits alone, as in skeletal muscle, or four
α subunits with four associated β subunits, as in smooth muscle and
neurons
(19–21).
Of the four types of β subunits that have been identified, the β1
subunit is predominantly expressed in smooth muscle
(13,
20). When coexpressed with
α subunits, the β1 subunit greatly increases the open probability
(Po) of BK channels over the physiological range of
)
(1–12).
When BK channels are open, K+ flows out of cells through the open
channels, driving the membrane potential more negative. This change in
membrane potential allows BK channels to modulate many important physiological
processes, such as smooth muscle contraction
(13–15),
frequency tuning of sensory hair cells
(16), and regulation of
neurotransmitter release (17,
18). BK channels can be
comprised of four α subunits alone, as in skeletal muscle, or four
α subunits with four associated β subunits, as in smooth muscle and
neurons
(19–21).
Of the four types of β subunits that have been identified, the β1
subunit is predominantly expressed in smooth muscle
(13,
20). When coexpressed with
α subunits, the β1 subunit greatly increases the open probability
(Po) of BK channels over the physiological range of
 and voltage
(22–27).
This increase in Po gives an apparent increase in the
Ca2+ sensitivity of the channel, because less
and voltage
(22–27).
This increase in Po gives an apparent increase in the
Ca2+ sensitivity of the channel, because less
 is required for 50%
activation
(22–27).
Decreasing the activation of BK channels in smooth muscle by knocking out the
β1 subunit results in chronic hypertension from increased contraction of
the arterial smooth muscle
(13,
14).
is required for 50%
activation
(22–27).
Decreasing the activation of BK channels in smooth muscle by knocking out the
β1 subunit results in chronic hypertension from increased contraction of
the arterial smooth muscle
(13,
14).
Despite the importance of the β1 subunit, the underlying mechanism by which the β1 subunit increases Po of the BK channel remains unclear, although progress has been made (22–24, 26, 27). Previous studies at both the single-channel and macrocurrent level have suggested that the major (80%) facilitating effect of the β1 subunit is independent of Ca2+, with a smaller (20%) Ca2+-dependent contribution (23, 24, 26). Consistent with the idea that the major action of the β1 subunit is not through changes in Ca2+ affinity (23, 24, 26), the Ca2+ bowl, a key site located on the C terminus associated with Ca2+ activation of BK channels (28, 29), is not required for the β1 subunit to increase the apparent Ca2+ sensitivity of the BK channel, but other structural features of the C terminus are required (27). Thus, the β1 subunit may act on the allosteric machinery coupling the Ca2+ binding sites to the gate rather than directly on the sites.
To explore this possibility, we take advantage of recent studies that
suggest that there are at least two major classes of divalent binding sites
located on the intracellular C terminus of the α subunit of the BK
channel. One of these classes is of low affinity (millimolar) and activates
the channel through the binding of Ca2+ or
Mg2+, and the other is of high affinity (micromolar) and
activates the channel through the binding of Ca2+. For
the high-affinity class, combined mutations at two separate sites, the
Ca2+ bowl and D362/D367, remove the high-affinity
Ca2+ sensitivity
(11). For the low-affinity
class, mutation of E399 removes the activation by millimolar concentrations of
Ca2+ or Mg2+
(10,
12). Mutation of these three
sites together produces a triple mutant (TM) channel with no
Ca2+ or Mg2+ sensitivity for
concentrations of 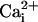 and
and
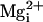 <10 mM
(11). Because the TM channel
has no Ca2+ and Mg2+ sensitivity,
it provides a unique platform to examine to what extent the β1 subunit
may require functional Ca2+ and
Mg2+ activating mechanisms to modulate the BK
channel.
<10 mM
(11). Because the TM channel
has no Ca2+ and Mg2+ sensitivity,
it provides a unique platform to examine to what extent the β1 subunit
may require functional Ca2+ and
Mg2+ activating mechanisms to modulate the BK
channel.
We found that the β1 subunit increased Po of the
TM channel in the absence of
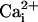 and
and
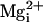 , just as it does for WT
channels, indicating that the triple mutation does not destroy the gating
machinery on which the β1 subunit acts. Surprisingly, we also found that
the β1 subunit restored some Ca2+ sensitivity to
the TM Ca2+-insensitive channel. This restoration was
still present after removing the only intracellular negative charge on the
β1 subunit, suggesting that the β1 subunit was not directly adding
an additional Ca2+ binding site. In contrast to the
restoration of Ca2+ sensitivity to the TM channels, the
β1 subunit did not restore any Mg2+ sensitivity. In
addition, the β1 subunit had no effect on the Mg2+
sensitivity of WT BK channels, compared with its pronounced effect of
increasing the apparent Ca2+ sensitivity. Taken
together, these results suggest that the β1 subunit couples
Ca2+, but not Mg2+, activating
mechanisms to the gating of BK channels, and that the action of the β1
subunit is not directly on the Ca2+ binding sites, but
rather on the allosteric machinery coupling the sites to the gate. Finally, we
made the observation that Mg2+ can inhibit channel
activity in the presence of the β1 subunit for physiological levels of
, just as it does for WT
channels, indicating that the triple mutation does not destroy the gating
machinery on which the β1 subunit acts. Surprisingly, we also found that
the β1 subunit restored some Ca2+ sensitivity to
the TM Ca2+-insensitive channel. This restoration was
still present after removing the only intracellular negative charge on the
β1 subunit, suggesting that the β1 subunit was not directly adding
an additional Ca2+ binding site. In contrast to the
restoration of Ca2+ sensitivity to the TM channels, the
β1 subunit did not restore any Mg2+ sensitivity. In
addition, the β1 subunit had no effect on the Mg2+
sensitivity of WT BK channels, compared with its pronounced effect of
increasing the apparent Ca2+ sensitivity. Taken
together, these results suggest that the β1 subunit couples
Ca2+, but not Mg2+, activating
mechanisms to the gating of BK channels, and that the action of the β1
subunit is not directly on the Ca2+ binding sites, but
rather on the allosteric machinery coupling the sites to the gate. Finally, we
made the observation that Mg2+ can inhibit channel
activity in the presence of the β1 subunit for physiological levels of
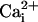 , in contrast to its
activation in the absence of the β1 subunit.
, in contrast to its
activation in the absence of the β1 subunit.
Materials and Methods
Clones, Mutagenesis, and Channel Expression. The WT Slo1 α subunit of the mouse BK channel (GenBank accession no. L16912) was provided by L. Salkoff (30), and the human β1 subunit (GenBank accession no. NM_004137) was provided by Merck Research Laboratories. The α subunit of the BK channel with mutations to remove three regulatory mechanisms (11), 5D5N in the Ca2+ bowl, D362A/D367A, and E399A (TM channel), and the human β1 subunit with mutations E13K and T14R were provided by X. Xia and C. Lingle (Washington University, St. Louis). Starting with the construct of the TM channel, we made two additional types of mutated channels: one by making a mutation in the pore region to decrease the tetraethylammonium (TEA) sensitivity (GYGDVYAKT to GYGDVVAKT) and the other by making a mutation on the C terminus by changing M513 to I513, using Stratagene's QuikChange Site-Directed Mutagenesis Kit. The mutations were checked by sequencing. The cDNA was transcribed in vitro by using the mMESSAGE mMACHINE Kit (Ambion, Austin, TX) to obtain cRNA for injection into the Xenopus oocytes. The oocytes were microinjected with 0.5–50 ng of cRNA 2–8 days before recording. For coexpression of α and β1 subunits, the cRNA for the β1 subunit was in 4–6 M excess over that for the α subunit to drive coassembly (22).
Electrophysiology and Solutions. Single-channel currents were
recorded with the patch clamp technique
(31) from inside-out patches
of membrane excised from Xenopus oocytes, except for two experiments
when TEA was applied to outside-out patches. (TEA was in the pipette for seven
additional experiments with TEA.) The pipette solution contained 158 mM KCl, 5
mM N-[tris(hydroxymethyl)methyl]-2-aminoethanesulfonic acid (TES),
and usually 10 μM GdCl3 to block the endogenous
stretch-activated channels
(32). The bath solution
contained 158 mM KCl, 5 mM TES, 1 mM EGTA, 1 mM N-(2-hydroxyethyl)
ethylenediamine-N,N′,N′-triacetic acid (HEDTA),
and sufficient added Ca2+ and Mg2+
to bring the free Ca2+ and Mg2+
levels to those indicated (23,
33). All solutions were
adjusted to pH 7.0. Solutions with no added Ca2+ had a
calculated free Ca2+ of
<10–8 M and will be referred to as 0
Ca2+ solutions because
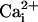 at these concentrations
has essentially no effect on the gating of the channel
(3,
24,
34). Voltages refer to the
intracellular potential. Experiments were performed at room temperature
(20–23°C).
at these concentrations
has essentially no effect on the gating of the channel
(3,
24,
34). Voltages refer to the
intracellular potential. Experiments were performed at room temperature
(20–23°C).
Data Analysis. Single-channel currents were typically filtered at 3–10 kHz and sampled at 200 kHz directly to disk by using pclamp8 (Axon Instuments, Union City, CA). Po and burst duration were then estimated from the digitized records by using custom programs (33, 35). Po vs. V plots derived from the single-channel data were fitted with a Boltzmann function (Eq. 1):
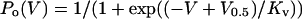 |
[1] |
where V0.5 is the voltage of half-maximal activation of
the channel and Kv is the voltage dependence of the
activation process in mV per e-fold change. Po vs.
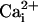 plots derived from the
single-channel data were fitted with a Hill equation (Eq. 2):
plots derived from the
single-channel data were fitted with a Hill equation (Eq. 2):
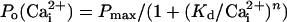 |
[2] |
where Pmax is the maximum Po for the
channels, Kd is the
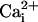 required for half
activation, and n is the Hill coefficient that reflects the slope of
the dose–response relationship. Data are expressed as the mean ±
SE.
required for half
activation, and n is the Hill coefficient that reflects the slope of
the dose–response relationship. Data are expressed as the mean ±
SE.
Results and Discussion
The β1 Subunit Restores Ca2+
Sensitivity to the TM Channel. We first examined whether the TM channel
is Ca2+-insensitive by using single-channel recording
techniques. Increasing 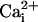 from 0 to 10 mM was found to have no effect on the activity of single BK
channels in seven experiments (example in
Fig. 1A), confirming
the Ca2+ insensitivity of the TM channel
(11).
from 0 to 10 mM was found to have no effect on the activity of single BK
channels in seven experiments (example in
Fig. 1A), confirming
the Ca2+ insensitivity of the TM channel
(11).
Fig. 1.

The β1 subunit partially restores Ca2+
sensitivity to TM Ca2+-insensitive BK channels.
(A and B) Single-channel currents recorded from a TM channel
in the absence (A) and presence (B)ofthe β1 subunit at
the indicated 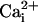 and +50 mV.
(C) Plots of Po vs.
and +50 mV.
(C) Plots of Po vs.
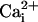 from 10 representative
experiments to show the range of restoration of Ca2+
sensitivity to the TM channel by the β1 subunit. (D) Plots of
Po vs. V
at 0
from 10 representative
experiments to show the range of restoration of Ca2+
sensitivity to the TM channel by the β1 subunit. (D) Plots of
Po vs. V
at 0  for TM channels with
and without the β1 subunit (data from six patches for each point). The
standard errors are often obscured by the symbol.
for TM channels with
and without the β1 subunit (data from six patches for each point). The
standard errors are often obscured by the symbol.
To examine the effect of the β1 subunit on the TM channel, we then
coexpressed β1 subunits with TM α subunits. Surprisingly, in the
presence of the β1 subunit the same change in
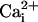 now greatly increased the
single-channel activity of the TM channel
(Fig. 1B). The degree
of restoration of Ca2+ sensitivity to the TM channel by
the β1 subunit was consistent for each channel but variable among
channels (data from 10 different experiments at +50 mV in
Fig. 1C), with maximum
Pos of 0.44 ± 0.10 (range 0.002–0.9), typical
Hill coefficients of 1.1 ± 0.1 (range 0.5–1.8), and
Kds of 38.1 ± 11.2 μM (range 14.7–108.2).
In contrast, such a wide variability in the response is not observed for WT
channels with β1 subunits, which typically have maximum
Pos of >0.9, Hill coefficients of 3.5 or 4.5, and
Kds of 1.2 or 2.6 μM for data obtained at either +60 or
+30 mV, respectively (23,
26). Thus, the β1 subunit
only partially restored Ca2+ sensitivity to the TM
Ca2+-insensitive channel, as reflected by the decreased
Hill coefficient, greater Kd values, and inability to
consistently drive the Po to the maximal level of >0.9
observed for WT channels with β1 subunits
(23).
now greatly increased the
single-channel activity of the TM channel
(Fig. 1B). The degree
of restoration of Ca2+ sensitivity to the TM channel by
the β1 subunit was consistent for each channel but variable among
channels (data from 10 different experiments at +50 mV in
Fig. 1C), with maximum
Pos of 0.44 ± 0.10 (range 0.002–0.9), typical
Hill coefficients of 1.1 ± 0.1 (range 0.5–1.8), and
Kds of 38.1 ± 11.2 μM (range 14.7–108.2).
In contrast, such a wide variability in the response is not observed for WT
channels with β1 subunits, which typically have maximum
Pos of >0.9, Hill coefficients of 3.5 or 4.5, and
Kds of 1.2 or 2.6 μM for data obtained at either +60 or
+30 mV, respectively (23,
26). Thus, the β1 subunit
only partially restored Ca2+ sensitivity to the TM
Ca2+-insensitive channel, as reflected by the decreased
Hill coefficient, greater Kd values, and inability to
consistently drive the Po to the maximal level of >0.9
observed for WT channels with β1 subunits
(23).
The reason for the variability in the restoration of
Ca2+ sensitivity for the TM channels is not known, but
Wang et al. (36) have
shown for WT channels that decreasing the number of β1 subunits per
channel results in less activation for a given
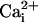 . Perhaps TM channels are
less likely than WT channels to have a full complement of four β1
subunits, giving rise to graded effects. The TM channels typically gate in
normal mode activity, so the variability in response is not likely to arise
from entry into other modes of gating (see ref.
33 for discussion of modes).
Although the reason for the variability is not known, the important point is
that the β1 subunit typically restored some Ca2+
sensitivity to the TM channels. The following sections explore possible
mechanisms for the restoration of Ca2+ sensitivity.
. Perhaps TM channels are
less likely than WT channels to have a full complement of four β1
subunits, giving rise to graded effects. The TM channels typically gate in
normal mode activity, so the variability in response is not likely to arise
from entry into other modes of gating (see ref.
33 for discussion of modes).
Although the reason for the variability is not known, the important point is
that the β1 subunit typically restored some Ca2+
sensitivity to the TM channels. The following sections explore possible
mechanisms for the restoration of Ca2+ sensitivity.
The Mutations Producing the TM Channel Do Not Facilitate the Action of
the β1 Subunit. Nimigean and Magleby
(23,
24) showed that the β1
subunit acts by increasing burst duration and Po by a
relatively fixed multiplicative amount in the absence and presence of
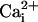 . If this multiplicative
effect were greatly facilitated in some manner by the three mutations
producing the TM channel, then an imperceptible Ca2+
sensitivity in the TM channel might become pronounced in the presence of the
β1 subunit. To test for this possibility we examined whether the β1
subunit in the absence of
. If this multiplicative
effect were greatly facilitated in some manner by the three mutations
producing the TM channel, then an imperceptible Ca2+
sensitivity in the TM channel might become pronounced in the presence of the
β1 subunit. To test for this possibility we examined whether the β1
subunit in the absence of 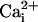 had a greater effect on burst duration and Po on the TM
channel than on the WT channel. We found that the β1 subunit increased
burst duration (5.2± 0.7-fold) and Po (8.7±
1.3-fold at +50 mV; n = 8) in the TM channel (example in
Fig. 1B, upper trace,
and results over a range of voltage in Fig.
1D), and that these effects were somewhat less than
reported for WT channels (24).
These results indicate that the mutations in the TM channel do not facilitate
the action of the β1 subunit on burst duration and
Po. If anything, the action of the β1 subunit on
burst duration and Po is reduced for the TM channel.
Consequently, the restoration of Ca2+ sensitivity to the
TM channel by the β1 subunit must arise by some other mechanism.
had a greater effect on burst duration and Po on the TM
channel than on the WT channel. We found that the β1 subunit increased
burst duration (5.2± 0.7-fold) and Po (8.7±
1.3-fold at +50 mV; n = 8) in the TM channel (example in
Fig. 1B, upper trace,
and results over a range of voltage in Fig.
1D), and that these effects were somewhat less than
reported for WT channels (24).
These results indicate that the mutations in the TM channel do not facilitate
the action of the β1 subunit on burst duration and
Po. If anything, the action of the β1 subunit on
burst duration and Po is reduced for the TM channel.
Consequently, the restoration of Ca2+ sensitivity to the
TM channel by the β1 subunit must arise by some other mechanism.
TM Channels with Restored Ca2+ Sensitivity Do Not Incorporate Subunits from Endogenous BK Channels. Endogenous BK channels have been reported at very low levels in Xenopus oocytes (37). Consequently, it is possible in our experiments where the β1 subunit restored Ca2+ sensitivity to the TM channels that either endogenous channels or endogenous subunits mixed with TM subunits contribute to the Ca2+ sensitivity. To check this possibility, we introduced a point mutation to the TEA binding site located at the outer mouth of the TM subunit. The subunit composition of the channels was then identified from the single-channel current amplitude in the presence of 1.5 mM external TEA (TEAo) (38, 39). For the conditions of these experiments (+50 mV, symmetrical 158 mM KCl), the single-channel current amplitude should be ≈13 pA without TEAo, and should be reduced to ≈9.3, 7.5, 4.0, 1.5, and 0 pA in the presence of 1.5 mM TEAo for channels with 4, 3, 2, 1, and 0 subunits having their TEA binding sites mutated (39).
When oocytes were injected with cRNA for TM channels with the TEA binding sites intact, the single-channel currents were 13 pA in the absence of TEAo and then were completely blocked with 1.5 mM TEAo (Fig. 2A), just as expected for channels with four intact TEA binding sites (38, 39). After injecting cRNA for TM channels with the TEA binding site mutated, the observed single-channel currents were ≈13 pA in the absence of TEAo and ≈9 pA with 1.5 mM TEAo (Fig. 2B), just what would be expected if all four subunits had the TEA binding site mutated (38, 39). Similar results were found in eight additional experiments with TEAo, where the single-channel amplitudes of the TM channels were ≈9 pA with 1.5 mM TEAo. This indicates that the channels being recorded from were homotetrameric channels in which each subunit had the pore mutation to reduce TEA sensitivity and consequently also had the TM. If one or more of the subunits were endogenous (without mutated TEA sites), then the single-channel currents should have been considerably less than 9 pA, as described above. Thus, the β1 subunit restores Ca2+ sensitivity to homotetrameric TM channels.
Fig. 2.

The β1 subunit restores the Ca2+ sensitivity to channels in which the four α subunits all have the TM, such that no endogenous α subunits from BK channels are present. (A) Single-channel currents from a TM channel in the presence of the β1 subunit are fully blocked by 1.5 mM TEAo, as indicated in the current amplitude histogram. Data are from the same outside-out patch. (B) Single-channel currents in the presence of the β1 subunit from a TM channel with the TEA binding sites mutated are partially blocked by 1.5 mM TEAo, reducing the current from ≈13 pA to ≈9 pA. Data are from the same outside-out patch. Similar results were obtained in eight additional experiments. If one or more of the subunits did not have the TEA site mutation, then the current would have been ≈7.5 pA or less (39).
The β1 Subunit Does Not Directly Provide a Ca2+ Binding Site. Another possible mechanism for the restoration of Ca2+ sensitivity to the TM channel is that the β1 subunit directly adds a novel Ca2+ binding site to the channel. To explore this possibility, we examined whether β1 subunits with the double mutation E13K and T14R to replace the only negative intracellular charge with two positive charges still restored Ca2+ sensitivity, because such charge alteration should destroy any potential Ca2+ binding site. After the double mutation E13K and T14R to the N terminus, we found that the β1 subunit still restored the Ca2+ sensitivity to the TM channel (Fig. 3). Thus, it is unlikely that the β1 subunit is directly adding a Ca2+ binding site to the TM channel.
Fig. 3.

The β1 subunit still restores Ca2+ sensitivity
to the TM channel after replacing the only intracellular negative charge on
the β1 subunit with a positive charge (E13K) and adding another adjacent
positive charge (T14R). (A) Representative single-channel currents
recorded from a TM channel in the presence of the doubly mutated β1
subunit. (B) Plots of Po vs.
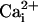 for TM channels with the
doubly mutated β1 subunit.
for TM channels with the
doubly mutated β1 subunit.
M513 Is Not Responsible for Restoring Ca2+ Sensitivity to the TM Channel by the β1 Subunit. In addition to the three sites (Ca2+ bowl, D362/D367, and E399) in the C terminus of the TM channel associated with Ca2+ sensitivity, an additional site, M513, has also been identified in the C terminus that greatly reduces Ca2+ sensitivity when mutated (8). The M513 site provides no Ca2+ sensitivity to the TM channel even though it has not been mutated, because the TM channel is Ca2+-insensitive (Fig. 1 A). Nevertheless, the possibility arises that the M513 site may be involved in the restoration of the Ca2+ sensitivity in the presence of the β1 subunit. To explore this possibility, we mutated the M513 site in the TM channel to obtain a quadra mutant channel and then examined whether the β1 subunit still restored Ca2+ sensitivity. The quadra mutant channel was found to be Ca2+-insensitive in the absence of the β1 subunit (Fig. 4A), just as the TM channel was (Fig. 1 A). The β1 subunit then restored Ca2+ sensitivity to the quadra mutant channel (Fig. 4B), just as it did to the TM channel (Fig. 1B). Thus, the M513 site is not responsible for restoring Ca2+ sensitivity to the TM channel by the β1 subunit.
Fig. 4.

The M513 site is not responsible for the restoration of the Ca2+ sensitivity to the TM channel by the β1 subunit. (A) Single-channel currents recorded from a Ca2+-insensitive quadra mutant channel made by adding the mutation M513I to the TM channel. (B) The β1 subunit restores Ca2+ sensitivity to the quadra mutant channel, +50 mV.
The β1 Subunit May Restore the Ca2+ Sensitivity by Restoring Coupling of Ca2+ Binding Sites to the Gating of the Channel. The mutations to specific sites in the TM and quadra mutant channels could remove the Ca2+ sensitivity by disrupting the Ca2+ binding sites or by disrupting the coupling between the Ca2+ sites and the gating machinery. If the mutations disrupt the Ca2+ binding sites, then the β1 subunit could act by directly restoring one or more of the mutated Ca2+ binding sites. If the mutations disrupt the coupling, then the β1 subunit could restore Ca2+ sensitivity by restoring the coupling between the Ca2+ binding sites and the gating machinery. It is unlikely that β1 subunits act directly on the Ca2+ binding sites, because the β1 subunit has little effect on either the Kd for high-affinity Ca2+ binding or the critical levels of Ca2+ required to first activate the WT channel (23, 26).
Consistent with the idea that the β1 subunit is unlikely to act through the Ca2+ binding sites, we found that the β1 subunit still increased burst duration and Po for WT channels in which each of the four sites in the quadra mutant channel were mutated individually (data not shown), similar to what we observed previously for a BK channel with a mutated Ca2+ bowl (27). Thus, the β1 subunit does not specifically require any one of the four sites identified on the C terminus to either increase the apparent Ca2+ sensitivity of the WT channel or to restore Ca2+ sensitivity to the triple or quadra mutant channel, suggesting that it may be acting on the coupling between the sites and the gating mechanism.
Nevertheless, it cannot be ruled out that the β1 subunit restores
Ca2+ sensitivity to the triple and quadra mutant
channels by unmasking additional Ca2+ binding sites on
either S0–S6 (40,
41) or the C terminus that are
silent in the triple and quadra mutant channels. A recent study
(41) has reported that BK
channels with the entire C terminus removed beyond S6 are still gated by
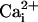 . In this case, then, the
triple and quadra mutations would have to disrupt the coupling between the
proposed Ca2+ sites on S0–S6 and the gating, and
the β1 subunit would then have to restore the coupling.
. In this case, then, the
triple and quadra mutations would have to disrupt the coupling between the
proposed Ca2+ sites on S0–S6 and the gating, and
the β1 subunit would then have to restore the coupling.
The β1 Subunit Does Not Restore
Ca2+ Sensitivity Through the Low-Affinity
Ca2+/Mg2+ Site.
To examine whether the β1 subunit might restore
Ca2+ sensitivity to the TM channel by recoupling the
low-affinity Ca2+/Mg2+ site to the
gating, we took advantage of the observation that
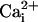 activates both the high-
and low-affinity sites, whereas
activates both the high-
and low-affinity sites, whereas
 activates only the
low-affinity E399 site (10,
12). If the restoration of the
Ca2+ sensitivity to the TM channel by the β1
subunit were through the low-affinity site, then the β1 subunit would be
expected to also restore Mg2+ sensitivity. If the
restoration of Ca2+ sensitivity were through a
Ca2+-specific high-affinity site, then the β1
subunit would not be expected to restore Mg2+
sensitivity.
activates only the
low-affinity E399 site (10,
12). If the restoration of the
Ca2+ sensitivity to the TM channel by the β1
subunit were through the low-affinity site, then the β1 subunit would be
expected to also restore Mg2+ sensitivity. If the
restoration of Ca2+ sensitivity were through a
Ca2+-specific high-affinity site, then the β1
subunit would not be expected to restore Mg2+
sensitivity.
We first verified that 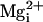 activates WT channels (10,
12). Increasing
activates WT channels (10,
12). Increasing
 from 1 to 5 to 10 mM
increased Po from 0.05 to 0.2 to 0.4 at 100 mV
(Fig. 5A). We also
verified (Fig. 5B)
that the Po of the TM channel is not sensitive to
from 1 to 5 to 10 mM
increased Po from 0.05 to 0.2 to 0.4 at 100 mV
(Fig. 5A). We also
verified (Fig. 5B)
that the Po of the TM channel is not sensitive to
 for
for
 ≤ 10 mM
(11). We then examined whether
the β1 subunit restored the lost Mg2+ sensitivity
to the TM channel. In contrast to the restoration of
Ca2+ sensitivity by the β1 subunit to the TM
channel (Fig. 1 B and
C), the β1 subunit did not restore the lost
Mg2+ sensitivity to the TM channel
(Fig. 5 C and
D). Hence, the β1 subunit does not restore
Ca2+ sensitivity through the low-affinity
Ca2+/Mg2+ activating
mechanism.
≤ 10 mM
(11). We then examined whether
the β1 subunit restored the lost Mg2+ sensitivity
to the TM channel. In contrast to the restoration of
Ca2+ sensitivity by the β1 subunit to the TM
channel (Fig. 1 B and
C), the β1 subunit did not restore the lost
Mg2+ sensitivity to the TM channel
(Fig. 5 C and
D). Hence, the β1 subunit does not restore
Ca2+ sensitivity through the low-affinity
Ca2+/Mg2+ activating
mechanism.
Fig. 5.

The β1 subunit does not restore the Mg2+
sensitivity to the TM channel, and it has little effect on the
Mg2+ sensitivity of WT BK channels. (A)
Single-channel currents recorded from WT channels at the indicated
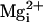 at +100 mV. (B
and C) Currents from TM channels in the absence (B) and presence
(C)ofthe β1 subunit. (D) Plots of
Po vs.
at +100 mV. (B
and C) Currents from TM channels in the absence (B) and presence
(C)ofthe β1 subunit. (D) Plots of
Po vs.
 for TM and WT channels
with and without the β1 subunit at +100 mV. (E) Currents from WT
channels in the absence and presence of the β1 subunit with 5.7 μM
for TM and WT channels
with and without the β1 subunit at +100 mV. (E) Currents from WT
channels in the absence and presence of the β1 subunit with 5.7 μM
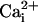 at –50 mV.
(F) Currents from WT channels in the absence and presence of the
β1 subunit with 0
at –50 mV.
(F) Currents from WT channels in the absence and presence of the
β1 subunit with 0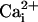 and
10 mM
and
10 mM 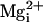 at +50 mV.
(G) Plots of Po vs. V from experiments
like those in E and F (n ≥ 5 for each point in
D and G). The open triangles typically obscure the filled
triangles.
at +50 mV.
(G) Plots of Po vs. V from experiments
like those in E and F (n ≥ 5 for each point in
D and G). The open triangles typically obscure the filled
triangles.
The β1 Subunit Has No Effect on Activation of the BK Channel
by Mg2+. If the β1 subunit does not act through
the low-affinity Ca2+/Mg2+
activating mechanism, as suggested in the previous section, then it might be
expected that the β1 subunit would not amplify the increased channel
activity induced by  for
WT channels, because Mg2+ activates only through the
low-affinity Ca2+/Mg2+ site
(9,
10,
12). This was found to be the
case. In contrast to the pronounced
(22–27)
large increase in burst duration and Po induced by the
β1 subunit for WT channels (example for 5.7 μM
for
WT channels, because Mg2+ activates only through the
low-affinity Ca2+/Mg2+ site
(9,
10,
12). This was found to be the
case. In contrast to the pronounced
(22–27)
large increase in burst duration and Po induced by the
β1 subunit for WT channels (example for 5.7 μM
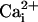 in
Fig. 5E), the β1
subunit had no effect on the burst duration and Po for WT
channels activated by 10 mM
in
Fig. 5E), the β1
subunit had no effect on the burst duration and Po for WT
channels activated by 10 mM
 (Fig. 5F). This
differential effect of the β1 subunit on facilitating the activation of
BK channels by
(Fig. 5F). This
differential effect of the β1 subunit on facilitating the activation of
BK channels by 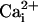 , but not
, but not
 , over a range of voltages
is shown in Fig. 5G.
With 5.7 μM
, over a range of voltages
is shown in Fig. 5G.
With 5.7 μM 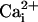 , the
β1 subunit shifted the Po vs. membrane potential
curve 75 mV to the left (Fig.
5G, from filled to open diamonds) so that less
depolarization was required to activate the channel. In contrast, with 10 mM
, the
β1 subunit shifted the Po vs. membrane potential
curve 75 mV to the left (Fig.
5G, from filled to open diamonds) so that less
depolarization was required to activate the channel. In contrast, with 10 mM
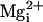 the β1 subunit had
no effect on the Po vs. V plots
(Fig. 5G, triangles).
A lack of effect of the β1 subunit on the Po vs.
V plots was also observed with 1, 5, 30, and 100 mM added
the β1 subunit had
no effect on the Po vs. V plots
(Fig. 5G, triangles).
A lack of effect of the β1 subunit on the Po vs.
V plots was also observed with 1, 5, 30, and 100 mM added
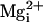 (Fig. 5D). Hence, in
contrast to the facilitating effect of the β1 subunit on burst duration
and Po in the presence of
(Fig. 5D). Hence, in
contrast to the facilitating effect of the β1 subunit on burst duration
and Po in the presence of
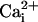 (22–27),
the β1 subunit had no effect on burst duration and Po
in the presence of
(22–27),
the β1 subunit had no effect on burst duration and Po
in the presence of 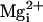 (Fig. 5 D, F, and
G).
(Fig. 5 D, F, and
G).
These results suggest that the β1 subunit does not act through the
low-affinity Ca2+/Mg2+ activating
mechanism, but rather acts through the high-affinity
Ca2+ activating mechanisms. This differential effect of
the β1 subunit on the high- and low-affinity activating mechanisms
supports the previous proposal that these mechanisms are separate and act
relatively independently to facilitate channel activity
(10,
12). This differential effect
also rules out the possibility that the β1 subunit acts at a common step
in the gating mechanism shared by the high- and low-affinity sites, for if it
did, the β1 subunit should have enhanced activation by both
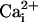 and
and
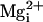 , rather than just
, rather than just
 . Since previous studies
have shown that the major action of the β1 subunit is not through changes
in the binding affinity of
. Since previous studies
have shown that the major action of the β1 subunit is not through changes
in the binding affinity of
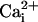 (23,
26), these results when taken
together with our observations suggest that the β1 subunit acts on the
allosteric linker at a location between the high-affinity
Ca2+ binding sites and any common elements in the
Ca2+ and Mg2+ gating
mechanisms.
(23,
26), these results when taken
together with our observations suggest that the β1 subunit acts on the
allosteric linker at a location between the high-affinity
Ca2+ binding sites and any common elements in the
Ca2+ and Mg2+ gating
mechanisms.
 Inhibits, Rather than Activates, BK Channels in the Presence of the β1
Subunit for Intermediate Levels of
Inhibits, Rather than Activates, BK Channels in the Presence of the β1
Subunit for Intermediate Levels of
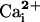 . Previous
studies in the absence of the β1 subunit have shown that
Mg2+ activates BK channels, with the maximal activation
at 0 and saturating
. Previous
studies in the absence of the β1 subunit have shown that
Mg2+ activates BK channels, with the maximal activation
at 0 and saturating  and
the minimal activation at intermediate levels of
and
the minimal activation at intermediate levels of
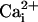 (10,
12). Since we found in the
experiments delineated in previous sections that the β1 subunit has
differential effects on the Ca2+ and
Mg2+ activating systems, we examined whether
Mg2+ still activates BK channels in the presence of the
β1 subunit.
(10,
12). Since we found in the
experiments delineated in previous sections that the β1 subunit has
differential effects on the Ca2+ and
Mg2+ activating systems, we examined whether
Mg2+ still activates BK channels in the presence of the
β1 subunit.
The key observation is shown in the single-channel records in
Fig. 6 A and
B. In the absence of the β1 subunit, adding 10 mM
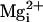 to 5.7 μM
to 5.7 μM
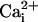 had minimal activating
effect on the channel. In the presence of the β1 subunit the activity
with 5.7 μM
had minimal activating
effect on the channel. In the presence of the β1 subunit the activity
with 5.7 μM  was
greatly increased (compare Fig. 6
B, upper trace, with A, upper trace), and adding
10 mM
was
greatly increased (compare Fig. 6
B, upper trace, with A, upper trace), and adding
10 mM 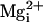 to the 5.7 μM
to the 5.7 μM
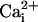 now greatly decreased
Po (Fig.
6B, lower trace). Results from experiments of this type
over a range of voltages and
now greatly decreased
Po (Fig.
6B, lower trace). Results from experiments of this type
over a range of voltages and
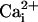 levels are presented in
Fig. 6 C and
D. In the absence of the β1 subunit with 5.7
μM
levels are presented in
Fig. 6 C and
D. In the absence of the β1 subunit with 5.7
μM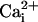 ,10mM
,10mM had a minimal activating effect on channel activity
(Fig. 6C, squares). In
the presence of the β1 subunit with 5.7 μM
had a minimal activating effect on channel activity
(Fig. 6C, squares). In
the presence of the β1 subunit with 5.7 μM
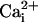 , 10 mM
, 10 mM
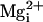 now inhibited channel
activity, as indicated by the 40 mV rightward shift in the
Po vs. V curves
(Fig. 6D, from filled
to open squares). In contrast to this inhibition by 10 mM
Mg2+ in the presence of the β1 subunit with 5.7
μM
now inhibited channel
activity, as indicated by the 40 mV rightward shift in the
Po vs. V curves
(Fig. 6D, from filled
to open squares). In contrast to this inhibition by 10 mM
Mg2+ in the presence of the β1 subunit with 5.7
μM 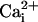 ,
,
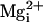 greatly increased channel
activity with 0 or 100 μM
greatly increased channel
activity with 0 or 100 μM
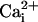 , shifting the
Po vs. V plots 50–60 mV to the left, and
this was the case in the presence (Fig.
6D) and absence (Fig.
6C) of the β1 subunit.
, shifting the
Po vs. V plots 50–60 mV to the left, and
this was the case in the presence (Fig.
6D) and absence (Fig.
6C) of the β1 subunit.
Fig. 6.

In the presence of the β1 subunit,
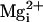 inhibits the activity of
WT channels for intermediate levels of
inhibits the activity of
WT channels for intermediate levels of
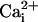 . (A)
Single-channel currents recorded from WT channels in the absence of the
β1 subunit at 5.7
μM
. (A)
Single-channel currents recorded from WT channels in the absence of the
β1 subunit at 5.7
μM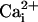 without and with 10
mM
without and with 10
mM 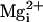 at –30 mV.
(B) As in A, except for the presence of the β1 subunit.
(C) Plots of Po vs. V for WT channels
with the β1 subunit with 0, 5.7, and 100
μM
at –30 mV.
(B) As in A, except for the presence of the β1 subunit.
(C) Plots of Po vs. V for WT channels
with the β1 subunit with 0, 5.7, and 100
μM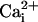 , each
with and without 10 mM
, each
with and without 10 mM  .
(D)Asin C, except for the presence of the β1 subunit.
(E) Plots of the voltage for half activation,
V0.5, vs.
.
(D)Asin C, except for the presence of the β1 subunit.
(E) Plots of the voltage for half activation,
V0.5, vs.
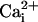 for WT channels with and
without the β1 subunit, as indicated. (n ≥ 3 for each point
in C–E.)
for WT channels with and
without the β1 subunit, as indicated. (n ≥ 3 for each point
in C–E.)
To define the range of 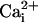 levels over which Mg2+ activates and inhibits the BK
channel in the absence and presence of the β1 subunit, we performed
experiments like those shown in Fig. 6
A–D for additional
levels over which Mg2+ activates and inhibits the BK
channel in the absence and presence of the β1 subunit, we performed
experiments like those shown in Fig. 6
A–D for additional
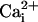 , and we plotted the
results as V0.5, the voltage required for half activation
of the channel, vs.
, and we plotted the
results as V0.5, the voltage required for half activation
of the channel, vs. 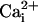 . The
more negative the V0.5, the greater the activity of the
channel at a fixed voltage. In the absence of the β1 subunit, 10 mM
Mg2+ always activated the channel, but with a negligible
effect at 5.7 μM
. The
more negative the V0.5, the greater the activity of the
channel at a fixed voltage. In the absence of the β1 subunit, 10 mM
Mg2+ always activated the channel, but with a negligible
effect at 5.7 μM 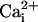 (Fig. 6E). In the
presence of the β1 subunit, 10 mM Mg2+ inhibited
channel activity for
(Fig. 6E). In the
presence of the β1 subunit, 10 mM Mg2+ inhibited
channel activity for 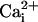 ranging from ≈2 to 30 μM (Fig.
6E, shaded area) and activated the channel for
Ca2+ <2 μM or >30 μM. This plot clearly
shows that the β1 subunit has differential effects on the
Ca2+ and Mg2+ activating
mechanisms.
ranging from ≈2 to 30 μM (Fig.
6E, shaded area) and activated the channel for
Ca2+ <2 μM or >30 μM. This plot clearly
shows that the β1 subunit has differential effects on the
Ca2+ and Mg2+ activating
mechanisms.
The reason for the inhibition by
 in the presence of the
β1 subunit is not clear, but it has been proposed that
Mg2+ has two effects on channel activity: activation of
the channel through the low-affinity Mg2+ sites, and
antagonism of the Ca2+-dependent activation of the
channel by displacement of Ca2+ from the high-affinity
Ca2+ sites
(10,
12). Whether the net result of
Mg2+ is activation or inhibition would depend on the
relative contributions of the low-affinity Mg2+ site and
high-affinity Ca2+ sites to the activation. On this
basis, our observations can be explained by this hypothesis as follows. At
very low or very high
in the presence of the
β1 subunit is not clear, but it has been proposed that
Mg2+ has two effects on channel activity: activation of
the channel through the low-affinity Mg2+ sites, and
antagonism of the Ca2+-dependent activation of the
channel by displacement of Ca2+ from the high-affinity
Ca2+ sites
(10,
12). Whether the net result of
Mg2+ is activation or inhibition would depend on the
relative contributions of the low-affinity Mg2+ site and
high-affinity Ca2+ sites to the activation. On this
basis, our observations can be explained by this hypothesis as follows. At
very low or very high 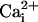 ,
Mg2+ would not displace significant
Ca2+ from the high-affinity sites, so that the
activation by Mg2+ on the Mg2+
sites would be readily apparent (Figs.
5 and
6). At intermediate levels of
,
Mg2+ would not displace significant
Ca2+ from the high-affinity sites, so that the
activation by Mg2+ on the Mg2+
sites would be readily apparent (Figs.
5 and
6). At intermediate levels of
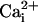 ,
Mg2+ would displace Ca2+ from the
high-affinity sites, decreasing the activation by
,
Mg2+ would displace Ca2+ from the
high-affinity sites, decreasing the activation by
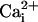 . In the absence of the
β1 subunit, adding 10 mM Mg2+ to 5.7 μM
. In the absence of the
β1 subunit, adding 10 mM Mg2+ to 5.7 μM
 would have little effect
because the reduced activation by
would have little effect
because the reduced activation by
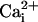 due to the displacement
of the
due to the displacement
of the  from the
high-affinity sites by Mg2+ would be about equal to the
increased activation by
from the
high-affinity sites by Mg2+ would be about equal to the
increased activation by  (Fig. 6 A, C, and
E) (10,
12). In the presence of the
β1 subunit, which greatly facilitates the activation of the channel by
(Fig. 6 A, C, and
E) (10,
12). In the presence of the
β1 subunit, which greatly facilitates the activation of the channel by
 but not by
but not by
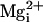 (Fig. 5), adding 10 mM
(Fig. 5), adding 10 mM
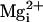 to 5.7
μM
to 5.7
μM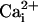 would now have an
inhibitory effect (Fig. 6 B, D,
and E), because the reduced activation by
would now have an
inhibitory effect (Fig. 6 B, D,
and E), because the reduced activation by
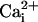 due to displacement of
the
due to displacement of
the 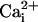 from the
high-affinity sites by
from the
high-affinity sites by 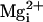 would now be greater than the activation by
would now be greater than the activation by
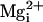 .
.
Because intracellular free Mg2+ can increase to
2–4 mM with ischemia
(42,
43), such an increased
 in the presence of the
β1 subunit could decrease the activation of BK channels for intermediate
levels of
in the presence of the
β1 subunit could decrease the activation of BK channels for intermediate
levels of 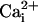 , decreasing the
protective ability of BK channels to prevent depolarization. This inhibitory
effect of Mg2+ on BK channels in the presence of the
β1 subunit would act through changes in gating and would be in addition
to the inhibitory effect of Mg2+ on the conductance
described previously (1,
42,
44).
, decreasing the
protective ability of BK channels to prevent depolarization. This inhibitory
effect of Mg2+ on BK channels in the presence of the
β1 subunit would act through changes in gating and would be in addition
to the inhibitory effect of Mg2+ on the conductance
described previously (1,
42,
44).
Conclusions. (i) Our results suggest that
Ca2+ and Mg2+ activate BK channels
through separate mechanisms, with the β1 subunit acting through the
Ca2+ activating mechanism rather than the
Mg2+ activating mechanism. The action of the β1
subunit appears not to be on the Ca2+ binding sites, but
on the allosteric machinery that couples the Ca2+ site
to the opening–closing transition. (ii) It has previously been
shown that  only
facilitates the action of BK channels
(10,
12). Here we show that, in the
presence of the β1 subunit,
only
facilitates the action of BK channels
(10,
12). Here we show that, in the
presence of the β1 subunit,
 can also inhibit the
activity of BK channels for physiologically relevant levels of
can also inhibit the
activity of BK channels for physiologically relevant levels of
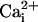 . This inhibition results
from the fact that the β1 subunit has differential effects on the
Ca2+ and Mg2+ activating
mechanisms.
. This inhibition results
from the fact that the β1 subunit has differential effects on the
Ca2+ and Mg2+ activating
mechanisms.
Acknowledgments
This work was supported by National Institutes of Health Grant AR32805, Florida Biomedical Research Program Grant BM029 (both to K.L.M.), and a fellowship from the Florida Affiliate of the American Heart Association (to X.Q.).
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: BK, large conductance Ca2+ and voltage-activated K+; TEA, tetraethylammonium; TEAo, external TEA; TM, triple mutant.
References
- 1.Golowasch, J., Kirkwood, A. & Miller, C. (1986) J. Exp. Biol. 124, 5–13. [DOI] [PubMed] [Google Scholar]
- 2.Horrigan, F. T. & Aldrich, R. W. (2002) J. Gen. Physiol. 120, 267–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Horrigan, F. T., Cui, J. & Aldrich, R. W. (1999) J. Gen. Physiol. 114, 277–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Horrigan, F. T. & Aldrich, R. W. (1999) J. Gen. Physiol. 114, 305–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rothberg, B. S. & Magleby, K. L. (2001) Biophys. J. 80, 3025–3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rothberg, B. S. & Magleby, K. L. (2000) J. Gen. Physiol. 116, 75–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rothberg, B. S. & Magleby, K. L. (1999) J. Gen. Physiol. 114, 93–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bao, L., Rapin, A. M., Holmstrand, E. C. & Cox, D. H. (2002) J. Gen. Physiol. 120, 173–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shi, J., Krishnamoorthy, G., Yang, Y., Hu, L., Chaturvedi, N., Harilal, D., Qin, J. & Cui, J. (2002) Nature 418, 876–880. [DOI] [PubMed] [Google Scholar]
- 10.Shi, J. & Cui, J. (2001) J. Gen. Physiol. 118, 589–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xia, X. M., Zeng, X. & Lingle, C. J. (2002) Nature 418, 880–884. [DOI] [PubMed] [Google Scholar]
- 12.Zhang, X., Solaro, C. R. & Lingle, C. J. (2001) J. Gen. Physiol. 118, 607–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brenner, R., Perez, G. J., Bonev, A. D., Eckman, D. M., Kosek, J. C., Wiler, S. W., Patterson, A. J., Nelson, M. T. & Aldrich, R. W. (2000) Nature 407, 870–876. [DOI] [PubMed] [Google Scholar]
- 14.Pluger, S., Faulhaber, J., Furstenau, M., Lohn, M., Waldschutz, R., Gollasch, M., Haller, H., Luft, F. C., Ehmke, H. & Pongs, O. (2000) Circ. Res. 87, E53–E60. [DOI] [PubMed] [Google Scholar]
- 15.Petkov, G. V., Bonev, A. D., Heppner, T. J., Brenner, R., Aldrich, R. W. & Nelson, M. T. (2001) J. Physiol. 537, 443–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fettiplace, R. & Fuchs, P. A. (1999) Annu. Rev. Physiol 61, 809–834. [DOI] [PubMed] [Google Scholar]
- 17.Robitaille, R. & Charlton, M. P. (1992) J. Neurosci. 12, 297–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang, Z. W., Saifee, O., Nonet, M. L. & Salkoff, L. (2001) Neuron 32, 867–881. [DOI] [PubMed] [Google Scholar]
- 19.Weiger, T. M., Holmqvist, M. H., Levitan, I. B., Clark, F. T., Sprague, S., Huang, W. J., Ge, P., Wang, C., Lawson, D., Jurman, M. E., et al. (2000) J. Neurosci. 20, 3563–3570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tanaka, Y., Meera, P., Song, M., Knaus, H. G. & Toro, L. (1997) J. Physiol. 502, 545–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garcia-Calvo, M., Knaus, H. G., McManus, O. B., Giangiacomo, K. M., Kaczorowski, G. J. & Garcia, M. L. (1994) J. Biol. Chem. 269, 676–682. [PubMed] [Google Scholar]
- 22.McManus, O. B., Helms, L. M., Pallanck, L., Ganetzky, B., Swanson, R. & Leonard, R. J. (1995) Neuron 14, 645–650. [DOI] [PubMed] [Google Scholar]
- 23.Nimigean, C. M. & Magleby, K. L. (1999) J. Gen. Physiol. 113, 425–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nimigean, C. M. & Magleby, K. L. (2000) J. Gen. Physiol. 115, 719–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ramanathan, K., Michael, T. H. & Fuchs, P. A. (2000) J. Neurosci. 20, 1675–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cox, D. H. & Aldrich, R. W. (2000) J. Gen. Physiol. 116, 411–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Qian, X., Nimigean, C. M., Niu, X., Moss, B. L. & Magleby, K. L. (2002) J. Gen. Physiol. 120, 829–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schreiber, M. & Salkoff, L. (1997) Biophys. J. 73, 1355–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bian, S., Favre, I. & Moczydlowski, E. (2001) Proc. Natl. Acad. Sci. USA 98, 4776–4781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Butler, A., Tsunoda, S., McCobb, D. P., Wei, A. & Salkoff, L. (1993) Science 261, 221–224. [DOI] [PubMed] [Google Scholar]
- 31.Hamill, O. P., Marty, A., Neher, E., Sakmann, B. & Sigworth, F. J. (1981) Pflugers Arch. 391, 85–100. [DOI] [PubMed] [Google Scholar]
- 32.Yang, X. C. & Sachs, F. (1989) Science 243, 1068–1071. [DOI] [PubMed] [Google Scholar]
- 33.McManus, O. B. & Magleby, K. L. (1988) J. Physiol. 402, 79–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Meera, P., Wallner, M., Jiang, Z. & Toro, L. (1996) FEBS Lett. 385, 127–128. [DOI] [PubMed] [Google Scholar]
- 35.McManus, O. B., Blatz, A. L. & Magleby, K. L. (1987) Pflugers Arch. 410, 530–553. [DOI] [PubMed] [Google Scholar]
- 36.Wang, Y. W., Ding, J. P., Xia, X. M. & Lingle, C. J. (2002) J. Neurosci. 22, 1550–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Krause, J. D., Foster, C. D. & Reinhart, P. H. (1996) Neuropharmacology 35, 1017–1022. [DOI] [PubMed] [Google Scholar]
- 38.Shen, K. Z., Lagrutta, A., Davies, N. W., Standen, N. B., Adelman, J. P. & North, R. A. (1994) Pflugers Arch. 426, 440–445. [DOI] [PubMed] [Google Scholar]
- 39.Niu, X. & Magleby, K. L. (2002) Proc. Natl. Acad. Sci. USA 99, 11441–11446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Braun, A. F. & Sy, L. (2001) J. Physiol. 533, 681–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Piskorowski, R. & Aldrich, R. W. (2002) Nature 420, 499–502. [DOI] [PubMed] [Google Scholar]
- 42.Wachter, C. & Turnheim, K. (1996) J. Membr. Biol. 150, 275–282. [DOI] [PubMed] [Google Scholar]
- 43.Li, H. Y., Dai, L. J. & Quamme, G. A. (1993) J. Lab. Clin. Med. 122, 260–272. [PubMed] [Google Scholar]
- 44.Ferguson, W. B. (1991) J. Gen. Physiol. 98, 163–181. [DOI] [PMC free article] [PubMed] [Google Scholar]